
Abstract
We have recently identified a pool of intracellular β1 adrenergic receptors (β1ARs) at the sarcoplasmic reticulum (SR) crucial for cardiac function. Here, we aim to characterize the integrative control of intracellular catecholamine for subcellular β1AR signaling and cardiac function. Using anchored Förster resonance energy transfer (FRET) biosensors and transgenic mice, we determined the regulation of compartmentalized β1AR-PKA signaling at the SR and plasma membrane (PM) microdomains by organic cation transporter 3 (OCT3) and monoamine oxidase A (MAO-A), two critical modulators of catecholamine uptake and homeostasis. Additionally, we examined local PKA substrate phosphorylation and excitation–contraction coupling in cardiomyocyte. Cardiac-specific deletion of MAO-A (MAO-A-CKO) elevates catecholamines and cAMP levels in the myocardium, baseline cardiac function, and adrenergic responses. Both MAO-A deletion and inhibitor (MAOi) selectively enhance the local β1AR-PKA activity at the SR but not PM, and augment phosphorylation of phospholamban, Ca2+ cycling, and myocyte contractile response. Overexpression of MAO-A suppresses the SR-β1AR-PKA activity and PKA phosphorylation. However, deletion or inhibition of OCT3 by corticosterone prevents the effects induced by MAOi and MAO-A deletion in cardiomyocytes. Deletion or inhibition of OCT3 also negates the effects of MAOi and MAO-A deficiency in cardiac function and adrenergic responses in vivo. Our data show that MAO-A and OCT3 act in concert to fine-tune the intracellular SR-β1AR-PKA signaling and cardiac fight-or-flight response. We reveal a drug contraindication between anti-inflammatory corticosterone and anti-depressant MAOi in modulating adrenergic regulation in the heart, providing novel perspectives of these drugs with cardiac implications.
Similar content being viewed by others

Introduction
During fight-or-flight responses, activation of the sympathetic nervous system releases catecholamines to stimulate βARs in the heart, enhancing heart rate, cardiac contractility, and cardiac output [30]. Blunted βAR and cardiac responses to catecholamines hallmark cardiac dysfunction in heart failure (HF) [2]. Restoring the βAR responsiveness represents a critical therapeutic strategy to rescue cardiac function. Our previous studies suggest targeting monoamine oxidase A (MAO-A) and organic transporter 3 (OCT3) as promising strategies for modulating the intracellular β1AR signaling in cardiomyocytes [57, 58]. However, a functional crosstalk of targeting cardiac MAO-A and OCT3 remains unknown.
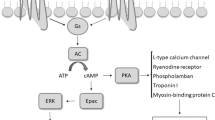
In addition to the plasma membrane, G protein-coupled receptors (GPCRs) are present and function at intracellular membranes, including endosomes, the Golgi, the sarcoplasmic reticulum (SR), nuclear membranes, and mitochondrial [2, 6, 11, 12, 34, 40, 43, 53, 55, 59]. We have recently identified a pool of β1ARs residing on the SR (SR-β1ARs), which associate with SR Ca2+ ATPase 2a (SERCA2a) but not ryanodine receptor 2 (RyR2). Stimulation of SR-β1ARs promotes local protein kinase A (PKA) activity for phosphorylation of phospholamban (PLB) and enhances Ca2+ cycling in excitation–contraction (E–C) coupling [57, 58]. The intracellular SR-β1ARs produce spatially biased signaling and distinct effects from the β1ARs at the PM, providing a framework to selectively fine-tune the local βAR signals and develop therapeutic strategies.
Both OCT3 and MAO-A can tune the cytoplasmic catecholamines by modulating catecholamine uptake or degradation, respectively. In a classic paradigm, catecholamines (norepinephrine, NE, and epinephrine, EPI) released from the sympathetic ganglia neurons activate the β1ARs on the PM (PM-β1ARs) to enhance heart rate, cardiac contractility, and cardiac output [30]. The majority (90%) of catecholamines are reabsorbed back into the cytoplasm in neurons. The myocardium absorbs about 5% of catecholamines through OCT3 [13]. In fact, the catecholamines in myocytes can activate the intracellular adrenergic receptors [36, 57, 61], including the SR-β1ARs, which synergize with the PM-β1AR signaling to promote E–C coupling [5, 36, 47, 57, 61]. Additionally, MAO-A oxidizes intracellular catecholamines, thereby negatively regulating the SR-β1ARs signaling [27, 58]. It is still unknown whether MAO-A and OCT3 functionally interact to coordinate spatial–temporal regulation of subcellular β1AR and cardiac fight-or-flight response.
Both OCT3 and MAO-A are associated with HF development, which is featured by desensitized βAR signaling [21, 27, 28, 58]. Increased MAO-A expression and activity correlate with reduced myocardium catecholamine contents, increased reactive oxidative stress (ROS), and impaired β1AR function in HF [27, 58]. On the other hand, elevated corticosterone (CORTI), an endogenous OCT3 inhibitor, is an independent risk factor for HF [38]. The actions of CORTI in the heart remain unclear. We have recently reported that activation of the SR-β1ARs is governed by the OCT3-mediated catecholamine transport into myocytes. CORTI suppresses the intracellular SR-β1AR signaling and inotropic response by inhibiting OCT3. Therefore, we hypothesize that the intracellular catecholamine levels, β1AR signaling, and cardiac function are dynamically controlled by MAO-A or OCT3; the drugs targeting these proteins may interact in modulating adrenergic stimulation of cardiac function.
In this study, we combined subcellular targeted biosensors with genetic and pharmacological approaches to investigate the interaction between OCT3 and MAO-A in modulating the intracellular SR-β1AR signaling and cardiac function. We offer compelling evidence to support that OCT3 and MAO-A calibrate the intracellular β1AR signaling and cardiac E–C coupling under catecholamine stimulation. Our data highlight the essential interaction between two classes of clinically used drugs, anti-inflammation CORTI, and anti-depressant MAOi, in modulating the intracellular β1AR signaling and cardiac function.
Materials and methods
Surgery and animals
C57BL/6J WT, OCT3 knockout (OCT3-KO), MAO-A flox (MAO-A-FF), MAO-A flox/MHC-cre + (cardiac-specific knockout, MAO-A-CKO), and β1AR-KO male and female mice at 2–4 months old were randomized and used in this study. AVMs from 3 to 6 months old rabbits were provided by Dr. Donald Bers’s Laboratory. Detailed descriptions of echocardiography, Ca2+ imaging, fluorescence resonance energy transfer (FRET), immunoblot, and myocardial infarction surgery are provided in the Online Supplemental Materials and Methods. All animal procedures were performed following the guidelines of the Institutional Animal Care and Use Committee (IACUC, Protocol 20234 and 20957) at the University of California at Davis and followed the NIH and ARRIVE guidelines. Animals were housed in the UC Davis animal facility with automatically controlled humidity (30–70%), temperature (22 °C), and lightning (12/12-h cycle). The facility husbandry staff was responsible for the feeding, watering, and care of animals. The animal strain information was listed in the Animal Resource Table. Mice were anesthetized with isoflurane (1–2%) in oxygen through a nose cone during echocardiography. Mice were humanely euthanized for tissue harvest and cell isolation under deep isoflurane anesthesia (3–5%). The hearts were quickly excised and snap-frozen in liquid nitrogen and then transferred to a − 80 °C freezer for long-term storage. Alternatively, hearts were rinsed promptly in a chilled buffer for cannulation to isolate adult ventricle myocytes (AVMs).
Statistical analysis
Pooled data were represented as the mean ± SD. Animals were randomized during the experiments. For the statistical analysis, all pairwise comparison was conducted by compare the mean of each column with the mean of every other column. Only p values of the interested comparisons were reported given the space limits. We performed fully blinded analyses with different persons carrying out the experiments and analysis. Representative figures/images reflected the average levels of each experiment. Normality of the data (n ≥ 6) was assessed using the Shapiro–Wilk test in GraphPad Prism 9 with significance at alpha = 0.05 (GraphPad Inc., San Diego, CA). If n < 6, a non-parametric Mann–Whitey test or Kruskal–Wallis test was performed where appropriate. Comparisons between two groups were performed by a two-tailed unpaired t test or paired t test. Comparisons between more than two groups were performed by one-way ANOVA or two-way ANOVA followed by Tukey’s post hoc using Prism 9.0 software (GraphPad). Paired t tests compare the means of the same group of mice and does not assume equal variance between two groups. For unpaired t tests, the variance is assumed to be equal. Paired two-tailed student t test or row-matched (RM) one-way AVONA analysis was performed to compare the mean of different measurements taken from the same animals. All comparisons and pairings were designed in original experimental protocol.
Results
Corticosterone prevents MAO-A inhibitor from enhancing cardiac fight-or-flight response
WT mice were challenged with a series of doses of β1-agonist dobutamine (DOB, intraperitoneal injection, i.p.) to mimic the fight-or-flight response under stress. At 100 μg/kg, DOB produced near-maximal increases in ejection fraction (EF) and heart rate (HR, Fig. 1, Online Figure I, and Online Table 1); and the concentration was chosen for β-agonists in the following in vivo experiments. Our recent study shows that MAOi enhances myocyte shortening by promoting the intracellular SR-β1AR signaling induced by catecholamines [58]. Here, treatment with MAOi elevated the baseline EF and enhanced the EPI-induced cardiac EF responses in WT mice (Fig. 1A, B, Online Figure IC, and Online Table 2). As a non-substrate of MAO [63], DOB increased cardiac EF and HR over the baselines, which were not affected by MAOi (Fig. 1A, C, Online Figure ID, and Online Table 3). These data indicate that inhibition of MAO-A enhances the catecholamine-triggered cardiac contractile responses. Interestingly, catecholamines are transported into cardiomyocytes by OCT3 [5, 57, 61]. We assessed whether OCT3 inhibitors modulate the effects of MAOi on cardiac function. Administration of an OCT3 inhibitor, CORTI, abolished the effects of MAOi on EF and HR after stimulation with EPI (Fig. 1D, F and Online Table 2). Administration of another OCT3 inhibitor, decynium-22 (200 μg/kg, i.p [17]), also suppressed the cardiac responses to EPI and MAOi treatments (Online Figure IIA, B and Online Table 4).

Corticosterone suppresses the MAOi-mediated enhancement of inotropic response in mouse hearts. M-mode echocardiography was recorded for 2 min at baseline and for 8 min after each drug administration. The maximal cardiac response was reported. A Representative echocardiography images of WT mouse before and after injection of EPI or DOB (100 µg/kg, i.p.) with and without MAOi pretreatment (0.4 mg/kg, i.p., 5 min). B, C Quantification of mouse cardiac EF and HR as treated in A. D Representative echocardiography images of WT mouse in response to EPI after pretreatment with MAOi and CORTI (200 μg/kg, i.p., 5 min). E, F Quantification of maximal EF and HR in D. Data are shown as mean ± SD of individual mice. n = 9/group, p values were obtained by row-matched (RM) one-way ANOVA followed by Tukey’s test
To exclude the impacts of CORTI and MAOi on the central nervous system in the observations above, we tested the direct impacts of CORTI and MAOi on isolated AVMs. MAOi enhanced the catecholamine (EPI and NE)-induced sarcomere shortening (SS%) in AVMs, but did not affect the responses to DOB stimulation (Fig. 2A, B). MAOi also enhanced the EPI- and NE-induced Ca2+ transient (CaT) amplitude and decay without affecting the responses to DOB (Fig. 2C, D and Online Figure III). Moreover, the effects of MAOi on myocyte contractile and Ca2+ responses to EPI stimulation were abolished by CORTI (Fig. 2D–G). Notably, CORTI and anti-depressant MAOi are widely used in clinical settings [51]. These data highlight the interaction between anti-inflammatory CORTI and anti-depressant MAOi in modulating contractile function in the myocardium.

Corticosterone prevents MAOi from enhancing catecholamine stimulation of excitation–contraction coupling in WT AVMs. AVMs were loaded with Ca2+ indicator (5 µmol/L Fluo-4 AM) and paced at 1 Hz. AVMs was pretreated with MAOi or CORTI for 5 min and followed by 6-min incubation of β-agonists (NE, EPI, or DOB). SS and Ca2+ were recorded for 2 min at baseline and for 6 min after agonist stimulation. The maximal SS and Ca2+ were reported. A, B Representative and quantification of SS% kinetics at the baselines and after stimulation with NE, EPI, or DOB in the absence or presence of MAOi. Dot plots represent the mean ± SD of the indicated number of AVMs from 6 WT mice. C Ca2+ transient (CaT) amplitude and decay (Tau) in response to NE, EPI, or DOB in the absence or presence MAOi. D, E Representative and quantification of SS% in response to EPI, MAOi, and CORTI cotreatment. Dot plots represent the mean ± SD of the indicated number of AVMs from 6 WT mice. F, G Representative CaT dynamics and quantification of CaT amplitude and Tau after EPI, MAOi, and CORTI cotreatment. Dot plots represent the mean ± SD of the indicated number of AVMs from 6 mice. NE = 0.1 µmol/L, EPI = 1 µmol/L, DOB = 1 µmol/L, MAOi = 5 µmol/L, and CORTI = 2 µmol/L. Data are shown as mean ± SD. p values were obtained by one-way ANOVA followed with Tukey’s test
OCT3 and MAO-A control the accessibility of catecholamines to activate SR-β1AR signaling in AVMs
Mechanistically, we hypothesized that OCT3 and MAO-A coordinate the activation of the SR-β1AR and PKA phosphorylation of PLB at Ser16 by controlling the levels of intracellular catecholamines. In this paradigm, catecholamines enter the cell through OCT3 and are subjected to MAO-A-mediated oxidation and inactivation (Fig. 3A and [57]). To validate the role of the β1AR in this regulation, we tested cardiac response in the β1AR-KO mice [62] (Online Figure IIC, D, Online Table 5). Deleting β1AR abolished the inotropic effects of EPI and MAOi, suggesting that the effects are mainly mediated by the β1AR. We then applied a pair of fluorescence resonance energy transfer (FRET)-based biosensors (A-kinase activity reporter 3, AKAR3) anchoring at either the PM or SR microdomains to monitor the local β1AR-PKA signaling in AVMs [2, 44]. Inhibiting MAO-A by clorgyline augmented the EPI-induced PKA activity at the SR without affecting the PKA activity at the PM (Fig. 3B, C). The β1AR-specific antagonist CGP20712a (0.3 µmol/L) but not the β2AR-specific antagonist ICI118551 (1 µmol/L) abolished the SR-PKA FRET response induced by EPI in the presence of MAOi (Online Figure IV), confirming that the β1AR is responsible for the effects of MAOi. In agreement, MAOi enhanced the EPI-induced PKA phosphorylation of PLB at Ser16 at the SR, but did not affect the EPI-induced PKA phosphorylation of LTCC at Ser1928 and RyR2 at Ser2808 on the PM (Fig. 3D, E). In controls, MAOi did not affect the PKA phosphorylation of PLB, LTCC, and RyR2 in response to DOB stimulation (Online Figure VA–C).

Corticosterone antagonizes the positive effects of MAOi on promoting SR-localized β1AR signaling and PLB phosphorylation. Mouse AVMs expressing AKAR3 biosensors were treated with drugs as indicated. YFP/CFP FRET ratio was recorded before and after agonist stimulation for total 400 s. The maximal FRET response after drug stimulation was plotted. AVMs was pretreated with MAOi or CORTI for 5 min and followed by 5-min incubation of β-agonists (EPI or DOB) for western blot. A Schematic of intracellular catecholamine hemostasis and local β1AR activation is modulated by OCT3 and MAO-A. B, C WT AVMs were stimulated with EPI after pretreatment with vehicle control (Ctrl) or MAOi. Time courses of FRET dynamics and quantification of local PKA activities at the PM and SR after EPI and MAOi administration were plotted. Dot plots represent the mean ± SD of the indicated number of AVMs from 4 WT mice. D, E Immunoblots show detection of phosphorylated Ca2+ handling proteins (p-Ser 16 of PLB, p-Ser 1928 of LTCC, and p-Ser 2808 of RyR2) in response to EPI in the presence of MAOi. n ≥ 3 WT mice. Data were shown as mean ± SD. F Schematic depicting the effects of CORTI on subcellular β1AR activation by inhibiting OCT3. G, H Time-course curves and quantification of local PKA activity at the PM and SR after EPI, MAOi, and CORTI coadministration were plotted. Dot plots represent the mean ± SD of the indicated number of AVMs from 4 WT mice. I, J Immunoblots show detection of phosphorylated LTCC (p-Ser 1928) and PLB (p-Ser 16) in response to coadministration of EPI, MAOi, and CORTI. n = 6 rabbits. Data were shown as mean ± SD. EPI = 1 µmol/L, MAOi = 5 µmol/L, and CORTI = 2 µmol/L. A.U. = arbitrary unit. p values were obtained by unpaired Student’s t test (B, C), non-parametric Kruskal–Wallis test followed with Dunn’s multiple comparisons test (E), or one-way ANOVA followed with Tukey’s test (G, H, J)
OCT3 transports catecholamines into myocytes. Thus, inhibition of OCT3 should prevent catecholamine from activating the SR-β1AR and suppress the responses to EPI stimulation in AVMs (Fig. 3F and [57, 61]). While MAOi enhanced the SR-PKA activity triggered by EPI in AVMs, CORTI suppressed the SR-PKA response (Fig. 3H). Accordingly, MAOi enhanced the EPI-induced PKA phosphorylation of PLB in AVMs, and the effects of MAOi were attenuated by CORTI (Fig. 3I, J). In comparison, neither MAOi nor CORTI affected the EPI-induced PKA activity and the PKA phosphorylation of LTCC at the PM in AVMs (Fig. 3B, G, I, and J). As controls, neither MAOi nor CORTI affected the PKA activity and phosphorylation of PLB and LTCC after stimulation with DOB (Online Figure VD, E). Our data highlight the essential role of OCT3-dependent entry of catecholamines for activation of the SR-β1AR, which is subjected to additional regulation by MAO-A.
Deletion of MAO-A enhances the local β1AR signaling at the SR and promotes E–C coupling and cardiac contraction
We further applied cardiac-specific deletion of MAO-A (Online Figure VIA, B) to verify the effects of MAOi in regulating the intracellular SR-β1AR signaling and contractile function. MAO-A-CKO mice displayed increased catecholamine concentrations in the myocardium but not in the plasma or brain (Fig. 4A). Consistently, MAO-A-CKO hearts exhibited increases in cAMP and PKA phosphorylation of PLB (Ser16), TnI (Ser23/24), and LTCC (Ser1928) (Fig. 4B, E, and F). Meanwhile, MAO-A displayed a proximity to PLB in AVMs, supporting a local and preferential effect on the SR-β1AR signaling (Online Figure VIC, D). Although catecholamine oxidation by MAO-A is known to generate reactive oxygen species (ROS) [26, 27], deleting MAO-A did not change the ROS levels in the myocardium (Fig. 4C), suggesting that ROS unlikely contributes to the enhanced adrenergic signaling in MAO-A-CKO hearts. MAO-A-CKO mice had a similar heart weight/body weight ratio to the FF control littermates (Fig. 4D). MAO-A-CKO mice displayed a modest increase in cardiac EF at the baselines, consistent with the elevated PKA phosphorylation of substrates. When challenged with EPI, MAO-A-CKO mice showed more robust inotropic and chronotropic responses relative to the FF controls (Fig. 4H, I, Online Figure VIE–G, and Online Table 6). In controls, DOB stimulation promoted similar increases in EF and HR in MAO-A-CKO and FF groups (Fig. 4H, I and Online Table 6). Together, these data confirmed that cardiac deletion of MAO-A reduces catecholamine oxidation and inactivation (Fig. 4G) and mimics the effects of MAOi in promoting inotropic and chronotropic responses.

Cardiac-specific deletion of MAO-A increases myocardium catecholamine contents and enhances contractile function. A Quantitative measurements of endogenous NE in mouse heart, plasma, and brain (n ≥ 6 mice per group). B Quantification of cyclic AMP levels in MAO-A-FF and CKO heart tissues (n = 8 FF, 7 CKO). C Quantification of ROS in mice hearts (n = 10 mice per group). D Heart weight/body weight ratio in MAO-A-FF and CKO hearts. n = 8. E, F Western blots show the phosphorylation of PLB (p-Ser16), TnI (p-Ser23/24), RyR2 (p-Ser2808), and LTCC (p-Ser1928) in MAO-A-FF and CKO heart tissues (n = 6/group). G Schematic shows that deletion of MAO-A enhances local β1AR signaling at the SR but not PM microdomain. H M-mode echocardiography was recorded for 2 min at baselines and for 8 min after each drug administration. The maximal cardiac response was reported. Representative echocardiography images of MAO-A-FF and CKO mice before and after EPI and DOB stimulation (100 µg/kg, i.p.). I Quantification of EF and HR before and after EPI or DOB stimulation n = 9. Data are shown as mean ± SD. p values were obtained by unpaired Student’s t test (A–D, and F) or one-way ANOVA with Tukey’s multiple comparison correction
At the cellular levels, EPI triggered rapid increases in the PM-PKA and SR-PKA activity in both MAO-A FF and CKO AVMs (Fig. 5A, C). Compared to the FF group, MAO-A-CKO AVMs had higher PKA activities at the SR and a left-shifted dose–response curve of SR-PKA activity induced by EPI (Fig. 5A, C and Online Figure VIIA). Deleting MAO-A, however, did not affect the dose–response curve of PM-PKA activity induced by EPI (Online Figure VIIB). Consequently, deleting MAO-A enhanced the EPI-induced phosphorylation of PLB at Ser16 without affecting the PKA phosphorylation of LTCC (Fig. 5E, F). EPI also promoted larger increases in SS% in MAO-A-CKO AVMs relative to the FF controls, associated with higher Ca2+ transient amplitudes and faster Ca2+ decay tau (Fig. 6A–E). In contrast, deleting MAO-A did not affect the DOB-induced PKA responses at either the PM or SR microdomains (Fig. 5B–D and Online Figure VIIC, D). Deleting MAO-A did not affect the DOB-induced responses in PKA phosphorylation, myocyte shortening, and Ca2+ transient, either (Figs. 5E, F and 6A–E). Together, these results confirm that MAO-A deletion enhances the SR-β1AR signaling and E–C coupling by attenuating catecholamine oxidation.

Cardiac-specific deletion of MAO-A selectively enhances PKA signals at the SR microdomain. MAO-A-FF and CKO AVMs expressing AKAR3 biosensors were treated with drugs as indicated. YFP/CFP FRET ratio was recorded before and after agonist stimulation for total 400 s. A–D Time courses of SR-PKA or PM-PKA FRET ratio changes after 1 µmol/L EPI (top) or DOB (bottom) stimulation in MAO-A-FF and CKO AVMs. The maximal increases in SR-PKA and PM-PKA in AVMs were plotted as mean ± SD of the indicated number of AVMs from 4 MAO-A-FF and 5 CKO mice. E, F Western blots show detection and quantification of p-Ser 16 of PLB and p-Ser 1928 of LTCC in isolated AVMs after EPI and DOB stimulation (1 µmol/L, 5 min). Dot plots represent mean ± SD of at least three independent experiments in AVMs from MAO-A-FF and CKO mice. Data were shown as mean ± SD. p values were obtained by unpaired Student’s t test (A–D) or non-parametric Kruskal–Wallis test followed by Dunn’s multiple comparisons test (E, F)

Deletion of MAO-A sensitizes myocyte E–C coupling response to catecholamine. MAO-A-FF and CKO AVMs were loaded with Ca2+ indicator, 5 µmol/L Fluo-4 AM, and paced at 1 Hz. AVMs SS and Ca2+ were recorded at the baseline and for 6 min after agonist stimulation. The maximal response was reported. A, C Representative and quantification of SS% at the baseline and after stimulation with 1 µmol/L EPI or DOB. Dot plots represent the mean ± SD of the indicated number of AVMs from 4 MAO-A-FF and 6 CKO mice. B, D Representative traces and quantification of CaT and Tau at the baseline and after stimulation with 1 µmol/L EPI or DOB. Dot plots represent the mean ± SD of the indicated number of AVMs from 4 MAO-A-FF and 6 CKO mice. E, F Agonist dose–response curves of SS% to EPI (EC50, CKO 82.9 ± 12.9 nmol/L, FF 62.9 ± 7.8 nmol/L) or DOB (EC50, CKO 42.5 ± 7.9 nmol/L, FF 20.8 ± 7.3 nmol/L) stimulation were analyzed in MAO-A-FF and CKO AVMs. Data represent the mean ± SD of the AVMs from 6 MAO-A-FF and 8 CKO mice. p values were obtained by one-way ANOVA (C, D) or two-way ANOVA (E, F) test followed by Tukey’s multiple comparison correction
Conversely, overexpressing MAO-A attenuated the SR-PKA activity and right-shifted the concentration–response curves of the SR-PKA activity after NE or EPI stimulation without affecting the PM-PKA (Online Figure VIIIA, B). MAO-A over-expression, however, did not affect the subcellular PKA activity in response to DOB stimulation (Online Figure VIIIA, B). Accordingly, MAO-A over-expression attenuated the PKA phosphorylation of PLB at ser16 but not LTCC at ser1928 induced by NE or EPI. In contrast, MAO-A over-expression did not affect the PKA-dependent phosphorylation induced by DOB (Online Figure VIIIC–E). Thus, an elevated MAO-A selectively inhibits the β1AR-PKA signaling and substrate phosphorylation at the SR in response to catecholamines.
OCT3 inhibition impairs the MAO-A-dependent augments of intracellular SR-β1AR signaling and cardiac contractility
While EPI stimulation induced the higher SR-PKA response in MAO-A-CKO AVMs relative to FF controls, inhibiting OCT3 by CORTI reduced the SR-PKA responses in both genotypes (Fig. 7A, B). In comparison, the PKA activity at the PM was comparable between MAO-A-CKO and FF AVMs, and was not affected by CORTI (Fig. 7E, F). While MAO-A-CKO AVMs exhibited higher increases in SS% and Ca2+ transient amplitude and faster Ca2+ decay tau after EPI stimulation, CORTI abrogated the these increases in both genotypes (Fig. 8A–D). In contrast, CORTI did not affect the PKA activity at either the PM or SR in responding to DOB (Fig. 7C, D, G, H) as DOB crosses the cell membrane independent of OCT3 (https://pubchem.ncbi.nlm.nih.gov). Moreover, the DOB-induced contractile and Ca2+ responses were not affected by CORTI in either MAO-A-CKO or FF AVMs (Fig. 8E–H).

Inhibiting OCT3 by corticosterone abrogates the amplification of SR-localized β1AR signaling in MAO-A-CKO. MAO-A-FF and CKO AVMs expressing AKAR3 biosensors were treated with drugs as indicated. YFP/CFP FRET ratio was recorded before and after agonist stimulation for total 400 s. A–D Time courses and quantification of changes in SR-PKA FRET ratio after EPI or DOB (1 µmol/L) stimulation in the presence of CORTI pretreatment (2 µmol/L, 5 min). E–H Time courses and quantification of changes in PM-PKA FRET ratio after EPI or DOB (1 µmol/L) stimulation in the presence of CORTI pretreatment (2 µmol/L, 5 min). Dot plots represent the mean ± SD of the indicated number of MAO-A-FF and CKO AVMs from 4 MAO-A-FF and 5 CKO mice. Data were shown as mean ± SD. p values were obtained by one-way ANOVA test followed by Tukey’s test

Corticosterone abolishes the efficacy of MAO-A deletion in promoting contractility and Ca2+ cycling. AVMs were incubated with 5 µmol/L Fluo-4 AM and paced at 1 Hz. SS and Ca2+ were recorded at baseline and for 6 min after agonist stimulation. The maximal SS and Ca2+ responses were reported. A, B Representative traces and maximal increases in SS% after EPI (1 µmol/L) stimulation with or without CORTI pretreatment (5 min). C, D Ca2+ dynamics and quantification of CaT and Tau before and after EPI (1 µmol/L) stimulation with or without CORTI pretreatment (2 µmol/L, 5 min). Dot plots represent the mean ± SD of the indicated number of AVMs 4 MAO-A-FF and 7 CKO mice. E, F Representative SS% curve and maximal SS% before and after DOB (1 µmol/L) stimulation with or without CORTI pretreatment (5 min). G, H Ca2+ dynamics and quantification of CaT and Tau before/after DOB (1 µmol/L) stimulation with or without CORT pretreatment (2 µmol/L, 5 min). Dot plots represent the mean ± SD of the indicated number of AVMs 4 MAO-A-FF and 7 CKO mice. p values were obtained by one-way ANOVA followed by Tukey’s test
We then validated the functional interaction between OCT3 and MAO-A using MAO-A CKO and OCT3-KO mice in vivo. While EPI triggered stronger increases in EF and HR in MAO-A CKO mice relative to FF controls, CORTI attenuated the inotropic and chronotropic responses in both genotypes (Fig. 9A–C and Online Table 7). Conversely, administration of EPI modestly enhanced cardiac EF and HR in OCT3-KO mice; and MAOi failed to improve the cardiac EF response to EPI stimulation (Fig. 9E–H and Online Table 8). Our results reveal that deletion and inhibition of MAO-A enhance the intracellular β1AR signaling and cardiac E–C coupling and contractility. Deletion and inhibition of OCT3 abrogate the MAO-A-dependent effects on the intracellular β1AR signaling and cardiac function.

MAO-A and OCT3 coordinate cardiac fight-or-flight response. M-mode echocardiography was recorded for 2 min at baselines and for 8 min after drug administration. The maximal cardiac response was reported. A Representative echocardiography images of MAO-A-CKO mouse heart before and after EPI stimulation (100 µg/kg, i.p.) with and without CORTI pretreatment (200 µg/kg, i.p., 5 min). B The schematic depicts that inhibiting OCT3 by CORTI suppresses the SR-localized β1ARs activation in MAO-A-CKO hearts. C The EF and HR of MAOA-A-FF and CKO mice before and after treated with EPI and CORTI described in A. n = 8 mice in each condition. D Representative echocardiography images of OCT3-KO mouse heart before and after EPI stimulation (100 µg/kg, i.p.) with and without MAOi (0.4 mg/kg, i.p.). E The schematic depicts that deletion of OCT3 prevents the effects of MAOi on enhancing the SR-localized β1AR signaling. F The EF and HR in WT and OCT3-KO mice before and after EPI and MAOi treatment described in D. n = 8 KO mice. Data were shown as mean ± SD. p values were obtained by one-way ANOVA followed by Tukey’s test
Furthermore, we characterized the interaction between MAO-A and OCT3 in mice with ischemic HF induced by myocardial infarction (MI, Online Figure IX). The ischemic HF was indicated by the Sirius Red staining and decreased EF (Online Figure IXA–C and Online Table 9). The MI hearts displayed increased MAO-A expression and reduced β1AR expression but no change in OCT3 expression relative to healthy controls, consistent with previous studies (Online Figure IXB, C and [31]). Application of MAOi promoted cardiac EF in the MI mice, whereas inhibiting OCT3 by CORTI prevented the effects of MAOi (Online Figure IXB, C and Online Table 10). These data indicate that the interaction between MAOi and OCT3 is maintained in the ischemic HF.
Discussion
The sympathetic regulation in the heart is achieved through the catecholamine-activated βARs and downstream cAMP-PKA signaling, which is diminished in cardiac diseases [29, 52]. We have recently identified a functional pool of β1ARs associated with SERCA2a on the SR that is essential for promoting SERCA2a function in E–C coupling [57]. In this study, cardiac knockout of MAO-A elevates catecholamine contents, enhances the local β1AR-cAMP-PKA signaling and PKA phosphorylation of PLB at the SR, and sensitizes myocardial contractile responsiveness to endogenous and exogenous catecholamines in vivo. However, suppressing catecholamine transport by genetic deletion of OCT3 negates the effects of MAO-A deficiency in promoting the SR-β1AR responsiveness and cardiac contraction. These data demonstrate that MAO-A and OCT3 gate the SR local β1AR signaling and fine-tune cardiac fight-or-flight responses. Additionally, CORTI, the OCT3 inhibitor, counteracts MAOi in modulating the catecholamine-driven intracellular SR-β1AR signaling and cardiac function. Our data thus unravel the contra-indicatory effects of anti-inflammatory CORTI and anti-depressant MAOi in regulating cardiomyocyte catecholamine homeostasis and cardiac contractile response, providing novel implications of those drugs in patients with cardiac diseases. Despite the promising therapeutic application indicated from both in vitro and mice model, systemic clinical trial-like studies using larger animal models, including pig or monkey model, are essential to confirm the safety and efficacy of inhibiting MAO-A or OCT3. For these confirmatory studies, a bigger sample size and pre-registered experimental protocol with a clearly defined endpoint should be deployed to improve the translation of this innovative hypothesis targeting intracellular β1-AR for managing heart function. Given this study belongs to the Stage II (hypothesis-generating exploratory studies) of preclinical research [9], the significant p value (p < 0.05) should be considered as establishing a new hypothesis.
Heterogeneity of cardiac adrenergic receptor signaling at the cell surface and intracellular membrane
The highly organized architecture in cardiomyocytes provides spatial context for compartmentalized microdomain and local cAMP/PKA signaling in regulating E–C coupling [50, 65]. Increasing evidence supports the key roles of subcellular distribution of βARs in precisely regulating local cAMP/PKA activity in cardiomyocytes [36, 57]. In a classic view, the βARs signal from the PM, including T-tubular and crest membrane. While the β1ARs are found on the entire cell surface, the β2ARs are exclusively targeted to the T-tubules [3]. The T-tubular membrane forms dyads together with the juxtamembrane of the SR, allowing tight coupling between LTCC and RyR2 and increasing [Ca2+]i during E–C coupling. The β1ARs and β2ARs at these membrane microdomains are critical for the local PKA phosphorylation of LTCC and RyR2 [4, 23, 56]. In comparison, the β1ARs at the crest membrane are believed to induce signals to other organelles, such as myofilaments and mitochondria [2]. Recent studies show functional βARs at the intracellular compartments, including the nuclei, the Golgi, and the SR [5, 36, 57]. In particular, the SR-localized b1ARs associate with SERCA2a and are critical for promoting the local PKA activity and phosphorylation of PLB and enhancing SERCA2a dependent Ca2+ reuptake into the SR [57]. These observations highlight the highly localized subcellular βAR signaling to coordinate E–C coupling machinery. The identification of specific β1AR signaling nano-domains at the dyad-associated junctional SR membrane [4] and the SERCA2a-associated longitudinal SR membrane [57] offers opportunities to explore the compartmentalized β1AR signaling in physio(patho)logical conditions.
In this study, we show that MAO-A and OCT3 are indispensable players in fine-tuning the catecholamine-induced SR-β1AR signaling in the heart. Catecholamines, including NE and EPI, are membrane impermeant and enter the cells via organic cation transporters OCT3 and plasma membrane monoamine transporter (PMAT) [14, 19]. Our data show that OCT3 deletion and inhibition can reduce the SR-b1AR signaling in the hearts, whereas the potential role of PMAT in modulating the SR-β1AR signaling in hearts remains to be examined. In comparison, many synthetic ligands, including isoproterenol, DOB, and β-blockers (e.g., carvedilol and alprenolol), can cross the membrane and reach the internal β1ARs by passive diffusion. Inside cardiomyocytes, catecholamines are subjected to the MAO-mediated oxidative degradation [7]. The SR is physically connected to mitochondria [45]. MAO-A is localized at the mitochondrial outer membrane, which displays proximity to PLB and regulates the SR-β1AR signaling in a local vicinity. Inhibition of MAO-A selectively enhances the SR-β1AR signaling without affecting the β1AR at the PM. Thus, OCT3 and MAO-A act as dual-level gauges to fine-tune intracellular catecholamine levels, the local PKA activity at the SR, and SERCA2a function during physiological stress. This setting could have two benefits: calibrating cardiac contractility according to the intensity of the sympathetic tone (i.e., the catecholamine levels) and avoiding catecholamine storms from overdriving cardiac βARs and subsequent arrhythmia.
MAO-A differentially modulates PM and intracellular adrenergic receptor signaling in HF
Desensitized βAR signaling and depressed contractile response are hallmarks of HF [6]. The desensitization of βAR signaling is correlated with the reduced β1AR density at the PM, catecholamine content, cAMP signal, and PKA activity in failing hearts [6, 27]. However, evidence indicates heterogeneity of PKA phosphorylation of substrates in failing hearts. For example, while the PKA phosphorylation of PLB is commonly reduced in HF patients, the phosphorylation of LTCC and RyR2 may not be changed or even increased [2, 4, 44]. The reason for the differential regulation of these substrates is not entirely understood. Previous studies have shown that the cardiac β1AR undergoes translocation from the PM to the intracellular compartments in HF, including increased association with SERCA2a on the SR [58]. However, the increased association between the β1AR and SERCA2a in HF fails to enhance the PKA phosphorylation of PLB. Recent studies reveal that despite excessive circulating plasma catecholamines in HF patients, cardiac NE content is decreased and accompanied by diminished NE uptake and elevated MAO-A expression [27, 47]. The increased MAO-A expression prevents the access of catecholamines to the SR-β1AR and effectively inhibits the intracellular receptor activity. Additionally, alternations of other signaling molecules, such as phosphodiesterase and phosphatases, also contribute to the remodeling of the local βAR signaling [65]. Moreover, evidence indicates that the up-regulated expressions of other adrenergic receptors, including αAR, β2AR, and β3AR, regulate cardiac contraction and heart rate in the failing human heart [24, 37, 60, 64]. While our observations are limited to mice and rabbits, future studies should be pursued to dissect the intracellular adrenergic signaling in human heart failure.
MAO-A underlies cross talk between depression and HF
Multiple dynamic processes regulate catecholamine homeostasis, including synthesis, storage, release, reuptake, and metabolism. Notably, the reuptake and metabolism of catecholamines are key regulators in multiple sympathoadrenal disorders [15, 47]. Impaired homeostasis of catecholamines is a hallmark of sympathetic dysregulation and contributes to the progression of neurological and cardiac diseases [18, 40, 66]. For example, increased MAO-A expression and catecholamine abnormalities are associated with heart diseases [18, 66], Parkinson’s diseases [40], and age-associated impaired lipolysis [41]. Thus, catecholamines occupy key positions in managing health and diseases [15]. Drugs targeting noradrenergic transmission (e.g., MAO inhibitors, inotropes, and β-blockers) are first-line treatments for sympathoadrenal disorders and contemplate the development of new therapeutic strategies [16, 34, 51].
Interestingly, depression is common in HF patients (42% of the patient [32]) and is underrecognized and linked to adverse outcomes, such as poor cardiac function and elevated mortality [8]. Effective treatments for HF patients with depression are urgently needed. However, despite the wide use of MAOi in the central nervous system, the role of MAO-A in the heart is not well understood. MAOi has been shown to increase both central and peripheral catecholamine levels [22], whereas an overdose of MAOi can lead to overaction of the sympathetic nervous system, hypertension, and tachycardia [48, 59]. Recent studies show that cardiac-specific MAO-A over-expression leads to chronic ventricle dysfunction [54]. MAO-A inhibition prevents cardiac remodeling and dysfunction in the pressure-overload-induced HF model [27]. In rodent hearts, MAO-A is also up-regulated in aging and HF and contributes to the increased reactive oxidative species [39]. Inhibition of MAO-A is beneficial in HF by preventing the production of reactive oxidative species. Our data demonstrate that MAOi enhances cardiac adrenergic response via regulating the intracellular βAR signaling, indicating that inhibition of MAO-A may offer additional benefit to the myocardium by enhancing the intracellular adrenergic signaling and rescuing the impaired SERCA2a function. Compared with conventional treatment of inotropes, which do not account for the subcellular localization of βARs and are known to cause oxidative injury and arrhythmias, restoring local catecholamines and subcellular adrenergic sensitivity may have considerable potential and advantages in treating heart diseases.
Contraindication between anti-inflammation and anti-depressant drugs in the heart
Catecholamine import and degradation by OCT3 and MAO-A determine the local concentration, duration, and physical spread of released catecholamines, critically modulating the magnitude, duration, and diversity of their effects in physio(patho)logical conditions [20]. Given their role in modulating neurotransmitters, MAO-A and OCT3 in the brain are also pathogenic factors of depressive disorders [11, 33, 35, 42, 51]. Here, we found elevated cardiac catecholamine levels in cardiac-specific MAO-A knockout mice. By contrast, OCT3 knockout mice demonstrated decreases in tissue levels of catecholamines in the hearts [53]. While MAO-A inhibition effectively enhances the SR-β1AR signaling and PKA phosphorylation of PLB, the efficacy of MAO-A inhibition is abrogated by inhibiting OCT3 with CORTI. These observations again support that a joint mechanism involving MAO-A and OCT3 exists in the central and peripheral disorders. Indeed, fludrocortisone, a corticosteroid similar to CORTI, has been used to antagonize the effect of MAOi in the clinical treatment of neuronal disorders [10]; and a high cortisol level is used as a predictor of nonresponse to MAOi treatment in patients [25]. Moreover, dexamethasone, a synthesized corticosteroid inhibiting OCT3, decreases MAO-A catalytic activity on monoamine substrates [49]. Acute treatment of dexamethasone decreases mouse heart diastolic function, which is highly regulated by SERCA function [1]. Our study thus provides a fresh perspective on the application of MAOi in managing comorbidity of HF and depression. However, cautions remain for MAOi treatment in patients with elevated CORTI levels, given that CORTI undermines the efficacies of MAOi.
In summary, we define MAO-A and OCT3 as essential regulators in calibrating access of catecholamines to the intracellular β1AR at the SR and fine-tuning cardiac fight-or-flight response. Our data support the potential utility of anti-depressant MAOi in rescuing β1AR signaling and inotropic responses in HF, whereas CORTI may exacerbate depressed cardiac ejection fraction.
Data availability
All experimental reagents used in the study are commercially available (see Online Methods). All raw data generated or analyzed during this study are included in this published article (and its supplementary files). All other data or resources are available from the corresponding author upon request.
References
Agnew EJ, Garcia-Burgos A, Richardson RV, Manos H, Thomson AJW, Sooy K, Just G, Homer NZM, Moran CM, Brunton PJ, Gray GA, Chapman KE (2019) Antenatal dexamethasone treatment transiently alters diastolic function in the mouse fetal heart. J Endocrinol 241:279–292. https://doi.org/10.1530/JOE-18-0666
Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK (2016) Genetically encoded biosensors reveal PKA hyperphosphorylation on the myofilaments in rabbit heart failure. Circ Res 119:931–943. https://doi.org/10.1161/CIRCRESAHA.116.308964
Bathe-Peters M, Gmach P, Boltz H-H, Einsiedel J, Gotthardt M, Hübner H, Gmeiner P, Lohse MJ, Annibale P (2021) Visualization of β-adrenergic receptor dynamics and differential localization in cardiomyocytes. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.2101119118
Berisha F, Götz KR, Wegener JW, Brandenburg S, Subramanian H, Molina CE, Rüffer A, Petersen J, Bernhardt A, Girdauskas E, Jungen C, Pape U, Kraft AE, Warnke S, Lindner D, Westermann D, Blankenberg S, Meyer C, Hasenfuß G, Lehnart SE, Nikolaev VO (2021) cAMP imaging at ryanodine receptors reveals β2-adrenoceptor driven arrhythmias. Circ Res 129:81–94. https://doi.org/10.1161/CIRCRESAHA.120.318234
Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve L-R, Ethier N, Trieu P, Allen BG, Hébert TE (2006) Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res 71:69–78. https://doi.org/10.1016/j.cardiores.2006.03.015
Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB (1982) Decreased catecholamine sensitivity and β-adrenergic-receptor density in failing human hearts. N Engl J Med 307:205–211. https://doi.org/10.1056/NEJM198207223070401
Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm Y-H, Brown CW, Elsworth J, Rodeheffer MS, Schultze JL, Dixit VD (2017) Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550:119–123. https://doi.org/10.1038/nature24022
Celano CM, Villegas AC, Albanese AM, Gaggin HK, Huffman JC (2018) Depression and anxiety in heart failure: a review. Harv Rev Psychiatry 26:175–184. https://doi.org/10.1097/HRP.0000000000000162
Chamuleau SAJ, van der Naald M, Climent AM, Kraaijeveld AO, Wever KE, Duncker DJ, Fernández-Avilés F, Bolli R, Transnational Alliance for Regenerative Therapies in Cardiovascular Syndromes (TACTICS) Group (2018) Translational research in cardiovascular repair: a call for a paradigm shift. Circ Res 122:310–318. https://doi.org/10.1161/CIRCRESAHA.117.311565
Cockhill LA, Remick RA (1987) Blood pressure effects of monoamine oxidase inhibitors–the highs and lows. Can J Psychiatry Rev Can Psychiatr 32:803–808. https://doi.org/10.1177/070674378703200915
Corbineau S, Breton M, Mialet-Perez J, Costemale-Lacoste J-F (2017) Major depression and heart failure: interest of monoamine oxidase inhibitors. Int J Cardiol 247:1–6. https://doi.org/10.1016/j.ijcard.2017.07.005
Crilly SE, Puthenveedu MA (2021) Compartmentalized GPCR signaling from intracellular membranes. J Membr Biol 254:259–271. https://doi.org/10.1007/s00232-020-00158-7
Dahl EF, Wright CD, O’Connell TD (2015) Quantification of catecholamine uptake in adult cardiac myocytes. Methods Mol Biol Clifton NJ 1234:43–52. https://doi.org/10.1007/978-1-4939-1755-6_5
Duan H, Wang J (2010) Selective transport of monoamine neurotransmitters by human plasma membrane monoamine transporter and organic cation transporter 3. J Pharmacol Exp Ther 335:743–753. https://doi.org/10.1124/jpet.110.170142
Eisenhofer G, Kopin IJ, Goldstein DS (2004) Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 56:331–349. https://doi.org/10.1124/pr.56.3.1
Francis GS, Bartos JA, Adatya S (2014) Inotropes. J Am Coll Cardiol 63:2069–2078. https://doi.org/10.1016/j.jacc.2014.01.016
Fraser-Spears R, Krause-Heuer AM, Basiouny M, Mayer FP, Manishimwe R, Wyatt NA, Dobrowolski JC, Roberts MP, Greguric I, Kumar N, Koek W, Sitte HH, Callaghan PD, Fraser BH, Daws LC (2019) Comparative analysis of novel decynium-22 analogs to inhibit transport by the low-affinity, high-capacity monoamine transporters, organic cation transporters 2 and 3, and plasma membrane monoamine transporter. Eur J Pharmacol 842:351–364. https://doi.org/10.1016/j.ejphar.2018.10.028
Ganguly PK, Sherwood GR (1991) Noradrenaline turnover and metabolism in myocardium following aortic constriction in rats. Cardiovasc Res 25:579–585. https://doi.org/10.1093/cvr/25.7.579
Gasser PJ (2021) Organic cation transporters in brain catecholamine homeostasis. Handb Exp Pharmacol 266:187–197. https://doi.org/10.1007/164_2021_470
Ghimire LV, Kohli U, Li C, Sofowora GG, Muszkat M, Friedman EA, Solus JF, Wood AJJ, Stein CM, Kurnik D (2012) Catecholamine pathway gene variation is associated with norepinephrine and epinephrine concentrations at rest and exercise. Pharmacogenet Genomics 22:254–260. https://doi.org/10.1097/FPC.0b013e328350a274
Grube M, Ameling S, Noutsias M, Köck K, Triebel I, Bonitz K, Meissner K, Jedlitschky G, Herda LR, Reinthaler M, Rohde M, Hoffmann W, Kühl U, Schultheiss H-P, Völker U, Felix SB, Klingel K, Kandolf R, Kroemer HK (2011) Selective regulation of cardiac organic cation transporter novel type 2 (OCTN2) in dilated cardiomyopathy. Am J Pathol 178:2547–2559. https://doi.org/10.1016/j.ajpath.2011.02.020
Happe K (2007) Monoamine oxidase inhibitors. In: Enna SJ, Bylund DB (eds) xPharm: the comprehensive pharmacology reference. Elsevier, New York, pp 1–3
Harvey RD, Hell JW (2013) CaV1.2 signaling complexes in the heart. J Mol Cell Cardiol 58:143–152. https://doi.org/10.1016/j.yjmcc.2012.12.006
Heusch G (2017) There is more to β-blockade than just blockade of β-receptors. J Am Coll Cardiol 70:193–195. https://doi.org/10.1016/j.jacc.2017.05.017
Janicak PG, Pandey GN, Sharma R, Boshes R, Bresnahan D, Davis JM (1987) Pretreatment dexamethasone suppression test as a predictor of response to phenelzine. J Clin Psychiatry 48:480–482
Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F (2014) Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol 73:34–42. https://doi.org/10.1016/j.yjmcc.2013.12.032
Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, Chen K, Gabrielson KL, Blakely RD, Shih JC, Pacak K, Kass DA, Lisa FD, Paolocci N (2010) Monoamine oxidase A mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res 106:193–202. https://doi.org/10.1161/CIRCRESAHA.109.198366
Li L, He M, Zhou L, Miao X, Wu F, Huang S, Dai X, Wang T, Wu T (2015) A solute carrier family 22 member 3 variant rs3088442 G→A associated with coronary heart disease inhibits lipopolysaccharide-induced inflammatory response. J Biol Chem 290:5328–5340. https://doi.org/10.1074/jbc.M114.584953
Lymperopoulos A, Rengo G, Koch WJ (2013) Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113:739–753. https://doi.org/10.1161/CIRCRESAHA.113.300308
Madamanchi A (2007) β-Adrenergic receptor signaling in cardiac function and heart failure. McGill J Med MJM 10:99–104
Manni ME, Rigacci S, Borchi E, Bargelli V, Miceli C, Giordano C, Raimondi L, Nediani C (2016) Monoamine oxidase is overactivated in left and right ventricles from ischemic hearts: an intriguing therapeutic target. Oxid Med Cell Longev 2016:4375418. https://doi.org/10.1155/2016/4375418
Marashdeh RAM, Parsons R, Sim TF (2019) Antidepressant prescribing patterns in heart failure patients at residential aged care facilities in Australia: a cross-sectional study. J Res Pharm Pract 8:69–74. https://doi.org/10.4103/jrpp.JRPP_18_63
Marcinkiewcz CA, Devine DP (2015) Modulation of OCT3 expression by stress, and antidepressant-like activity of decynium-22 in an animal model of depression. Pharmacol Biochem Behav 131:33–41. https://doi.org/10.1016/j.pbb.2015.01.004
Metra M, Teerlink JR (2017) Heart failure. Lancet Lond Engl 390:1981–1995. https://doi.org/10.1016/S0140-6736(17)31071-1
Naoi M, Maruyama W, Shamoto-Nagai M (2018) Type A monoamine oxidase and serotonin are coordinately involved in depressive disorders: from neurotransmitter imbalance to impaired neurogenesis. J Neural Transm 125:53–66. https://doi.org/10.1007/s00702-017-1709-8
Nash CA, Wei W, Irannejad R, Smrcka AV (2019) Golgi localized β1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCε to regulate cardiac hypertrophy. Elife. https://doi.org/10.7554/eLife.48167
O’Connell TD, Jensen BC, Baker AJ, Simpson PC (2014) Cardiac alpha1-adrenergic receptors: novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol Rev 66:308–333. https://doi.org/10.1124/pr.112.007203
Ohtani T, Mano T, Hikoso S, Sakata Y, Nishio M, Takeda Y, Otsu K, Miwa T, Masuyama T, Hori M, Yamamoto K (2009) Cardiac steroidogenesis and glucocorticoid in the development of cardiac hypertrophy during the progression to heart failure. J Hypertens 27:1074–1083. https://doi.org/10.1097/HJH.0b013e328326cb04
Petrak J, Pospisilova J, Sedinova M, Jedelsky P, Lorkova L, Vit O, Kolar M, Strnad H, Benes J, Sedmera D, Cervenka L, Melenovsky V (2011) Proteomic and transcriptomic analysis of heart failure due to volume overload in a rat aorto-caval fistula model provides support for new potential therapeutic targets—monoamine oxidase A and transglutaminase 2. Proteome Sci 9:69. https://doi.org/10.1186/1477-5956-9-69
Pifl C, Hornykiewicz O, Blesa J, Adánez R, Cavada C, Obeso JA (2013) Reduced noradrenaline, but not dopamine and serotonin in motor thalamus of the MPTP primate: relation to severity of parkinsonism. J Neurochem 125:657–662. https://doi.org/10.1111/jnc.12162
Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, Mendes R, Gres V, Kubasova N, Morris I, Arús BA, Larabee CM, Vasques M, Tortosa F, Sousa AL, Anandan S, Tranfield E, Hahn MK, Iannacone M, Spann NJ, Glass CK, Domingos AI (2017) Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med 23:1309–1318. https://doi.org/10.1038/nm.4422
Plieger T, Melchers M, Felten A, Lieser T, Meermann R, Reuter M (2019) Moderator effects of life stress on the association between MAOA-uVNTR, depression, and burnout. Neuropsychobiology 78:86–94. https://doi.org/10.1159/000499085
Purgert CA, Izumi Y, Jong Y-JI, Kumar V, Zorumski CF, O’Malley KL (2014) Intracellular mGluR5 can mediate synaptic plasticity in the hippocampus. J Neurosci Off J Soc Neurosci 34:4589–4598. https://doi.org/10.1523/JNEUROSCI.3451-13.2014
Reddy GR, West TM, Jian Z, Jaradeh M, Shi Q, Wang Y, Chen-Izu Y, Xiang YK (2018) Illuminating cell signaling with genetically encoded FRET biosensors in adult mouse cardiomyocytes. J Gen Physiol 150:1567–1582. https://doi.org/10.1085/jgp.201812119
Rossini M, Filadi R (2020) Sarcoplasmic reticulum-mitochondria kissing in cardiomyocytes: Ca2+, ATP, and undisclosed secrets. Front Cell Dev Biol 8:532. https://doi.org/10.3389/fcell.2020.00532
Schomig A, Haass M, Richardt G (1991) Catecholamine release and arrhythmias in acute myocardial ischaemia. Eur Heart J 12:38–47. https://doi.org/10.1093/eurheartj/12.suppl_F.38
Schroeder C, Jordan J (2012) Norepinephrine transporter function and human cardiovascular disease. Am J Physiol-Heart Circ Physiol 303:H1273–H1282. https://doi.org/10.1152/ajpheart.00492.2012
Shalom Feinberg S (2019) Spontaneous MAOI hypertensive reaction, not likely armodafinil–tranylcypromine interaction. J Neurol Sci 398:1. https://doi.org/10.1016/j.jns.2019.01.003
Soliman A, Udemgba C, Fan I, Xu X, Miler L, Rusjan P, Houle S, Wilson AA, Pruessner J, Ou X-M, Meyer JH (2012) Convergent effects of acute stress and glucocorticoid exposure upon MAO-A in humans. J Neurosci 32:17120–17127. https://doi.org/10.1523/JNEUROSCI.2091-12.2012
Surdo NC, Berrera M, Koschinski A, Brescia M, Machado MR, Carr C, Wright P, Gorelik J, Morotti S, Grandi E, Bers DM, Pantano S, Zaccolo M (2017) FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat Commun 8:1–14. https://doi.org/10.1038/ncomms15031
Thomas SJ, Shin M, McInnis MG, Bostwick JR (2015) Combination therapy with monoamine oxidase inhibitors and other antidepressants or stimulants: strategies for the management of treatment-resistant depression. Pharmacotherapy 35:433–449. https://doi.org/10.1002/phar.1576
Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J (2009) The sympathetic nervous system in heart failure. J Am Coll Cardiol 54:1747–1762. https://doi.org/10.1016/j.jacc.2009.05.015
Vialou V, Balasse L, Callebert J, Launay J-M, Giros B, Gautron S (2008) Altered aminergic neurotransmission in the brain of organic cation transporter 3-deficient mice. J Neurochem 106:1471–1482. https://doi.org/10.1111/j.1471-4159.2008.05506.x
Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, De Paulis D, Couderc B, Spreux-Varoquaux O, Tortosa F, Garnier A, Knauf C, Valet P, Borchi E, Nediani C, Gharib A, Ovize M, Delisle M-B, Parini A, Mialet-Perez J (2013) p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal 18:5–18. https://doi.org/10.1089/ars.2011.4373
Wang W, Qiao Y, Li Z (2018) New insights into modes of GPCR activation. Trends Pharmacol Sci 39:367–386. https://doi.org/10.1016/j.tips.2018.01.001
Wang W, Zhang H, Gao H, Kubo H, Berretta RM, Chen X, Houser SR (2010) β1-Adrenergic receptor activation induces mouse cardiac myocyte death through both L-type calcium channel-dependent and -independent pathways. Am J Physiol Heart Circ Physiol 299:H322–H331. https://doi.org/10.1152/ajpheart.00392.2010
Wang Y, Shi Q, Li M, Zhao M, Reddy Gopireddy R, Teoh J-P, Xu B, Zhu C, Ireton KE, Srinivasan S, Chen S, Gasser PJ, Bossuyt J, Hell JW, Bers DM, Xiang YK (2021) Intracellular β1-adrenergic receptors and organic cation transporter 3 mediate phospholamban phosphorylation to enhance cardiac contractility. Circ Res 128:246–261. https://doi.org/10.1161/CIRCRESAHA.120.317452
Wang Y, Zhao M, Shi Q, Xu B, Zhu C, Li M, Mir V, Bers DM, Xiang YK (2021) Monoamine oxidases desensitize intracellular β1AR signaling in heart failure. Circ Res 129:965–967. https://doi.org/10.1161/CIRCRESAHA.121.319546
Waring WS, Wallace WAH (2007) Acute myocarditis after massive phenelzine overdose. Eur J Clin Pharmacol 63:1007–1009. https://doi.org/10.1007/s00228-007-0360-y
West TM, Wang Q, Deng B, Zhang Y, Barbagallo F, Reddy GR, Chen D, Phan KS, Xu B, Isidori A, Xiang YK (2019) Phosphodiesterase 5 associates with β2 adrenergic receptor to modulate cardiac function in type 2 diabetic hearts. J Am Heart Assoc 8:e012273. https://doi.org/10.1161/JAHA.119.012273
Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S, O’Connell TD (2008) Nuclear α1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res 103:992–1000. https://doi.org/10.1161/CIRCRESAHA.108.176024
Xu B, Li M, Wang Y, Zhao M, Morotti S, Shi Q, Wang Q, Barbagallo F, Teoh JP, Reddy GR, Bayne EF, Liu Y, Shen A, Puglisi JL, Ge Y, Li J, Grandi E, Nieves-Cintron M, Xiang YK (2020) GRK5 controls SAP97-dependent cardiotoxic β1 adrenergic receptor-CaMKII signaling in heart failure. Circ Res 127:796–810. https://doi.org/10.1161/CIRCRESAHA.119.316319
Yan M, Webster LT, Blumer JL (2002) Kinetic interactions of dopamine and dobutamine with human catechol-O-methyltransferase and monoamine oxidase in vitro. J Pharmacol Exp Ther 301:315–321. https://doi.org/10.1124/jpet.301.1.315
Yang W, Wei X, Su X, Shen Y, Jin W, Fang Y (2019) Depletion of β3-adrenergic receptor induces left ventricular diastolic dysfunction via potential regulation of energy metabolism and cardiac contraction. Gene 697:1–10. https://doi.org/10.1016/j.gene.2019.02.038
Zaccolo M, Zerio A, Lobo MJ (2021) Subcellular organization of the cAMP signaling pathway. Pharmacol Rev 73:278–309. https://doi.org/10.1124/pharmrev.120.000086
Zelis R, Clemson B, Baily R, Davis D (1992) Regulation of tissue noradrenaline in the rat myocardial infarction model of chronic heart failure. Cardiovasc Res 26:933–938. https://doi.org/10.1093/cvr/26.10.933
Funding
This work was supported by the National Institutes of Health grant R01-HL147263 (YKX), and a VA Merit grant 01BX005100 (YKX). YW and CZ are recipients of the American Heart Association postdoctoral fellowship. YKX is an established American Heart Association investigator.
Author information
Authors and Affiliations
Contributions
YKX and YW conceived of the idea and designed the research; YW, MMZ, BX, SMB, CQZ, AJX, NK, and FDL collected data; JCS, AJ, JWH, and NP provided critical research resources; YW, SMB, MMZ, HBN, NP, and YKX analyzed and interpreted data; YW and YKX drafted the manuscript; all authors critically revised the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Zhao, M., Xu, B. et al. Monoamine oxidase A and organic cation transporter 3 coordinate intracellular β1AR signaling to calibrate cardiac contractile function. Basic Res Cardiol 117, 37 (2022). https://doi.org/10.1007/s00395-022-00944-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-022-00944-5