
Abstract
The interactions between Emiliania huxleyi and E. huxleyi virus (EhV) regulate marine carbon and sulfur biogeochemical cycles and play a prominent role in global climate change. As a large DNA virus, EhV has developed a novel “virocell metabolism” model to meet its high metabolic needs. Although it has been widely demonstrated that EhV infection can profoundly rewire lipid metabolism, the epigenetic regulatory mechanisms of lipid metabolism are still obscure. MicroRNAs (miRNAs) can regulate biological pathways by targeting hub genes in the metabolic processes. In this study, the transcriptome, lipidome, and miRNAome were applied to investigate the epigenetic regulation of lipid metabolism in E. huxleyi cells during a detailed time course of viral infection. Combined transcriptomic, lipidomic, and physiological experiments revealed reprogrammed lipid metabolism, along with mitochondrial dysfunction and calcium influx through the cell membrane. A total of 69 host miRNAs (including 1 known miRNA) and 7 viral miRNAs were identified, 27 of which were differentially expressed. Bioinformatic prediction revealed that miRNAs involved in the regulation of lipid metabolism and a dual-luciferase reporter assay suggested that phosphatidylinositol 3-kinase (PI3K) gene might be a target of ehx-miR5. Further qPCR and western blot analysis showed a significant negative correlation between the expression of ehx-miR5 and its target gene PI3K, along with the lower activity of its downstream components (p-Akt, p-TOR, SREBP), indicating that lipid metabolism might be regulated by ehx-miR5 through the PI3K-Akt-TOR signaling pathway. Our findings reveal several novel mechanisms of viral strategies to manipulate host lipid metabolism and provide evidence that ehx-miR5 negatively modulates the expression of PI3K and disturbs lipid metabolism in the interactions between E. huxleyi and EhV.
Similar content being viewed by others


Introduction
The coccolithophore Emiliania huxleyi frequently forms large oceanic algal blooms in subpolar waters, which are significant contributors to the global production and export of calcium carbonate (calcite) [1,2,3]. The collapse of massive E. huxleyi blooms is frequently terminated by the infection of a specific large double-stranded DNA (dsDNA) virus (E. huxleyi virus, EhV) [4, 5]. E. huxleyi–EhV interactions therefore play a profound role in determining carbon flow in the ocean and global climate processes [6,7,8]. As a relatively large dsDNA virus, EhV has evolved complex and dynamic interactions with its host cells [9] by which it can reshape its host’s metabolic network, generating the virocell metabolic state that supports its specific requirement [10]. For example, EhV depends on host metabolic networks to supply essential building blocks such as amino acids, nucleotides, and fatty acids (FAs) for the production of new virions [6, 11, 12]. Recent studies demonstrated that, in addition to being passive consumers of host metabolic products, EhVs could expand the metabolic capabilities of their hosts by introducing virus-encoded auxiliary metabolic genes (vAMGs) to support their specific requirements [10]. In particular, virus-encoded enzymes induce a metabolic switch in sphingolipid metabolism to synthesize unique virus-specific glycosphingolipids, which are required for virus assembly and egress [13, 14]. The realization of these multiple metabolic strategies in the virocells may involve the regulation of gene expression at multiple levels, ranging from transcriptional, posttranscriptional, translational, to posttranslational. The regulation of transcriptional level is the principal regulating means of gene expression; however, posttranscriptional (such as via microRNAs (miRNAs)) regulation plays an important role in the process of gene expression.
miRNAs are small noncoding RNAs that are involved in posttranscriptional regulation of their target genes in a sequence-specific manner [15]. As one of the most abundant classes of gene regulators, miRNAs are involved in many biological processes, such as cell proliferation, tumorigenesis, apoptosis, and inflammation [16, 17]. Furthermore, miRNAs control plant development, growth, stress adaptation, and other physiological processes [18, 19]. Recent studies have indicated that miRNAs also play novel roles in regulating metabolic pathways through the control of target genes involved in metabolic processes, and especially in controlling lipid and glucose metabolism [20, 21]. Emerging evidence demonstrates that miRNAs are critical regulators of lipid synthesis, FA oxidation, and lipoprotein formation and secretion [22,23,24,25]. Dysregulation of miRNAs disrupts the gene regulatory network, leading to metabolic syndrome and its related diseases in human beings [26]. To date, more than 30 miRNAs in mammalian cells have been identified to play crucial roles in the regulation of lipid metabolism through various pathways [21], such as FA oxidation, FA biosynthesis, cholesterol, and triacylglycerol [17, 25, 27]. In marine phytoplankton, miRNAs are involved in regulating phosphorus utilization in the dinoflagellate Prorocentrum donghaiense in response to phosphorus stress [28]. Moreover, both in higher animals and plants, miRNAs have been shown to play an important role in the virus-host arms race, including cellular antiviral responses and/or in the promotion of viral infection through complex regulatory pathways [29, 30]. This regulatory network represents a new and central role for miRNAs in lipid and antiviral regulation. Besides, it has been confirmed that viruses can also encode miRNAs. miRNAs are particularly useful tools for DNA viruses since they are nonimmunogenic for host cells, and thus miRNAs can potentially manipulate host cells without evoking host immune responses [30]. Zhang et al. [31] firstly reported the existence of miRNAs in the coccolithophore E. huxleyi, indicating that this form of posttranscriptional regulation might be a major mechanism controlling gene expression. Indeed, it has been found that miRNAs can be produced in the form of extracellular vesicles during EhV infection, which might serve as signal molecules to target sphingolipid metabolism and cell-cycle pathways [32]. To date, the regulatory roles of both host and viral miRNAs in metabolic pathways (especially in lipid metabolism) in the E. huxleyi–EhV system are largely unexplored.
In this study, we combined lipidome, transcriptome, and miRNAome analysis in E. huxleyi (BOF92) and its specific lytic virus (EhV99B1), a marine model host–virus system. Our data provide the most comprehensive insights to date into how EhVs employ diverse strategies to manipulate host lipid metabolism with some novel mechanisms while demonstrating a potential miRNA regulatory mechanism of lipid metabolism in the E. huxleyi–EhV system.
Materials and methods
Algal culture and viral infection
The E. huxleyi strain BOF92 and EhV (EhV99B1) used in this study were isolated from the west coast of Norway (60°24′N, 5°19′E). Cultures of E. huxleyi BOF92 were grown in f/2-Si medium at 16 °C with a 14:10 h light/dark illumination cycle. A light intensity of ~100 μM photons·m−2·s−1 was provided by cool white fluorescent lights. Exponentially growing cells (~7.5 × 105 cell·mL−1) were infected with a 1:50 volumetric ratio of virus lysate to culture (multiplicity of infection of ~1:1 viral particle per cell).
Sample collection
At ca. 0–96 h post inoculation (hpi), samples (2 mL) for algal and viral counts were fixed with 1.0% and 0.5% glutaraldehyde (final concentration) respectively, frozen in liquid nitrogen, and stored at −80 °C until analysis. At ca. 0, 6, 12, 24, 48, and 60 hpi, i.e., the early, middle, and late stages in the second lytic cycle when the majority of the cells were infected, 100 mL of the cultures was harvested by centrifugation at 3000 rpm for 10 min at 4 °C, and the cell pellets were fixed (4% formaldehyde + 1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4) for 4 h at 4 °C for sample preparation for transmission electron microscopy (TEM). The cell pellets of 1000 mL of cultures at the time points indicated were collected for RNA isolation, protein extraction, biochemical experiments (three biological replicates), and total lipids isolation (six biological replicates).
RNA isolation, library construction, and sequencing
Total RNA was extracted using TRIzol Reagent kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA quality was assessed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and the RNA integrity numbers of the samples were all above 6.0. Then, these isolated RNAs were used for sequencing and quantitative PCR analysis. Equal amounts of RNA masses extracted from the triplicate samples were pooled for RNA sequencing.
A total of 3 μg of RNA was used to generate paired-end RNA-seq library using NEBNext Ultra II Directional RNA Second Strand Synthesis Module (NEB, Ipswich, MA, USA). Small RNA libraries were generated using NEBNext Multiplex Small RNA Library Prep Set for Illumina (NEB, Ipswich, MA, USA) with the same amount of total RNA mass as used in RNA-seq. The libraries were purified (AMPure XP system), quantified using the Agilent Bioanalyzer 2100 system, and then sequenced using HiSeq 4000 and HiSeq 2500 for mRNA and small RNA sequencing, respectively. The raw data were deposited to NCBI in SRA (BioProject accession number: PRJNA705400). The Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GJZP00000000. The version described in this paper is the first version, GJZP01000000. The library preparation and deep sequencing were performed by Genedenovo Biotechnology Co., Ltd (Guangzhou, China).
Bioinformatics analysis
Raw reads for RNA-seq and miRNA sequencing were cleaned up by trimming adaptor sequences, removing poly-N-containing reads, and filtering low-quality reads (Q score ≤20 for ≥50% of nucleotides in each read) using fastp (version 0.18.0) [33]. All downstream analyses were based on clean data with high quality.
Host transcriptome de novo assembly was carried out using the short read assembly program Trinity [34], with the two sets of sequence reads as the input. Basic annotation of unigenes included protein functional annotation, pathway annotation, KOG functional annotation, and Gene Ontology (GO) annotation using the BLASTx program with an E-value threshold of 1e–5. Protein functional annotations could then be obtained according to the best alignment results. The gene abundances were calculated and normalized to reads per kilobase per million mapped reads (RPKM) [35]. For the assembly of viral transcriptome, an index of the reference genome was built, and paired-end clean reads were mapped to the reference genome (https://www.ncbi.nlm.nih.gov/nuccore/FN429076.1) using HISAT v2.2.4 [36] with “-rna-strandness RF” and other parameters set as a default. The mapped reads of each sample were assembled by using StringTie v1.3.1 [37, 38] in a reference-based approach. For each transcription region, a fragment per kilobase of transcript per million mapped reads value was calculated to quantify its expression abundance and variations. RNA differential expression analysis was performed by DESeq2 software [39] between two different groups. Genes with a false discovery rate below 0.05 and absolute fold change (FC) ≥2 were considered differentially expressed (DE) genes.
For the miRNA sequencing dataset, reads less than 18 nt in length were removed. All clean tags were aligned with small RNAs in the GenBank database [40] (Release 209.0) and Rfam database [41] (Release 11.0) to identify and remove rRNA, scRNA, snoRNA, snRNA, and tRNA. To avoid omitting the miRNAs located in intergenic regions or introns, all clean tags were aligned with both the genome of E. huxleyi CCMP1516 (https://www.ncbi.nlm.nih.gov/assembly/GCF_000372725.1) and the de novo transcriptome. The tags mapped to repeat sequences were removed. All clean tags were then searched against the miRBase database [42] (Release 22) to identify existing miRNAs. To date, the miRNA sequences of some species of phytoplankton have not been included in the miRBase database. The miRNA alignment with other species was a dependable way to identify the known miRNAs. According to their miRNA positions and hairpin structures predicted by Mireap_v0.2 software [43], novel miRNA candidates were identified. The default parameters of Mireap_v0.2 software were as follows: miRNA sequence length, 18–25 nt; miRNA reference sequence length, 20–23 nt; maximal copy number of miRNAs on reference, 20; maximal free energy allowed for a miRNA precursor, 18 kcal/mol; space between miRNA and miRNA*, 16–300 nt; maximal bulge between miRNA and miRNA*, 4 nt; maximal asymmetry of miRNA/miRNA* duplex, 4 nt; flank sequence length of miRNA precursor, 20 nt. The miRNA expression level was calculated and normalized to transcripts per million. After normalization, the miRNA expression profiles between the two miRNA libraries were compared. Differential expression analysis of two conditions was performed using the DESeq2. DE miRNAs were defined as those with an FC ≥2 and p value <0.05. Based on the sequences of the known miRNAs and novel miRNAs, the CDS and 3′ UTR sequence of each unigene transcript were used to predict both animal- and plant-type targets. miRanda [44] (miRanda-3.3a, -sc 140 -en -15 -strict) was used to predict targets with animal miRNA binding characteristics, while plant-type targets were predicted by patmatch software [45] (Version 1.2) with default parameters. The targets with Pearson correlation >−0.3 with their miRNAs were not included in the subsequent analysis.
Based on the high correlation of genes, gene modules were divided by weighted gene coexpression network analysis (WGCNA) [46], a systems biology method for describing the correlation patterns among genes across multiple samples. The DE unigenes expressing in more than half (nine) samples and with relatively high expression levels (the sum of RPKM >18) were retained, getting 26,473 genes for subsequent analysis. After filtering genes, gene expression values were imported into WGCNA, an R package [46], to construct coexpression modules using the automatic network construction function blockwiseModules with default settings, except that the power was 16 and minModuleSize was 100.
Pathway enrichment analysis identified significantly enriched metabolic pathways or signal transduction pathways in the analyzed genes compared with the whole genome background. All the module genes and miRNA target genes were considered in our analysis. The formula used to calculate the p value is based on the hypergeometric distribution test. The calculated p value was corrected by Q value, taking Q ≤ 0.05 as a threshold. Pathways meeting this condition were defined as significantly enriched pathways.
Lipidomics analysis
Nontargeted lipidomics analysis was performed with an ACQUITY ultra-performance liquid chromatograph (Waters, USA) coupled with a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific, USA). The methods for sample preparation, equipment, and mass spectrometry (MS) analysis were based on our previously published article [12]. The lipid internal standards utilized in this study are provided in the Supplementary information.
For data processing, the identification of lipid species was performed based on accurate m/z, tandem mass spectrometry (MS/MS) fragmentation patterns, and retention behavior using the LIPID MAPS database, and LipidSearch and Xcalibur software (Thermo, USA). Then, the lipids were quantified at an m/z tolerance of ±5 ppm and a retention time extraction window of ±15 s using Trace Finder software (Thermo, USA), and normalized to the peak areas of internal standards. Quality evaluation of lipid profiling and multivariate analysis with unit variance scaling were performed by Simca (v14.1) software (Umetrics, Sweden). Heatmaps for lipid profiles were generated and visualized by the R program. Lipid metabolic pathways were analyzed with reference to the LIPID MAPS and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. The lipidomics data have been deposited to the EMBL-EBI MetaboLights database [47] with the identifier MTBLS5178. Abbreviations for lipid classes used in this study are as follows: FA, fatty acid; OAHFA, (O-acyl)−1-hydroxy fatty acid; MG, monoglyceride; MGDG, monogalatosyl diglyceride; DG, diglyceride; TG, triacylglycerol; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; PA, phosphatidic acid; PI, phosphatidylinositol; PS, phosphatidylserine; CL, cardiolipin; Cer, ceramide; SM, sphingomyelin; GSL, glycosphingolipid; WE, wax ester; and CmE, cholesterol.
Quantitative real-time PCR
Reverse transcription quantitative PCR (qRT-PCR) was performed to determine the transcriptional levels of several genes involved in lipid metabolism. The primers used for qPCR are listed in Supplementary Table S1 (mRNAs) and Supplementary Table S2 (miRNAs). For miRNA qRT-PCR, a miRcute Plus miRNA First-Strand cDNA Kit (TIANGEN, Beijing, China) was used to facilitate cDNA synthesis. Then, the RT product was used as the template for qPCR. All qPCRs were performed on a Roche LightCycler 480II/96 Real-time PCR System (Roche, Switzerland) using Universal SYBR Green Supermix (Vazyme, Nanjing, China) in 96-well plates according to the manufacturer’s recommendations. U6 snRNA and CDKA [48] were used as references to calibrate the expression of the miRNAs and mRNA, respectively. Two technical and three biological replicates were conducted for both viral infection and control culture samples. The comparative threshold (2−ΔΔCt) [49] method was used to assess the relative expression levels.
Dual-luciferase reporter assay
Dual-luciferase reporter assays were performed using recombinant vectors carrying either the wild-type or mutated sequence of the phosphatidylinositol 3-kinase (PI3K) gene cotransfected into HepG2 cells with miRNA or NC mimics. The complete CDS of PI3K containing the putative binding sites of ehx-miR5 was cloned into the psi-CHECK2 vector to obtain the wild-type vectors. Mutated vectors were obtained via the method by which mutations were prepared in the seed sequences where ehx-miR5 was predicted to be completely complementary with it on the wild-type vector. All constructs were verified by DNA sequencing. For the transfection experiments, HepG2 cells were cultured in 24-well plates with 500 μL of medium and grown to approximately 70% confluence. HepG2 cells were cotransfected with 600 µg recombinant vectors and 20 pmol of miRNA mimics (or NC mimics) by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Cells transfected with recombinant vectors alone were employed as blank group. After 48 h of transfection, the cells were prepared for detecting relative luciferase activity following the protocols of the Dual-Luciferase Reporter Assay Kit (Vazyme, Nanjing, China). Each trial contained three replicates, and the experiments were repeated twice.
Western blotting
A standard western blotting protocol was used as described previously [50]. Briefly, control and virus-infected cells were lysed in cell lysis buffer. The concentration of total protein was determined using a BCA Protein Assay Reagent kit (SuperSignal West Dura Trial kit, Pierce, Covance, USA). Equal amounts (40–60 μg) of protein were electrophoresed, transferred to nitrocellulose membranes, and incubated with the primary antibody. Peroxidase-conjugated secondary antibody and a WesternBright ECL western blotting detection kit (Advansta, Menlo Park, CA, USA) were used to detect the signals with the Western Blotting System and analyzed with the Azure c400 Imaging System (Azure Biosystems, USA). In addition, subcellular fractionation experiments were used to estimate the expression of the molecules in the nuclear fraction including lipin 1 and sterol regulatory element-binding protein (SREBP). Nucleoproteins were separated from cells using the nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China) (specific procedures are shown in the Supplementary information). CDKA and histone 3 (H3) were used as the loading controls. The primary antibodies used are shown in Supplementary Table S3.
Statistical analysis
The results are displayed as the mean and standard deviation from at least three independent experiments. Statistical analysis was performed by Student’s t test (for two groups) or analysis of variance (for more than two groups) followed by SNK’s or Duncan’s tests, and a p value or corrected p value less than 0.05 was considered statistically significant. All analyses were conducted in SPSS 25.0.
Results and discussion
Infection dynamics of E. huxleyi and neutral lipid accumulation
We exposed E. huxleyi BOF92 to its lytic virus EhV99B1 and followed its dynamics over the time course of infection. Cultures infected with the viruses showed growth arrest and subsequent lysis (Supplementary Fig. S1A), along with a loss of brown pigmentation with the extension of infection time (Supplementary Fig. S1B–G). TEM revealed that lipid bodies accumulated in the cytoplasm and around the cell membrane at 24 hpi compared with 0 hpi (Supplementary Fig. S1H–M). At 48 hpi, the observation of viral particles within intracytoplasmic vacuoles and the assembled virions indicated the development of viral infection (Supplementary Fig. S1K, L). Prolific virus release could be observed at 72 hpi (Fig. 1M). Moreover, fluorescence microscopy imaging showed a strong punctuated BODIPY 493/505-stained fluorescence signal in cells infected with lytic viruses that was not observed in uninfected cells (Supplementary Fig. S1N), suggesting the accumulation of neutral lipids in lipid droplets during lytic infection.
Virus-driven lipid metabolism reprogramming
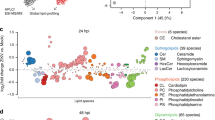
De novo transcriptome was conducted to identify critical genes involved in lipid metabolism during viral infection. The sequencing statistics (Supplementary Table S4), assembly quality statistics (Supplementary Table S5), and annotation information (Supplementary Table S6 and Supplementary Fig. S2) were provided in Supplementary information. Principal component and sample correlation analysis showed that one sample (Con_0h-3) was an extreme outlier (Supplementary Fig. S3), which was not included in the following analysis. We cannot rule out the possibility of potential contamination introduced during RNA-seq library construction. A total of 26,473 unigenes were clustered into nine co-expressed modules (MEs) (genes not belonging to any other ME were clustered into MEgery) by WGCNA (Supplementary Fig. S4), and MEs with different expression trends were distinguished by different colors (Fig. 1A). KEGG enrichment analysis of the unigenes in different MEs (Fig. 1B) showed that FA metabolism (MEyellow), sphingolipid metabolism (MEyellow), glycerolipid metabolism (MEyellow), and glycerophospholipid metabolism (MEturquoise) were significantly altered with the duration of infection (Fig. 1B, noted by red arrows), which were supported by our nontargeted lipidomic analysis results (Supplementary Figs. S5–S9, Supplementary Table S7, and Supplementary Dataset 1). Moreover, the PI3K-Akt and mTOR signaling pathways, one of the most important pathways participating in the regulation of glucose and lipid metabolism in mammalian cells [51] were enriched in MEturquoise (Fig. 1B, noted by blue arrows). In addition, a relaxed cut-off (FC ≥1.5) was also used to determine DE genes, and 30,048 DE genes were screened, which enriched several more pathways besides those that existed based on FC ≥2.0 (data not shown here). The analysis of the viral gene expression profiles revealed that almost all genes were explosively upregulated at 48 hpi (Supplementary Fig. S10), including the vAMGs participating in sphingolipid and glycerolipid metabolism. Thirty-one genes (including six viral genes) (Supplementary Table S8) participating in lipid metabolism were validated by qRT-PCR (Supplementary Fig. S11). Integrated transcriptomic and lipidomic pathway analysis (Supplementary Fig. S12–S14 and Supplementary Dataset 2) revealed TG accumulation and a metabolic shift toward viral sphingolipid metabolism (more details are provided in Supplementary results), which were consistent with those of the previous reports [11,12,13].

A Global gene expression profiles of host genes during infection. Different modules were generated by WGCNA and distinguished with different colors. B Significantly enriched KEGG pathways (Q < 0.05) related to host gene clusters as displayed in (A). Colors refer to clusters as indicated in (A).
Cardiolipins and calcium-mediated lipid accumulation in EhV-infected cells
The sum of CLs in our lipidomic data showed a significant decrease at the late stages of virus infection (48–60 hpi), whereas we noticed a significant accumulation of CLs at 24 hpi, accompanied by increased abundance in PGs, the precursor of CL synthesis and the CL synthase gene (CRLS) levels from 6 to 24 hpi (Supplementary Fig. S13). CL is the signature lipid of the mitochondrial inner membrane and any change in CL may cause alterations in mitochondrial function [52]. Several important mitochondrial indexes were detected and the results revealed gradually increased cellular reactive oxygen species levels (Supplementary Fig. S15A), reduced mitochondrial membrane potential (Supplementary Fig. S15B), and ATP levels (Supplementary Fig. S15C) during EhV infection. Compared with that at 0 and 6 hpi, the ATP concentrations barely changed at 24 hpi, although it declined significantly at 48 hpi (Supplementary Fig. S15C), suggesting that CLs alleviated the impaired mitochondrial electron transport function to transiently maintain the essential ATP level.
In addition, cellular calcium influx is important for transcriptional programming of lipid metabolism, including lipid storage and lipolysis [53,54,55]. To test whether the influx of calcium is required for cellular lipogenesis and lipid storage by controlling calcium homeostasis, we detected the Ca2+ influx across the cell membrane at 0–24 hpi by the Non-invasive Micro-test Technology (Supplementary materials and methods). The results showed a significant influx of extracellular Ca2+ at 12 and 24 h after viral infection (Supplementary Fig. S15D). We speculated that exogenous Ca2+ influx might lead to an increase in mitochondrial Ca2+ level and maintenance of mitochondrial calcium homeostasis and TCA cycle metabolites, hence providing indispensable energy for lipogenesis, CL remodeling, and lipid droplet storage during EhV infection. Altogether, our findings suggested that cellular calcium ions might be important for maintaining lipid homeostasis in the middle phase (24 hpi) of virus-infected E. huxleyi cells.
Identification and quantification of host and viral miRNAs
For miRNA identification, given that miRNAs might be derived from coding regions or noncoding regions, the genome of E. huxleyi CCMP1516 and our de novo transcriptome were both considered to be the mapping reference. Approximately 57.45% of the small clean tags were left after the filtration to clean reads (Supplementary Dataset 3). As a result, 69 host mature miRNAs and 7 viral miRNAs were identified, 1 host being known and 68 being novel miRNAs (the details are shown in Supplementary Dataset 4). The lengths of these miRNAs ranged from 20 to 23 nt with a peak at 21 nt (Fig. 2A), which agreed with typical miRNAs in plants with length peaks at 21 nt. The first nucleotide of these miRNAs had a bias for C (Fig. 2B), consistent with the first nucleotide bias of miRNAs identified in E. huxleyi CCMP1516 [31]. Twenty host mature miRNAs were differentially expressed in at least one comparative group and because all viral miRNAs were only expressed at 48 and 60 hpi, these seven miRNAs were all considered DE miRNAs (Supplementary Dataset 5). These DE miRNAs were named according to general miRNA nomenclature and the known miRNA (miR-574-5p) was named ehx-miR5 (Supplementary Dataset 5, the hairpin structures of DE miRNAs are shown in Supplementary Fig. S16). All DE miRNAs were verified by qRT-PCR (Supplementary Table S9), and the qPCR results were subject to regression analysis (R = 0.87, Supplementary Fig. S17).

A Length and number distribution of miRNAs. B First nucleotide bias of miRNAs.
Bioinformatic prediction of miRNA functions reveals that miRNAs might mediate the PI3K-AKT signaling pathway
Plant- and animal-type target genes were both analyzed in this study. Based on plant-like binding characteristics, 382 unigenes were targeted by host DE miRNAs, while virus-derived miRNAs only targeted three unigenes (unigene0025568, transketolase 1; unigene0047618, phenylalanine--tRNA ligase beta subunit; unigene0055743, N-acetylglutamate synthase). The 382 plant-type target genes were subjected to GO and KEGG functional analysis. Most GO terms did not seem to be related to lipid metabolism (Supplementary Fig. S18A). In KEGG annotation (Supplementary Fig. S18B), eight unigenes possibly participated in lipid metabolism (Supplementary Table S10), but the pathways to which these unigenes belonged were not significantly enriched. To date, the miRNA binding characteristics in E. huxleyi are largely unknown, although the existence of miRNAs has been confirmed [31]. Nevertheless, a previous study demonstrated that target hybridization to nucleotides of the seed region was sufficient to induce moderate repression of expression in another species of unicellular algae, Chlamydomonas reinhardtii, which might imply that the base-pairing requirements for small RNA-mediated repression were more similar to those of metazoans in C. reinhardtii [56]. Therefore, miRNA function analysis was subsequently focused on animal-type target genes.
Animal-type target gene analysis with stringent criteria against the de novo transcriptome yielded 8237 genes targeted by host DE miRNAs and 3886 genes as the targets of viral miRNAs. KEGG enrichment analysis showed that these target genes were significantly enriched in a wide variety of metabolic pathways (Fig. 3). For host miRNAs, lipid-related metabolism, such as glycerolipid metabolism, sphingolipid metabolism, and FA elongation, were significantly enriched (Fig. 3A). We also found that steroid biosynthesis, terpenoid backbone biosynthesis, sphingolipid metabolism, autophagy, etc. were targeted by viral miRNAs (Fig. 3B). The high expression of viral miRNAs at the late infection stage might regulate host metabolism to meet the special metabolic requirements of viruses or prevent the premature of host autophagy [30]. Target genes of host miRNAs were also enriched in the PI3K-Akt signaling pathway. In multicellular organisms, the PI3K-Akt signaling pathway combined with its downstream pathway (e.g., mTOR signaling pathway, which was also significantly influenced according to our transcriptomic data) played an important role in regulating FA and glycerolipid metabolism [51, 57, 58]. We speculated that lipid metabolism in the E. huxleyi–EhV system might be regulated via the miRNA-targeted PI3K-Akt signaling pathway. Therefore, further proofs of this speculation are explored in the following sections.

A Enrichment analysis of host DE miRNA targets. B Enrichment analysis of viral DE miRNA targets. The horizontal axis represents fold enrichment; the vertical axis represents significant pathways (Q < 0.05); the size of solid circles represents gene number in each pathway; and ColorRamp represents enrichment significance.
PI3K is a potential target of ehx-miR5 in the CDS region
Based on the bioinformatic prediction of miRNA targets in virus-infected E. huxleyi, we found PI3K gene was targeted by ehx-miR5 with perfect pairing on the CDS region by screening our de novo transcriptome database. BLASTn analysis of the PI3K fragment based on the genome of E. huxleyi CCMP1516 revealed that no 3′-poly A tail was detected in the PI3K fragment. Therefore, the complete CDS and 3′-UTR regions of PI3K were acquired by the 3′-RACE approach, but there was no target site on the 3′-UTR (data not shown). A previous study demonstrated that endogenous miRNAs in the green alga C. reinhardtii predominantly regulated gene expression through CDS-targeting [59]. Moreover, the expression levels of ehx-miR5 and PI3K were both verified by qRT-PCR and they displayed a negative correlation (the Pearson correlation coefficient was −0.837) (Fig. 4A, B). Thus, a dual-luciferase reporter assay was employed to further validate the prediction. The coding sequences of PI3K were cloned into the psi-CHECK2 vector to construct a recombinant vector (wild-type vector, WT vector) and mutations were made in the CDS seed sequences to construct another vector (mutant type vector, MT vector) (Fig. 4C). The constructed vectors were identified by restriction enzyme digestion (Supplementary Fig. S19) and nucleotide sequencing. The dual-luciferase reporter assay showed that relative luciferase activity significantly decreased by 27.1% in PI3K group compared with the control mimics (p < 0.05), and the results were not significantly different for transfection with the mutant vector (Fig. 4D). These results indicated that PI3K gene might be a target of ehx-miR5.

A, B qRT-PCR results of PI3K (A) and ehx-miR5 (B). C Putative binding sites of ehx-miR5 on the CDS region of PI3K predicted by miRanda; mutations are written in red. D The relative luminescence ratio in the PI3K CDS luciferase reporter assay. Blank groups were used as controls. The results were analyzed by Student’s t test. *p < 0.05.
miRNA-targeted PI3K-Akt-TOR signaling pathway is inhibited during late infection
In animal cells, the PI3K-Akt-mTOR signaling pathway has been proven to regulate lipid biosynthesis by promoting the nuclear accumulation of the mature form of SREBP [51, 57], a nuclear transcription factor that can promote the transcription of acetyl-CoA carboxylase (ACC), fatty acid synthase (FAS) and glycerol-3-phosphate O-acyltransferase (GPAT) [57, 60, 61]. Despite the important role of the PI3K-Akt-TOR signaling pathway in lipogenesis, its regulatory mechanism in phytoplankton (especially in virus-infected E. huxleyi) remains largely unexplored. Based on the de novo transcriptomic data, a complete gene annotation of the PI3K-Akt-TOR signaling pathway was obtained, facilitating the subsequent verification by western blotting and quantitative analysis (Fig. 5). The protein expression levels of PI3K gradually decreased starting from 12 until 60 hpi compared with the uninfected group (0 hpi) (Fig. 5A–E). The levels of Akt phosphorylation at Ser473 decreased significantly at 48 and 60 hpi, while the decreased phosphorylation of Thr308 was only observed at 60 hpi, concurrent with no significant effect on the total Akt levels (Fig. 5A–E). The phosphorylation levels of the downstream TOR at the Ser2448 site showed a significant increase at 6 and 12 hpi and then drastically decreased at 24–60 hpi, while only at 60 hpi did the total TOR levels moderately decrease (Fig. 5F–H), indicating that TOR signaling was impeded during late viral infection. These observations suggest that the PI3K-Akt-TOR signaling pathway is activated during early infection and inhibited during late infection.

A–E Representative bands by western blot for PI3K, Akt, p-Akt (Thr308), and p-Akt (Ser473) (A) and their quantitative analysis by ImageJ (B–E). F–H Representative western blots of TOR and p-TOR (Ser2448) (F), and their quantitative analysis (G, H). *p < 0.05, **p < 0.01.
Viral infection regulates the nucleoprotein levels of lipin 1 and SREBP
It has been demonstrated that activated TOR can phosphorylate lipin 1, a phosphatidic acid phosphatase, and phosphorylated lipin 1 cannot enter the nucleus to reduce SREBP promoter activity and nuclear SREBP protein abundance [58]. Based on the experimental results above, we speculated that the expression of the lipogenesis-related transcription factor SREBP and genes related to lipid synthesis was inhibited during EhV infection. We therefore examined the levels of intracellular nucleoprotein lipin 1 and SREBP by western blotting. The expression levels of lipin 1 increased markedly at 48 and 60 hpi, exhibiting a completely opposite trend with SREBP levels (Fig. 6A–C). The high expression levels of SREBP in early infection (0–12 hpi) favored the lipid-related gene transcription, including ACC, FAS, and GPAT, as verified by qRT-PCR (Fig. 6D–I), which further promoted TG accumulation during early EhV infection (Supplementary Fig. S13). Therefore, we believe that the expression of lipogenesis-related enzymes is regulated by TOR via lipin 1 and SREBP.

A Representative bands for lipin 1 and SREBP and B, C indicate their quantification. **p < 0.01. D–I qRT-PCR results of SREBP-mediated genes.
The potential miRNA regulatory mechanism for lipid metabolism in the E. huxleyi–EhV system
Based on the results above, we concluded a potential miRNA-based regulatory mechanism for lipid metabolism through the PI3K-Akt-TOR signaling pathway in virus-infected E. huxleyi cells (Fig. 7). During early infection (0–12 hpi), the relatively low expression of ehx-miR5 resulted in high expression of PI3K, which facilitated the activation of Akt by phosphorylation. Relying on the activated Akt, TOR was phosphorylated and then phosphorylated lipin 1, preventing the entry of lipin 1 into the cell nucleus. As a result, SREBP promoted genes transcription (Fig. 7A). During late infection (24–60 hpi), the high expression of ehx-miR5 led to low levels of PI3K, suppressing the phosphorylation of Akt, TOR, and lipin 1, and finally lipin 1 entered the cell nucleus, causing decreased levels of SREBP abundance and activity (Fig. 7B). Overall, these observations are basically in line with the expectation that the animal-like regulatory mechanism of lipid metabolism possibly exists in virus-infected E. huxleyi through PI3K-Akt-TOR-lipin 1-SREBP in an orchestrated manner.

During early infection, low expression levels of ehx-miR5 lead to high expression of PI3K. The activated PI3K-Akt-TOR signaling prevents the entry of lipin 1 into the cell nucleus by phosphorylation, and hence SREBP can promote gene transcription (A). In contrast, during late infection, PI3K-Akt-TOR signaling is inhibited under the effect of ehx-miR5. Dephosphorylated lipin 1 enters the cell nucleus and decreases the abundance and activity of SREBP (B).
The regulatory functions of miRNAs in lipid metabolism in animals are widespread and conserved processes. They cannot only regulate hub genes that directly participate in lipid metabolism, such as FA biosynthesis and degradation, and glycerolipid biosynthesis, but also indirectly mediate lipid metabolism through activating signal pathways (such as AMPK and PPARα signaling pathways) [21, 25, 27, 62]. miRNA (miR-378) can target p110α, a catalytic subunit of PI3K and hence a key transducer of the insulin signaling pathway, and thus controls glucose and lipid homeostasis through enhancing the liver’s response to feeding/fasting cues mediated by insulin [63]. Besides, some ATP-binding cassette (ABC) transporters are repressed by miRNAs, reducing the efflux of lipids from cells. Virus-derived miRNAs identified in our study were also predicted to target ABC transporters (Fig. 3B), indicating a potential strategy for the virus to accumulate lipids in E. huxleyi cells [64,65,66]. In this study, due to the limitation of experimental conditions, we were not able to directly validate the miRNA’s regulatory effect on PI3K by stable transfection in E. huxleyi cells, but we are confident that the results of our work are meaningful for exploring the miRNA-targeted lipid metabolism in marine phytoplankton infected by viruses.
Conclusions
We applied lipidomic, transcriptomic approaches, and experimental verification to reveal the reprogrammed lipid metabolic responses to viral infection in the coccolithophores E. huxleyi that are regulated at transcriptional, miRNA-mediated posttranscriptional, and translational levels. Moreover, our results demonstrate the possible regulatory role of miRNA in lipid metabolism through the PI3K-Akt-TOR signaling pathway. In addition, our findings also further indicated that cellular calcium ions might be important for maintaining lipid homeostasis in virus-infected E. huxleyi cells. These findings shed new light on the epigenetic regulation between marine algae-virus interactions and enable us to deepen the understanding of the chemical “arms race” among marine microbes.
Data availability
The generated raw data of the de novo transcriptome were deposited to NCBI in SRA (BioProject accession number: PRJNA705400). The Transcriptome Shotgun Assembly (TSA) project has been deposited at DDBJ/EMBL/GenBank under the accession GJZP00000000. The version described in this paper is the first version, GJZP01000000. The lipidomics data have been deposited to the EMBL-EBI MetaboLights database with the identifier MTBLS5178.
References
Holligan PM, Fernández E, Aiken J, Balch WM, Boyd P, Burkill PH, et al. A biogeochemical study of the coccolithophore, Emiliania huxleyi, in the North Atlantic. Glob Biogeochem Cycles. 1993;7:879–900.
Taylor AR, Brownlee C, Wheeler G. Coccolithophore cell biology: chalking up progress. Ann Rev Mar Sci. 2017;9:283–310.
Iglesias-Rodriguez MD, Halloran PR, Rickaby REM, Hall IR, Colmenero-Hidalgo E, Gittins JR, et al. Phytoplankton calcification in a high-CO2 world. Science. 2008;320:336–40.
Bratbak G, Egge J, Heldal M. Viral mortality of the marine alga Emiliania huxleyi (Haptophyceae) and termination of algal blooms. Mar Ecol Ser. 1993;93:39–48.
Wilson WH, Tarran GA, Schroeder D, Cox M, Oke J, Malin G. Isolation of viruses responsible for the demise of an Emiliania huxleyi bloom in the English Channel. J Mar Biolog Assoc UK. 2002;82:369–77.
Rosenwasser S, Mausz MA, Schatz D, Sheyn U, Malitsky S, Aharoni A, et al. Rewiring host lipid metabolism by large viruses determines the fate of Emiliania huxleyi, a bloom-forming alga in the ocean. Plant Cell. 2014;26:2689–707.
Laber CP, Hunter JE, Carvalho F, Collins JR, Hunter EJ, Schieler BM, et al. Coccolithovirus facilitation of carbon export in the North Atlantic. Nat Microbiol. 2018;3:537–47.
Johns CT, Grubb AR, Nissimov JI, Natale F, Knapp V, Mui A, et al. The mutual interplay between calcification and coccolithovirus infection. Environ Microbiol. 2019;21:1896–915.
Bidle KD, Vardi A. A chemical arms race at sea mediates algal host-virus interactions. Curr Opin Microbiol. 2011;14:449–57.
Rosenwasser S, Ziv C, Creveld SG, van, Vardi A. Virocell metabolism: metabolic innovations during host–virus interactions in the ocean. Trends Microbiol. 2016;24:821–32.
Malitsky S, Ziv C, Rosenwasser S, Zheng S, Schatz D, Porat Z, et al. Viral infection of the marine alga Emiliania huxleyi triggers lipidome remodeling and induces the production of highly saturated triacylglycerol. N Phytol. 2016;210:88–96.
Zeng J, Liu S, Cai W, Jiang H, Lu X, Li G, et al. Emerging lipidome patterns associated with marine Emiliania huxleyi-virus model system. Sci Total Environ. 2019;688:521–8.
Ziv C, Malitsky S, Othman A, Ben-Dor S, Wei Y, Zheng S, et al. Viral serine palmitoyltransferase induces metabolic switch in sphingolipid biosynthesis and is required for infection of a marine alga. Proc Natl Acad Sci USA. 2016;113:E1907–16.
Schleyer G, Shahaf N, Ziv C, Dong Y, Meoded RA, Helfrich EJN, et al. In plaque-mass spectrometry imaging of a bloom-forming alga during viral infection reveals a metabolic shift towards odd-chain fatty acid lipids. Nat Microbiol. 2019;4:527–38.
Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141:1202–7.
He Y, Sun Y, Zhang X. Noncoding miRNAs bridge virus infection and host autophagy in shrimp in vivo. FASEB J. 2017;31:2854–68.
Shao Y, Li C, Xu W, Zhang P, Zhang W, Zhao X. miR-31 links lipid metabolism and cell apoptosis in bacteria-challenged Apostichopus japonicus via targeting CTRP9. Front Immunol. 2017;8:1–16.
Li S, Castillo‐González C, Yu B, Zhang X. The functions of plant small RNAs in development and in stress responses. Plant J. 2017;90:654–70.
Manavella PA, Yang SW, Palatnik J. Keep calm and carry on: miRNA biogenesis under stress. Plant J. 2019;99:tpj.14369.
Feng J-L, Yang Z-J, Chen S-P, El-Kassaby YA, Chen H. High throughput sequencing of small RNAs reveals dynamic microRNAs expression of lipid metabolism during Camellia oleifera and C. meiocarpa seed natural drying. BMC Genomics. 2017;18:546.
Agbu P, Carthew RW. MicroRNA-mediated regulation of glucose and lipid metabolism. Nat Rev Mol Cell Biol. 2021;22:425–38.
Thibault PA, Wilson JA. MicroRNA-lipid one-upmanship. Nat Chem Biol. 2015;11:905–6.
Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat Commun. 2015;6:7466.
Sedgeman LR, Michell DL, Vickers KC. Integrative roles of microRNAs in lipid metabolism and dyslipidemia. Curr Opin Lipido. 2019;30:165–71.
Assmann TS, Cuevas-Sierra A, Riezu-Boj JI, Milagro FI, Martínez JA. Comprehensive analysis reveals novel interactions between circulating microRNAs and gut microbiota composition in human obesity. Int J Mol Sci. 2020;21:9509.
Yang Z, Cappello T, Wang L. Emerging role of microRNAs in lipid metabolism. Acta Pharm Sin B. 2015;5:145–50.
Yu X, Wang H, Wu J, Wang S, Tang Y-J, Tang Y, et al. Non-coding RNAs derailed: The many influences on the fatty acid reprogramming of cancer. Life Sci. 2019;231:116509.
Shi X, Lin X, Li L, Li M, Palenik B, Lin S. Transcriptomic and microRNAomic profiling reveals multi-faceted mechanisms to cope with phosphate stress in a dinoflagellate. ISME J. 2017;11:2209–18.
Bruscella P, Bottini S, Baudesson C, Pawlotsky J-M, Feray C, Trabucchi M. Viruses and miRNAs: More Friends than Foes. Front Microbiol. 2017;8:1–11.
Tagawa T, Serquiña A, Kook I, Ziegelbauer J. Viral non-coding RNAs: stealth strategies in the tug-of-war between humans and herpesviruses. Semin Cell Dev Biol. 2021;111:135–47.
Zhang X, Gamarra J, Castro S, Carrasco E, Hernandez A, Mock T, et al. Characterization of the small RNA transcriptome of the marine Coccolithophorid, Emiliania huxleyi. PLoS One. 2016;11:e0154279.
Schatz D, Rosenwasser S, Malitsky S, Wolf SG, Feldmesser E, Vardi A. Communication via extracellular vesicles enhances viral infection of a cosmopolitan alga. Nat Microbiol. 2017;2:1485–92.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–5.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11:1650–67.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2007;35:D21–5.
Griffiths-Jones S. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–41.
Griffiths-Jones S. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4.
Wang R, Xu L, Zhu X, Zhai L, Wang Y, Yu R, et al. Transcriptome-wide characterization of novel and heat-stress-responsive microRNAs in Radish (Raphanus Sativus L.) using next-generation sequencing. Plant Mol Biol Rep. 2015;33:867–80.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1.
Yan T, Yoo D, Berardini TZ, Mueller LA, Weems DC, Weng S, et al. PatMatch: a program for finding patterns in peptide and nucleotide sequences. Nucleic Acids Res. 2005;33:W262–6.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 2008;9:559.
Haug K, Cochrane K, Nainala VC, Williams M, Chang J, Jayaseelan KV, et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020;48:D440–D444.
Zhang E, Wu S, Cai W, Zeng J, Li J, Li G, et al. Validation of superior reference genes for qRT‐PCR and western blot analyses in marine Emiliania huxleyi –virus model system. J Appl Microbiol. 2021;131:257–71.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8.
Liu J, Cai W, Fang X, Wang X, Li G. Virus-induced apoptosis and phosphorylation form of metacaspase in the marine coccolithophorid Emiliania huxleyi. Arch Microbiol. 2018;200:413–22.
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–36.
Schlame M, Greenberg ML. Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862:3–7.
Arruda AP, Hotamisligil GS. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015;22:381–97.
Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, et al. Store-operated Ca2+ entry controls induction of lipolysis and the transcriptional reprogramming to lipid metabolism. Cell Metab. 2017;25:698–712.
Ding L, Yang X, Tian H, Liang J, Zhang F, Wang G, et al. Seipin regulates lipid homeostasis by ensuring calcium‐dependent mitochondrial metabolism. EMBO J. 2018;37:1–17.
Yamasaki T, Voshall A, Kim E-J, Moriyama E, Cerutti H, Ohama T. Complementarity to an miRNA seed region is sufficient to induce moderate repression of a target transcript in the unicellular green alga Chlamydomonas reinhardtii. Plant J. 2013;76:1045–56.
Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–20.
Barger SR, Penfield L, Bahmanyar S. Coupling lipid synthesis with nuclear envelope remodeling. Trends Biochem Sci. 2022;47:52–65.
Chung BYW, Deery MJ, Groen AJ, Howard J, Baulcombe DC. Endogenous miRNA in the green alga Chlamydomonas regulates gene expression through CDS-targeting. Nat Plants. 2017;3:787–94.
Ericsson J, Jackson SM, Kim JB, Spiegelman BM, Edwards PA. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1- and sterol regulatory element-binding protein-responsive gene. J Biol Chem. 1997;272:7298–305.
Coleman RA. It takes a village: channeling fatty acid metabolism and triacylglycerol formation via protein interactomes. J Lipid Res. 2019;60:490–7.
Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98.
Liu W, Cao H, Ye C, Chang C, Lu M, Jing Y, et al. Hepatic miR-378 targets p110α and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat Commun. 2014;5:5684.
Wagschal A, Najafi-Shoushtari SH, Wang L, Goedeke L, Sinha S, deLemos AS, et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat Med. 2015;21:1290–7.
Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–75.
Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci USA. 2010;107:17321–6.
Acknowledgements
We are grateful to thank Prof. Gunnar Bratbak (Department of Biology, University of Bergen) for providing the E. huxleyi BOF92 strain and the E. huxleyi virus 99B1 strain. We would like to thank Guangzhou Genedenovo Biotechnology Co., Ltd, for assisting in sequencing. This study was supported by the National Natural Science Foundation of China (Grant No. 42076086) and Fujian Province Natural Science Foundation of China (Grant No. 2019J01696).
Author information
Authors and Affiliations
Contributions
EQZ analyzed the RNA-seq data, performed the experiments, and drafted the manuscript. JJG and JZ carried out lipidomics analysis based on UPLC-Q-Exactive-MS method. ZHW contributed to the algal culture, virus preparation, and experiment performance. JL performed the flow cytometry assay, Ca2+ influx detection, and data analysis. GLL participated in critical discussion, figure organization, English editing, and manuscript revision. JWL designed the investigation, guided the study, and finished the final version of the manuscript. All authors contributed to the discussion, commented on the manuscript, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, E., Gao, J., Wei, Z. et al. MicroRNA-mediated regulation of lipid metabolism in virus-infected Emiliania huxleyi. ISME J 16, 2457–2466 (2022). https://doi.org/10.1038/s41396-022-01291-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-022-01291-y
This article is cited by
-
The consequences of viral infection on protists
Communications Biology (2024)
