
Abstract
Dye-sensitized solar cells (DSSCs) are cost-effective, sustainable, and versatile electricity producers, allowing them to be incorporated into a variety of devices. In this work, we explore the usage of pharmacophore modeling to identify metal-free dyes for DSSCs by means of virtual screening. Pharmacophore models were built based on experimentally tested sensitizers. Virtual screening was performed against a large dataset of commercially available compounds taken from the ZINC15 library and identified multiple virtual hits. A subset of these hits was subjected to DFT and time-dependent-DFT calculations leading to the identification of two compounds, TSC6 and ASC5, with appropriate molecular orbitals energies, favorable localization, and reasonable absorption UV–vis spectra. These results suggest that pharmacophore models, traditionally used in drug discovery and lead optimization, successfully predicted electronic properties, which are in agreement with the theoretical requirements for sensitizers. Such models may therefore find additional usages as modeling tools in materials sciences.
Similar content being viewed by others
Introduction
Chemoinformatics and materials-informatics are rapidly developing fields that apply computational techniques to the discovery of new compounds1,2,3. In particular, machine learning (ML) methods often referred to as quantitative structure-activity relationship (QSAR) modeling are used to derive predictive models for a variety of molecular properties. In the field of drug discovery, one of the common tools for predicting the binding of a ligand to its bio-target based on the three-dimensional (3D) complementarity between the two is pharmacophore modeling. In the present study, we apply pharmacophore modeling to a materials informatics problem, namely, the study of metal-free dyes for dye-sensitized solar cells (DSSCs).
A pharmacophore model is a set of functional groups defined by their identity (see below) and the spatial relationship (e.g., distances) between them, shared by a set of compounds with a specific activity and absent from compounds devoid of this activity4,5,6. The functional groups considered in pharmacophore modeling typically include negatively ionizable centers (NIC) and positively ionizable centers (PIC), i.e., atomic centers which can carry negative or positive charge under specific conditions, hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), hydrophobic interactions (HI), and aromatic rings (AR)5,7. The different features and their representation in LigandScout8 are shown in Fig. 1. Pharmacophore models are best derived from protein-ligand complexes. However, when this information is unavailable they could also be derived by analyzing isolated ligands (ligand-based pharmacophores). This is in fact the case in the present study, since the data available to us do not contain any information on the 3D complementarity between the dyes and the semiconductor onto which they are adsorbed. In such cases, the procedure for deriving pharmacophore models consists of the following steps5,9,10:
-
(1)
Obtain a set of active compounds.
-
(2)
For each of the compounds, perform a conformational search to identify a diverse set of conformations (a conformational ensemble). Pharmacophore modeling assumes that each compound is represented by its so-called bioactive conformation, namely, the conformation adopted by the compound when performing its activity11. In the case of ligand-based pharmacophore modeling, this information is typically unavailable. A diverse conformational ensemble holds the potential of containing the bioactive conformation12.
-
(3)
For each conformation of each ligand, identify all pharmacophoric features (NIC, PIC, HBA, HBD, HI, AR) and the distances between them.
-
(4)
Superimpose the conformational ensembles of the active compounds in order to identify a pharmacophore (i.e., a set of features and distances) common to all or most. Typically, a set of active compounds will give rise to several pharmacophores.
-
(5)
Validate the resulting pharmacophore(s) for its ability to pick active compounds from within a pool of inactive or random, yet presumed to be inactive compounds. As with all machine learning techniques, validation should be performed on an external dataset, namely a dataset not used for the construction of the pharmacophore.

Pharmacophore features and their color codes.
In recent years, pharmacophore models have been mainly used in computer-aided drug design to provide chemical insight into ligand-proteins interactions and for virtual screening (VS). Virtual screening is a process in which a computational model developed to predict a certain activity is used to screen large libraries of compounds in search for those predicted to possess this activity13. Pharmacophore models have been widely used in VS efforts in drug discovery projects14,15,16,17 and in the present study we wish to investigate their usefulness for the discovery of dyes for DSSCs.
DSSCs, pioneered by O’Regan and Grätzel18, emerged out of growing interest around the world in renewable energy, specifically in solar energy. DSSCs traditionally consist of five components: (1) A photo-anode, (2) A mesoporous semiconductor metal oxide film layer (commonly TiO2), (3) A molecular sensitizer (dye), (4) An electrolyte/hole transporter medium (HTM), (5) A counter electrode. Current is generated upon illumination, when a photon is absorbed by the sensitizer, causing electron injection from the photo-excited sensitizer into the semiconductor’s conduction band. Completion of the circuit is done by regeneration of the dye by the electrolyte solution, which is then reduced at the counter electrode18,19,20. The dye is a critical component for the cell’s function, as it is responsible for both photon harvesting and electron injection into the semiconductor surface21,22. Therefore, the dye’s properties govern the sunlight to electricity conversion capability of the cell, which is estimated by sunlight-to-power conversion efficiency (PCE) calculated from the generated photocurrent density (Jsc), the open-circuit potential (Voc), the fill factor (FF) and the incident illumination intensity (Pin) as follows19,23:
A compound can be utilized as a dye in a DSSC, as long as it meets the following criteria20,24:
-
(1)
The compound should have an absorption spectra covering ultraviolet–visible (UV–vis) and near-infrared (NIR) regions. Its molar extinction coefficient should be as high as possible.
-
(2)
The highest occupied molecular orbital (HOMO) should lie below the energy level of the redox electrolyte and farthest away from the conduction band of the semiconductor. The lowest unoccupied molecular orbital (LUMO) should be located close to the semiconductor’s surface, and higher than the semiconductor conduction band potential.
-
(3)
The sensitizer should have a hydrophobic periphery in order to enhance the cell’s stability, minimizing direct interactions between electrolyte and anode.
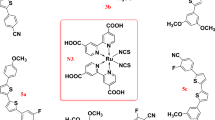
In their seminal work, O’Regan and Grätzel18, achieved a solar conversion efficiency of 7.1% with a ruthenium-based complex. Nazeeruddin et al.25 discovered that using cis-di(thiocyanato)bis(2,2-bipyridyl-4,4′-dicarboxylate)ruthenium(II) dye, known as N3 (Fig. 2) in DSSC leads to a PCE of 10%. Subsequently, additional ruthenium-based dyes were reported, such as black dye and N719 (Fig. 2), exhibiting efficiencies of more than 11%26,27,28,29. Ruthenium is a rare, expensive metal, and ruthenium-based sensitizers are difficult to synthesize and have relatively low molar extinction coefficients. Thus, extensive research efforts were dedicated to metal-free dyes for DSSC-based devices. Even though organic dyes typically have lower PCE than metal-based dyes, they have several notable advantages including low price, ease of synthesis, tunable (e.g., by chemical modification) photovoltaic and electrochemical properties and high molar extinction coefficients20,22,24,30. Significant progress in the development of organic dyes for DSSCs came with the usage of dyes with an electron donor-conjugated π system-electron acceptor (D-π-A) architecture30. This architecture enables a charge-separated resonance structure, as shown in Fig. 3, which can create an electron-hole pair22. Within this design, the acceptor is also responsible for the electron injection into the conduction band of the semiconductor and a wide absorption spectrum can be achieved. This architecture has been often manipulated to generate additional structural patterns20,31,32,33, and these structures make metal-free sensitizers easily tunable and flexible34. Usage of metal-free dyes in DSSCs was leveraged by Hara et al.35,36 and Yanagida et al.37 who used oligoenes containing dialkylaminophenyl as donors and cyanoacrylic acid (CAA) as the acceptor. These dyes achieved PCE of up to 6.8% in DSSCs with iodide/triiodide electrolyte. Many different organic dyes have been reported, utilizing the versatility of the D-π-A framework with different chemical groups38,39,40,41,42. In 2017, a DSSC incorporating a metal-free dye, achieving an efficiency of 11.18% with TTAR-b8 as sensitizer and \({{{\mathrm{I}}}}^ - /{{{\mathrm{I}}}}_3^ -\) as the electrolyte was reported43. Through molecular engineering, existing sensitizers could be modified in order to enhance desirable properties and new candidates for metal-free DSSCs could be synthesized and tested30,32,44. However for this paradigm to be applicable, a better understanding of the relations between molecular structures and photovoltaic properties is needed29,45. We propose that pharmacophore models could provide this understanding.

Molecular structures of ruthenium-based dyes: N3, N719 and Black dye.

Illustration of D-π-A architecture with TiO2.
At first sight, it might seem counter-intuitive to construct pharmacophore models for DSSCs from molecular features, typically used for analyzing ligand-protein interactions, for the study of photovoltaic (PV) properties. Indeed previous efforts to model the activities of DSSCs mainly relied on quantum mechanical (QM) calculations21,26,44. However, these calculations are time consuming and as a result, could not be performed on large sets of compounds, thereby precluding the usage of the resulting models for virtual screening. Other studies relied on machine learning methods including evolutionary algorithms46,47,48,49, multiple linear regression (MLR)48,49,50, partial least squares (PLS)45,47,50,51, gradient boosted regression trees52, and neural networks52 to model a variety of PV properties with mixed levels of successes. Of note, most of these models were based on two-dimensional (2D) descriptors thereby excluding all information pertaining to the 3D structures of the compounds. This information however is automatically accounted for by pharmacophore models. Moreover, we hypothesized that using the standard pharmacophoric features would account for key properties of active dyes in particular in what pertains to their aromaticity (via the AR feature), hydrophobicity (via the HI feature), and charge distribution (via the NIC, PIC, HBA, and HBA features).
Given a validated pharmacophore model, it could be applied in virtual screening to accelerate the discovery of new organic sensitizers. Pharmacophore-based VS is advantageous over other methods due to its low computational complexity and since it allows for finding hits that have chemical features similar to those of known active dyes, yet with molecular scaffolds that haven’t been tested for that purpose17.
Results
Pharmacophore modeling
Ligand-based pharmacophore models were generated for the four largest scaffold families found in the filtered database, which was derived from the dye-sensitized solar cell database (DSSCDB) published by Venkatraman et al.53, namely, carbazoles, indolines, phenothiazines, and triphenylamines (Table 1). Radial distribution function (RDF) based clustering (see Methods section) was employed on the most active/least active seven and most active/least active ten compounds from each family. Every clustering process resulted in one or two significant clusters (containing more than two compounds) from which pharmacophore models were derived. In addition, models were also built from the entire scaffold-based subsets described above. Pharmacophore alignment (PA) score-based clustering was also applied in several cases. In detail, 11 models were created for the carbazole family (eight based on highest activity compounds and three based on lowest activity compounds), nine for the indoline family (seven based on highest activity compounds and two based on lowest activity compounds), eight for the phenothiazine family (six based on highest activity compounds and two based on lowest activity compounds), and 17 for the triphenylamine family (13 based on highest activity compounds and four based on lowest activity compounds). Models derived from inactive compounds represent a collection of undesired pharmacophoric features. A list of all models based on division by scaffold is given in Supplementary Table 1.
In addition, the most active dyes (MAD) and the least active dyes (LAD), irrespective of their scaffold as well as the most active dyes from each scaffold were also clustered with both RDF and PA methods. Every clustering process resulted in 1–3 different significant clusters from which additional models were derived. In total, 33 different pharmacophore models were created using this approach. Out of them, 26 models were based on the most active dyes with one model being derived from the entire MAD set, and others being derived based on specific scaffolds within the MAD set or clusters derived from them. Few models were derived by fusing models generated from the individual scaffolds with MAD-based pharmacophore models. Seven additional pharmacophore models were developed using the LAD. A list of all models based on MAD and LAD is given in Supplementary Table 2.
Pharmacophore validation and selection
Each of the above-described pharmacophore models was evaluated for its ability to retrieve known active dyes taken from the DSSCDB which were embedded within a pool of structurally similar, i.e., having the same scaffold, random compounds (presumed to be inactive) retrieved from the ZINC1554 database (see Methods section). The MAD and LAD pharmacophore models were derived from dyes with different scaffolds and were therefore validated using random compounds containing these scaffolds. The validation sets for each scaffold are given in Table 2.
As elaborated in the “Methods” section, the performances of the different models were evaluated by several metrics including the area under the curve (AUC), the enrichment factor at 1% of the library (EF1%), the Matthews correlation coefficient (MCC), and the percentage of retrieved high activity (%HA) and low activity (%LA) compounds. These metrics highlight different aspects of models’ performances. Thus models with high EF1% retrieved true positives (TPs) early in the VS process whereas models with high AUC values demonstrated overall good performances. The MCC values were taken into consideration in order to select the models that could perform the best with imbalanced data. The %HA and %LA measures were also considered to ensure that compounds mined during VS would have features that better correspond with high-activity dyes (and not with low-activity dyes).
Values of all these metrics for the 78 pharmacophore models derived in this work are presented in Supplementary Table 3. Based on AUC and EF1% values, coupled with a visual inspection, we have selected a subset consisting of 18 high activity-based and three low activity-based models for virtual screening. The latter were used to eliminate false positive (FP) compounds that may result from the VS procedure with the active compounds-based pharmacophores. High activity-based and low activity-based models are presented in Tables 3 and 4, respectively and their validation statistics is provided in Table 5. In Tables 3 and 4, each model is shown aligned to one compound from the dataset used for its construction.
Virtual screening
The ZINC15 in-stock library comprised of more than 13.8 M compounds was screened against all 18 high activity-based pharmacophore models. Since all these models differed from each other both in their features as well as in their performances, we have adopted a consensus approach focusing on compounds that matched at least nine models. 5636 compounds met this criterion. Next, these compounds were screened against the three filtering pharmacophores (i.e., pharmacophores built based on low PCE dyes). This filtering process resulted in a final hitlist of 446 compounds that did not match any of the three low activity-based models (see Supplementary Table 4). Finally, these compounds were screened again against the 18 high activity-based pharmacophore models, in order to calculate their total score (TS) and average score (AS) values (see Eqs. 9 and 10 in the “Methods” section). The ten compounds with the highest TS and AS values (altogether 20 compounds) are listed in Table 6. These compounds were subjected to quantum mechanics (QM) calculations as discussed below.
As can be seen, most hits contain a carboxylic acid moiety, and some also contain rhodanine moieties, both of which can function as electron acceptor in DSSCs’ sensitizers55. In addition, most compounds possess interesting moieties that could function as potential donors such as barbiturate, triazine, and other aromatic moieties. However, most compounds lack a continuous conjugated π-bridge moiety to complete the D-π-A architecture. This is presumably due to limited emphasis given to the π-linkers in the pharmacophore models. In addition, most compounds lack a hydrophobic moiety in their periphery in accord with the absence of a corresponding hydrophobic feature in the pharmacophore models. This may seem puzzling since many of the active dyes are decorated by long alkyl chains, but could be rationalized by the vast conformational space characteristic of such flexible chains which may well challenge the alignment process and prevent the identification of a fixed pharmacophoric feature.
Because of these potential deficiencies of the top-ranking hits, we have visually inspected the 5636 compounds that matched at least 50% of the pharmacophores and selected an additional 15 of them for QM calculations. These compounds are shown in Fig. 4.

Manually selected compounds for QM calculations.
QM calculations
In order to computationally estimate whether the compounds identified by the pharmacophore-based VS procedure have the proper electronic properties for DSSCs, we have subjected the final list of 35 structures (shown in Table 6 and Fig. 4) to density functional theory (DFT) and time-dependent-DFT (TD-DFT) calculations to predict their HOMO/LUMO energies and UV–vis spectra. QM results of these compounds are reported in Supplementary Table 5. In order to provide a baseline for comparison, we have performed DFT and TD-DFT calculations on the 16 most active compounds reported in the DSSCDB (see the Methods section for more details) for which experimentally determined absorption spectra are available. The experimentally determined PCEs and λmax values together with the calculated HOMO, LUMO, and the λmax values are reported in Supplementary Table 6.
Discussion
Pharmacophore validation and selection
Looking at the various pharmacophore models in Tables 3 and 4, the prominence of the above-discussed D-π-A architecture is clearly evident. More specifically, in all models, barring models 1 and 54, the acceptor (A) is presented by at least one HBA feature, occasionally with one or two additional HBA features (for example in models 46 and 51) or NIC (models 6 and 78). This pattern matches the chemical characteristics of acceptors in metal-free sensitizers, which also serve as anchoring groups to the semiconductor component, and conventionally consists of carboxylic acid, cyanoacrylic acid or rhodanine24,55. The donor (D) is an electron-rich fragment in the sensitizer56,57, generally aromatic32,37,58. The alignment process during the pharmacophore generation determines whether an aromatic moiety will be represented by an aromatic or hydrophobic feature thus, the donor is represented either by AR or by HI or both. Similarly, π-bridges can be seen in some models (for example models 3 or 21); however, most models emphasize the donor and acceptor groups. This may be due to the sensitivity of the AR feature to the orientation of aromatic units59. Importantly, all these models have no more than five mandatory pharmacophoric features, highlighting the simplicity of the pharmacophore concept.
As can be seen from Table 5, all models achieved AUC above 0.5, indicating their above-random predictive ability for compounds of the same scaffold, with several models achieving AUC close to 1 which points to a perfect prediction (e.g., 21, 39, and 55). MCC values vary, yet are all above zero, suggesting at least some predictive ability, with some models achieving MCC values around 0.9, indicating an almost perfect predictive ability (e.g., 39 and 54). With respect to EF1% the highest values were found for the most selective models, e.g., models 6, 54, and 58, which did not retrieve any random compound during the validation process. All the selected phenothiazine (59, 61, 65, and 66) and triphenylamine-based (21, 39, 44–46, 51, 52, and 72) models have the same EF1% values since all retrieved the same number of active compounds within the first 1% of the library. With respect to models’ ability to retrieve high activity (HA) vs. low activity (LA) compounds, 15 out of the 18 models that were generated from dyes of high PCE values retrieved at least 60% of the HA dyes. However, some of these models also retrieved LA dyes. In particular, models like 54 and 55, which exhibited high AUC and decent %HA rate, retrieved all the dyes of low activity. On the other hand, models 3 and 7, both with MCC values lower than 0.5, retrieved only 12.5% and 27.5% of the low activity dyes, respectively. Based on the results it can be concluded that most selected pharmacophore models can successfully differentiate between photoactive sensitizers and structurally similar random compounds.
Regarding the three filtering models, generated from dyes of low PCE, it can be seen that apart from model 67, the two other models have relatively low MCC, yet higher than zero, suggesting that they can still differentiate between low activity dyes and decoys. Models 65 and 67 were selected because of their high %LA and their remarkable true negatives (TNs) retrieval rate (see Supplementary Table 3). Model 66 was selected because of its low %HA and despite its low MCC value. Model 67 may be too general, but could still be useful for filtering TNs with good confidence.
QM calculations
The results of our calculations on the 35 selected hits suggest that all met the criterion for the HOMO lying below the energy level of the electrolyte medium, and most of them (31 out of 35 compounds) met the criterion for the LUMO lying above the semiconductor’s conduction band potential (−4.85 eV for \({{{\mathrm{I}}}}^ - /{{{\mathrm{I}}}}_3^ -\) medium and −4.0 eV for TiO2 as semiconductor according to Fedowski et al.33). Notably, all compounds which did not meet the latter condition were manually selected, and none came from the list of highest scoring hits. Two compounds, TSC6 and ASC5, also met the condition for the HOMO and LUMO to lie furthest away from each other, as the LUMO should lie closest to the semiconductor surface, localized on the acceptor (A) part, while the HOMO should lie furthest from it, on the donor (D) part, in order to accelerate recombination of the oxidized dye (after electron injection) from the electrolyte medium and improve the cell’s efficiency20,60,61,62. These two compounds are presented in Fig. 5 alongside a compound taken from the DSSCDB, namely TA-DM-CA with experimentally determined LUMO energy and absorption spectrum, and a reported PCE of 9.67%63, positioning it within the top three most active metal-free sensitizers, that was also subjected to DFT and TD-DFT calculations as part of the baseline set. In TSC6, the HOMO is localized on the carbazole fragment, which is one of the most common donors for metal-free sensitizers, as can be seen in the DSSCDB and Table 1. At the same time, the LUMO is localized on the carboxylic acid, a highly common acceptor group in DSSCs’ sensitizers64,65, as well as on an amide carbonyl group which is a part of the succinimide moiety and could enhance the anchoring strength of the compound to the semiconductor surface66. The HOMO of ASC5 is localized on a benzene ring, further from the LUMO that is localized on the nitrophenyl unit. Nitro units have previously been utilized as acceptor groups in DSSCs67, although with rather low photovoltaic activity due to relatively low absorption onto TiO268. As expected, the calculated HOMO/LUMO energies of TA-DM-CA met the necessary criteria. However the calculated values deviated from the experimental values in particular for the LUMO which according to DFT calculations was estimated to be at −0.658 eV but found at −3.02 eV according to the experimental work of Im et al. Experimentally, the LUMO energy was estimated from the compound’s oxidation potential in combination with its optical band gap, estimated from the edge of the absorption spectrum63,69,70. Despite this discrepancy, we note that the HOMO and LUMO energies calculated for TSC6 and ASC5 lie well within the energy ranges of these orbitals calculated for the active dyes in the DSSCDB (Supplementary Table 6).

a HOMO/LUMO energy diagrams and structures. b Absorption spectrum for TSC6. c Absorption spectrum for ASC5. d Absorption spectrum for TA-DM-CA.
TD-DFT calculations showed that all 35 compounds (including TSC6 and ASC5) had absorption spectra in the UV–vis region, peaking for most compounds between 200 and 400 nm in the middle and near UV regions. The lack of rich π linkers in the selected dyes may be the reason for the spectra to be narrower than traditional organic sensitizers’ absorption spectra, specifically in the red and near IR region. The manually selected compounds (Fig. 4) exhibited broader spectra in the visible range due to their extended pi-conjugated frameworks in comparison with the compounds selected by scoring.
It is also important to note that the absorption spectrum of TA-DM-CA predicted by TD-DFT calculations was found to be not as broad as the experimentally determined absorption spectrum in ethanol, which may indicate that these calculations underestimate the spectra of the VS-selected candidates. This assumption is backed up by the known tendency of the CAM-B3LYP functional to yield blue-shifted spectra in comparison with experiment71,72. Indeed a comparison between experimentally and calculated λmax values for active dyes (Supplementary Table 6) suggest the latter to produce an average blue shift of 104 ± 76 nm. Using this value, we revised the estimation of the maximal absorption wavelengths in the UV–vis region for compounds TSC6, ASC5, and TA-DM-CA from the QM-calculated values of λ = 286 nm, λ = 293 nm and λ = 293 nm respectively to λ = 390 nm, λ = 397 nm and λ = 397 nm respectively. The experimentally determined λmax value for TA-DM-CA is 433 nm, in reasonable proximity to the revised prediction.
Based on the above results, we suggest that TSC6 and ASC5 are plausible candidates to be used as dyes in DSSCs, yet their performances should be verified experimentally.
As can be seen from Supplementary Table 5, other compounds met the required criteria for the HOMO and LUMO energies and displayed acceptable absorption spectra, e.g., TSC4, ASC3, MSC1, and MSC9, however, their predicted LUMO is localized on functional groups that are not considered traditional anchoring groups, and their ability to function in DSSCs requires further investigation.
In order to further validate the pharmacophore-based VS approach, we have selected a random set of 20 compounds from within those that matched more than three yet less than nine pharmacophore models, and subjected them to the same QM calculations. The results (Supplementary Table 7) demonstrated that out of these 20 compounds, two did not meet the HOMO/LUMO energies criteria (whereas all 20 highest-ranking compounds did), and none of them exhibited a proper separation between the HOMO and the LUMO. In addition, several compounds did not have a sustainable pi-conjugated framework. Finally, the average calculated λmax value for the 20 randomly selected compounds was lower than that for the 20 highest-ranking ones (255 nm versus 297 nm, respectively). While these are perhaps not staggering differences, we note that the 20 random compounds still matched a number of pharmacophore models and consequently may well display some photovoltaic activity. Selecting for comparison compounds that did not match any pharmacophore model would have been inappropriate since such compounds are likely too different from solar cell dyes to provide any meaningful conclusions.
In an attempt to achieve a better candidate for experimental evaluation, two modifications of TSC6 were suggested and subjected to DFT and TD-DFT calculations. In the first, the hydroxyl group on the phenyl unit was substituted with cyanoacrylic acid (CAA), and in the second, the succinimide unit was replaced with a pyrroledione moiety, turning the single C(sp3)-C(sp3) into a double bond. The DFT and TD-DFT results of the hypothesized compounds (termed TSC6-CAA and TSC6-US) are shown in Fig. 6.

a HOMO/LUMO energy diagrams and structures. b Absorption spectrum for TSC6-CAA. c Absorption spectrum for TSC6-US.
It can be seen that these modifications induced a slight favorable shift in the absorption spectrum toward the visible range in both compounds, with maximal absorption wavelengths estimated at λ = 308 nm and λ = 320 nm for TSC6-CAA and TSC6-US, respectively, and at λ = 412 nm and λ = 424 nm following the spectral shift correction. This may also indicate a possible contribution of the CAA moiety in organic sensitizers to the increase in their light absorption, in addition to its role as a highly common acceptor (A). While the HOMO in both compounds remained localized on the carbazole units, the LUMO in TSC6-US is mainly localized on the pyrroledione unit, which may hinder its applicability as a sensitizer due to a potential suboptimal attachment to the TiO2 layer. In TSC6-CAA the LUMO is localized on the CAA as expected, maintaining the amide carbonyl localization observed for TSC6 (Fig. 5).
In conclusion, we have developed an approach to identify potential candidates for metal-free dyes for DSSCs by VS using 3D pharmacophore models. The pharmacophore models were built from a dataset of experimentally tested dyes, and then used to screen the in-stock portion of the ZINC15 database which contains over 13.8 M compounds, in order to retrieve those that match the relevant pharmacophoric features.
The inclusion of multiple pharmacophore models, as well as models which were based on poor-performing dyes, enabled us to focus on hits with a higher probability of being active. Indeed, QM calculations performed on a small subset of the highest-ranking hits identified two with the appropriate electronic properties. In this respect, we wish to emphasize that while the QM results on HOMO/LUMO energies and λmax values do not always match the experimental data, in this work we demonstrated that values of these parameters calculated for our two most promising hits nicely fall within the range of these parameters calculated for experimentally active dyes. Yet the results of this research require experimental validation in order to unambiguously confirm the power of pharmacophore models as predictors of dyes for DSSCs.
The methodology presented in this work could be improved in several ways. First, an implicit assumption of ligand-based pharmacophore modeling is that each compound is represented by its active conformation. In this respect, identifying the exact conformation(s) of the dye when bound to the TiO2 layer can significantly improve the resulting pharmacophore models and the subsequent VS. Second, the pharmacophore models could be improved by adding additional features, for example, to account for the π-bridges. Third, the selection process of models for VS could be improved. Fourth, higher-level QM methods for improving the characterization of the selected VS hits could be evaluated. Finally, while in this work the construction of the pharmacophore models was based on dyes’ PCE, Jsc and Voc are also possible properties for modeling.
Yet regardless of these potential improvements, we suggest that, the ability of pharmacophore models to identify dyes with favorable (predicted) characteristics for DSSCs constructs an important bridge between the spatial arrangement of simple chemical moieties and electronic characteristics. We posit that traversing this bridge expands the usage of ligand-based pharmacophore modeling beyond its chemoinformatic/drug design ‘natural habitat’ into the realm of materials sciences with many potentially exciting applications.
Methods
Dataset
A dataset consisting of 1463 dyes was compiled from the dye-sensitized solar cell database (DSSCDB)53. The DSSCDB contains over 4000 experimental observations of DSSCs’ properties spanning different classes of compounds. Every entry in the DSSCDB contains information on photovoltaic properties (e.g., Jsc, Voc, FF, PCE) in addition to molar extinction coefficient, wavelength at maximal absorption, maximal emission wavelength, molecular keyword (scaffold), and additional data about the cell. The DSSCDB was preprocessed to create a homogenous dataset containing only metal-free dyes with no duplicate structures. Entries with co-sensitizers or co-adsorbents were excluded. Furthermore, in order to maintain uniformity, entries with light simulator conditions different from AM 1.5 G 100 Mw cm−2 (sun simulator conditions), semiconductors different from TiO2 or electrolytes different from iodide/triiodide were also excluded. The dataset was also divided into smaller subsets based on molecular scaffolds (Table 1).
Conformer generation
Conformers of dataset’s compounds were generated by the iCon73 conformer generator, implemented in LigandScout 4.48. BEST settings were applied; hence 200 conformers at most were generated for every compound.
Clustering
Each class of scaffolds was further clustered using the LigandScout 4.4 clustering tool to generate more structurally homogenous subclasses. Two clustering methods were employed:
-
(1)
Radial distribution function (RDF) similarity—An RDF gives the probability of finding an object at a distance r from another object and is defined according to Eq. (2):
$$g\left( r \right) = \mathop {\sum }\limits_{i = 1}^{N - 1} \mathop {\sum }\limits_{j > i}^N e^{ - B\left( {r - r_{ij}} \right)^2}$$(2)where rij is the distance between objects i and j, and N is the number of objects. Here the objects are the pharmacophoric features. An individual RDF is calculated for each ligand based on its pharmacophoric features and clustering is performed based on the similarity between the RDFs74. This is the default method used by LigandScout.
-
(2)
Pharmacophore alignment (PA) score—This method uses the ligands’ pharmacophore scores to calculate the similarity between them. Clustering is based on this similarity.
Pharmacophore modeling
Pharmacophore models were generated based on two types of input data and according to the steps outlines in the introduction:
-
(1)
Compounds with the best photovoltaic performance (as determined by PCE) in each class (scaffold-based) were taken to build pharmacophore models. Compounds with lowest PCE in each class were also taken to generate models with undesired features.
-
(2)
The most active dyes (MAD) in the entire dataset (PCE ≥ 8.5%, 12 entries) irrespective of their class membership were used to generate pharmacophore models, and the least active dyes (LAD) in the dataset (PCE ≤ 0.1%, 13 entries) were used in an attempt to find models with undesired features. Additional pharmacophore models were derived by dividing the MAD and LAD groups into different scaffold-based subsets and clusters within these sets.
Some of the final models were manually refined to better match their constituting compounds. Manual refinement was done in several ways: aligning different pharmacophores in order to extract shared feature models or merging them, adding new features or modifying the positions of existing features. Shared feature models and merged feature models were built by aligning two or more pharmacophores in order to build more inclusive and exclusive models, respectively. In a shared feature model, common features are extracted to generate a single model with features shared by all its constituting dyes, while in a merged feature model, features are combined to generate a single model containing unique elements from each of its constituting dyes.
Pharmacophore validation
Pharmacophore models were validated in small-scale VS experiments by testing their ability to identify active/inactive compounds from within a pool of random compounds (assumed to be inactive). Compounds with experimentally tested photovoltaic activity were obtained from the DSSCDB whereas random compounds were obtained from the ZINC1554 database. In order to make the validation process as rigorous as possible, random compounds were selected to have the same scaffolds as those characterizing the corresponding active compounds. To this end we have used the substructure search tool which is part of the ZINC15 database.
The performances of the various pharmacophore models in the VS experiments were validated using several metrics. First, the area under the curve (AUC) metric was calculated by generating receiver operating characteristic (ROC) curves75 for each model. A ROC curve is obtained by plotting model sensitivity (Se) against (1-model specificity (Sp)), termed (\(\overline {{{{\mathrm{Sp}}}}}\)). The relevant equations are:
Where TP, FN, TN, FP are the numbers of true positives, false negatives, true negatives and false positives, respectively. The AUC is calculated as follows76:
Where Se(x) is the sensitivity at rank position x and \(\overline {{{{\mathrm{Sp}}}}} \left( x \right)\) is the (1-model specificity) at rank position x77. AUC values range in the interval [0, 1], with 1 indicating perfect prediction and 0 indicating complete inverse prediction. An AUC value above 0.5 suggests that the model is better than a random assigner.
Another important metric for model validation is the enrichment factor (EF). It is defined as the ratio of TPs in a certain rank position x, normalized by the total ratio of active ligands given to the model78,79.
where TPx% represents the number of TPs in the top x% of the ranked dataset, TPtot represents the total number of active ligands in the dataset, Nx% is the number of compounds in rank position x% and Ntot is the total number of compounds. EF values provide insight into the model’s ability to find active compounds compared to a random selection at a certain position rank80. In the present work, we used x = 1% thereby focusing on model’s ability to retrieve active compounds early in the screening process.
Models were also evaluated by means of the Matthews correlation coefficient (MCC), a common measure of the success of binary classifications which takes into account true and false positives and negatives and is considered to be a balanced measure even when the data distribution is imbalanced81,82,83.
MCC takes values between −1 and 1, with 1 indicating perfect prediction and −1 indicating perfect inverse prediction. A MCC value of 0, suggests that the prediction has no correlation to the data.
Finally, models were evaluated by calculating the percentages of the retrieved high activity (%HA) and low activity (%LA) compounds. A model developed based on high-activity compounds only, should ideally retrieve only high-activity compounds whereas a model developed based on low activity compounds only, should ideally retrieve only low activity compounds. In the present study, HA and LA compounds were defined as those having PCE values >6.3% and <2.0%, respectively. These numbers correspond to the average PCE values across the entire dataset +/− one standard deviation.
Virtual screening and hits retrieval
Virtual screening was performed with a subset of the pharmacophore models that performed well during the validation stage, using LigandScout 4.4 virtual screening tool. Each high activity-based pharmacophore model was used for screening the in-stock catalogue of ZINC15, which contains over 13.8 M commercially available compounds, ready for shipment. Conformations of compounds in the screening library were generated by iCon with FAST settings applied; hence 25 conformations at most were generated for each compound. The compounds from all screening processes were crosschecked and combined into a single hitlist composed only of compounds that matched at least half of the pharmacophore models used for screening. The hitlist was then subjected to further screening against selected pharmacophore models based on low activity-dyes in order to filter out potential low-activity candidates that may have been identified in the first stage of the screening process. Following filtration, the remaining compounds were scored based on their ranks in the high activity-based pharmacophore models and the overall performances of these models. Scoring of compounds was done by the following formulae:
where TS(z) is the total score of compound z, score(p,z) is the score of the best matching conformation of compound z against pharmacophore model p, MCC(p) is the MCC value of pharmacophore p as calculated in the validation stage, AS(z) is the averaged score of z and Nz is the number of pharmacophore models that matched compound z. Thus, compounds matching many models with high MCC values have high TS values, and compounds having a better overlap with the pharmacophore models’ features have high AS values. Overall, this ranking procedure assigns higher ranks to compounds that well-match multiple higher-performing pharmacophore models.
Using the ZINC database as a source for inactive compounds in the validation stage and as a source for potentially active compounds in the VS stage merits some discussion. Experience with VS campaigns suggests that the percentage of compounds with any specific type of activity in screened databases is in the range of 0.5–0.7%84,85. Thus, it is reasonable to assume that most compounds found within the ZINC database are indeed inactive with respect to their PV properties. However, those compounds that, following the screening procedure, surface to the top of the list have a reasonable chance of being active.
QM calculations
QM calculations were executed in the Jaguar86 software package as implemented in Maestro 12.5.139. HOMO and LUMO energies were computed in the gas phase employing DFT methods with the B3LYP exchange-correlation functional, using the 6–31 G(d, p) basis set. UV–vis absorption spectra were calculated as well, employing TD-DFT methods with the CAM-B3LYP87 functional and the same basis set. Similar calculations with the same functional and basis set are reported in the literature, some providing results in good agreement with experimental values46,48,65,88,89,90. Input geometries for the QM calculations were selected by aligning the compounds to their respective pharmacophore models in order to emulate their photoactive conformations.
Data availability
The preprocessed DSSCDB data that was used to generate the pharmacophore models is available as Supplementary File 1.
References
Engel, T. Basic overview of chemoinformatics. J. Chem. Inf. Model. 46, 2267–2277 (2006).
Yosipof, A., Shimanovich, K. & Senderowitz, H. Materials Informatics: Statistical Modeling in Material Science. Mol. Inform. 35, 568–579 (2016).
Ramprasad, R., Batra, R., Pilania, G., Mannodi-Kanakkithodi, A. & Kim, C. Machine learning in materials informatics: recent applications and prospects. npj Comput. Mater. 3, 54 (2017).
Dhanjal, J. K., Sharma, S., Grover, A. & Das, A. Use of ligand-based pharmacophore modeling and docking approach to find novel acetylcholinesterase inhibitors for treating Alzheimer’s. Biomed. Pharmacother. 71, 146–152 (2015).
Seidel, T., Bryant, S. D., Ibis, G., Poli, G. & Langer, T. in Tutorials in Chemoinformatics (ed. Varnek, A.) 279–309 (John Wiley & Sons, Ltd, 2017).
Qing, X. et al. Pharmacophore modeling: advances, limitations, and current utility in drug discovery. J. Receptor. Ligand Channel Res. 7, 81–92 (2014).
Zuccotto, F. Pharmacophore features distributions in different classes of compounds. J. Chem. Inf. Comput. Sci. 43, 1542–1552 (2003).
Wolber, G. & Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 45, 160–169 (2005).
Khedkar, S. A., Malde, A. K., Coutinho, E. C. & Srivastava, S. Pharmacophore modeling in drug discovery and development: an overview. Med. Chem. 3, 187–197 (2007).
Kiani, Y. S., Kalsoom, S. & Riaz, N. In silico ligand-based pharmacophore model generation for the identification of novel Pneumocystis carinii DHFR inhibitors. Med. Chem. Res. 22, 949–963 (2013).
Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today 15, 444–450 (2010).
Schwab, C. H. Conformations and 3D pharmacophore searching. Drug Discov. Today Technol. 7, e245–e253 (2010).
Lionta, E., Spyrou, G., Vassilatis, D. K. & Cournia, Z. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 14, 1923–1938 (2014).
Ambure, P., Kar, S. & Roy, K. Pharmacophore mapping-based virtual screening followed by molecular docking studies in search of potential acetylcholinesterase inhibitors as anti-Alzheimer’s agents. BioSystems 116, 10–20 (2014).
Kandakatla, N. & Ramakrishnan, G. Ligand based pharmacophore modeling and virtual screening studies to design novel HDAC2 inhibitors. Adv. Bioinforma. 2014, 1–11 (2014).
Gaurav, A. & Gautam, V. Pharmacophore based virtual screening approach to identify selective PDE4B inhibitors. Iran. J. Pharm. Res. 16, 910–923 (2017).
Tahir, R. A., Hassan, F., Kareem, A., Iftikhar, U. & Sehgal, S. A. Ligand-based pharmacophore modeling and virtual screening to discover novel CYP1A1 inhibitors. Curr. Top. Med. Chem. 19, 2782–2794 (2019).
O’Regan, B. & Grätzel, M. A low-cost high-efficiency solar cell based on dye-sensitized collodial TiO2 films. Nature 353, 737–740 (1991).
Grätzel, M. Dye-sensitized solar cells. J. Photochem. Photobiol. C. Photochem. Rev. 4, 145–153 (2003).
Mishra, A., Fischer, M. K. R. & Bäuerle, P. Metal-Free organic dyes for dye-sensitized solar cells: From structure: property relationships to design rules. Angew. Chem. Int. Ed. 48, 2474–2499 (2009).
Li, H. et al. A cascaded QSAR model for efficient prediction of overall power conversion efficiency of all-organic dye-sensitized solar cells. J. Comput. Chem. 36, 1036–1046 (2015).
Cole, J. M. et al. Data mining with molecular design rules identifies new class of dyes for dye-sensitised solar cells. Phys. Chem. Chem. Phys. 16, 26684–26690 (2014).
Gong, J., Sumathy, K., Qiao, Q. & Zhou, Z. Review on dye-sensitized solar cells (DSSCs): advanced techniques and research trends. Renew. Sustain. Energy Rev. 68, 234–246 (2017).
Sharma, K., Sharma, V. & Sharma, S. S. Dye-sensitized solar cells: fundamentals and current status. Nanoscale Res. Lett. 13, 381–426 (2018).
Nazeeruddin, M. K. et al. Conversion of light to electricity by cis-X2Bis(2,2’-bipyridyl-4,4’-dicarboxylate)ruthenium(II) charge-transfer sensitizers on nanocrystalline TiO2 electrodes. J. Am. Chem. Soc. 115, 6382–6390 (1993).
Nazeeruddin, M. K. et al. Combined experimental and DFT-TDDFT computational study of photoelectrochemical cell ruthenium sensitizers. J. Am. Chem. Soc. 127, 16835–16847 (2005).
Giribabu, L., Kanaparthi, R. K. & Velkannan, V. Molecular engineering of sensitizers for dye-sensitized solar cell applications. Chem. Rec. 12, 306–328 (2012).
Grätzel, M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg. Chem. 44, 6841–6851 (2005).
Clifford, J. N., Martínez-Ferrero, E., Viterisi, A. & Palomares, E. Sensitizer molecular structure-device efficiency relationship in dye sensitized solar cells. Chem. Soc. Rev. 40, 1635–1646 (2011).
Ahmad, S., Guillén, E., Kavan, L., Grätzel, M. & Nazeeruddin, M. K. Metal free sensitizer and catalyst for dye sensitized solar cells. Energy Environ. Sci. 6, 3439–3466 (2013).
Chen, S. G., Jia, H. L., Ju, X. H. & Zheng, H. G. The impact of adjusting auxiliary donors on the performance of dye-sensitized solar cells based on phenothiazine D-D-π-A sensitizers. Dye. Pigment. 146, 127–135 (2017).
Mao, M. et al. Effects of donors of bodipy dyes on the performance of dye-sensitized solar cells. Dye. Pigment. 141, 148–160 (2017).
Ferdowsi, P. et al. Molecular design of efficient organic D-A-π -A dye featuring triphenylamine as donor fragment for application in dye-sensitized solar cells. ChemSusChem 11, 494–502 (2018).
Kanaparthi, R. K., Kandhadi, J. & Giribabu, L. Metal-free organic dyes for dye-sensitized solar cells: recent advances. Tetrahedron 68, 8383–8393 (2012).
Hara, K. et al. Novel polyene dyes for highly efficient dye-sensitized solar cells. Chem. Commun. 252–253. https://doi.org/10.1039/B210384B (2003).
Hara, K. et al. Novel conjugated organic dyes for efficient dye-sensitized solar cells. Adv. Funct. Mater. 15, 246–252 (2005).
Kitamura, T. et al. Phenyl-conjugated oligoene sensitizers for TiO2 solar cells. Chem. Mater. 16, 1806–1812 (2004).
Ito, S. et al. High-efficiency organic-dye-sensitized solar cells controlled by nanocrystalline-TiO2 electrode thickness. Adv. Mater. 18, 1202–1205 (2006).
Horiuchi, T., Miura, H., Sumioka, K. & Uchida, S. High efficiency of dye-sensitized solar cells based on metal-free indoline dyes. J. Am. Chem. Soc. 126, 12218–12219 (2004).
Choi, H. et al. Highly efficient and thermally stable organic sensitizers for solvent-free dye-sensitized solar cells. Angew. Chem. Int. Ed. 47, 327–330 (2008).
Hwang, S. et al. A highly efficient organic sensitizer for dye-sensitized solar cells. Chem. Commun. 4887–4889. https://doi.org/10.1039/b709859f (2007).
Qin, H. et al. An organic sensitizer with a fused dithienothiophene unit for efficient and stable dye-sensitized solar cells. J. Am. Chem. Soc. 130, 9202–9203 (2008).
Ezhumalai, Y. et al. Metal-free branched alkyl tetrathienoacene (TTAR)-based sensitizers for high-performance dye-sensitized solar cells. J. Mater. Chem. A 5, 12310–12321 (2017).
Eriksson, S. K. et al. Geometrical and energetical structural changes in organic dyes for dye-sensitized solar cells probed using photoelectron spectroscopy and DFT. Phys. Chem. Chem. Phys. 18, 252–260 (2016).
Venkatraman, V. & Alsberg, B. K. A quantitative structure-property relationship study of the photovoltaic performance of phenothiazine dyes. Dye. Pigment. 114, 69–77 (2015).
Venkatraman, V., Foscato, M., Jensen, V. R. & Alsberg, B. K. Evolutionary de novo design of phenothiazine derivatives for dye-sensitized solar cells. J. Mater. Chem. A 3, 9851–9860 (2015).
Venkatraman, V., Abburu, S. & Alsberg, B. K. Artificial evolution of coumarin dyes for dye sensitized solar cells. Phys. Chem. Chem. Phys. 17, 27672–27682 (2015).
Kar, S., Roy, J., Leszczynska, D. & Leszczynski, J. Power conversion efficiency of arylamine organic dyes for dye-sensitized solar cells (DSSCs) explicit to cobalt electrolyte: understanding the structural attributes using a direct QSPR approach. Computation 5, 2–18 (2017).
Kar, S., Roy, J. K. & Leszczynski, J. In silico designing of power conversion efficient organic lead dyes for solar cells using todays innovative approaches to assure renewable energy for future. npj Comput. Mater. 3, 22 (2017).
Krishna, J. G., Ojha, P. K., Kar, S., Roy, K. & Leszczynski, J. Chemometric modeling of power conversion efficiency of organic dyes in dye sensitized solar cells for the future renewable energy. Nano Energy 70, 104537–104559 (2020).
Krishna, J. G. & Roy, K. QSPR modeling of absorption maxima of dyes used in dye sensitized solar cells (DSSCs). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 265, 120387 (2022).
Greenman, K. P., Green, W. H. & Gómez-Bombarelli, R. Multi-fidelity prediction of molecular optical peaks with deep learning. Chem. Sci. 13, 1152–1162 (2022).
Venkatraman, V., Raju, R., Oikonomopoulos, S. P. & Alsberg, B. K. The dye-sensitized solar cell database. J. Cheminform. 10, 18–26 (2018).
Sterling, T. & Irwin, J. J. ZINC 15 - ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Kumara, N. T. R. N., Lim, A., Lim, C. M., Petra, M. I. & Ekanayake, P. Recent progress and utilization of natural pigments in dye sensitized solar cells: a review. Renew. Sustain. Energy Rev. 78, 301–317 (2017).
Srinivasan, V. et al. A diminutive modification in arylamine electron donors: Synthesis, photophysics and solvatochromic analysis-towards the understanding of dye sensitized solar cell performances. Phys. Chem. Chem. Phys. 17, 28647–28657 (2015).
Gabrielsson, E. et al. Convergent/divergent synthesis of a linker-varied series of dyes for dye-sensitized solar cells based on the D35 donor. Adv. Energy Mater. 3, 1647–1656 (2013).
Zhou, N. et al. Metal-free tetrathienoacene sensitizers for high-performance dye-sensitized solar cells. J. Am. Chem. Soc. 137, 4414–4423 (2015).
Wolber, G., Seidel, T., Bendix, F. & Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 13, 23–29 (2008).
Clifford, J. N. et al. Molecular control of recombination dynamics in dye-sensitized nanocrystalline TiO2 films: free energy vs distance dependence. J. Am. Chem. Soc. 126, 5225–5233 (2004).
Argazzi, R., Bignozzi, C. A., Heimer, T. A., Castellano, F. N. & Meyer, G. J. Long-lived photoinduced charge separation across nanocrystalline TiO2 interfaces. J. Am. Chem. Soc. 117, 11815–11816 (1995).
Hirata, N. et al. Supramolecular control of charge-transfer dynamics on dye-sensitized nanocrystalline TiO2 films. Chem. Eur. J. 10, 595–602 (2004).
Im, H. et al. High performance organic photosensitizers for dye-sensitized solar cells. Chem. Commun. 46, 1335–1337 (2010).
Hug, H., Bader, M., Mair, P. & Glatzel, T. Biophotovoltaics: natural pigments in dye-sensitized solar cells. Appl. Energy 115, 216–225 (2014).
Obotowo, I. N., Obot, I. B. & Ekpe, U. J. Organic sensitizers for dye-sensitized solar cell (DSSC): properties from computation, progress and future perspectives. J. Mol. Struct. 1122, 80–87 (2016).
Tingare, Y. S. et al. New oxindole-bridged acceptors for organic sensitizers: Substitution and performance studies in dye-sensitized solar cells. Molecules 25, 2159–2171 (2020).
Cong, J. et al. Nitro group as a new anchoring group for organic dyes in dye-sensitized solar cells. Chem. Commun. 48, 6663–6665 (2012).
Zhang, L. & Cole, J. M. Can nitro groups really anchor onto TiO2? Case study of dye-to-TiO2 adsorption using azo dyes with NO2 substituents. Phys. Chem. Chem. Phys. 18, 19062–19069 (2016).
Wang, Z.-S. et al. Thiophene-functionalized coumarin dye for efficient dye-sensitized solar cells: electron lifetime improved by coadsorption of deoxycholic acid. J. Phys. Chem. C. 111, 7224–7230 (2007).
Ying, W. et al. New pyrido[3,4-b]pyrazine-based sensitizers for efficient and stable dye-sensitized solar cells. Chem. Sci. 5, 206–214 (2014).
Ahn, D.-H. & Song, J.-W. Assessment of long-range corrected density functional theory on the absorption and vibrationally resolved fluorescence spectrum of carbon nanobelts. J. Comput. Chem. 42, 505–515 (2021).
Nayyar, I. H., Masunov, A. E. & Tretiak, S. Comparison of TD-DFT methods for the calculation of two-photon absorption spectra of oligophenylvinylenes. J. Phys. Chem. C 117, 18170–18189 (2013).
Poli, G., Seidel, T. & Langer, T. Conformational sampling of small molecules with iCon: performance assessment in comparison with OMEGA. Front. Chem. 6, 229 (2018).
Wegner, J. K., Fröhlich, H. & Zell, A. Feature selection for descriptor based classification models. 2. Human intestinal absorption (HIA). J. Chem. Inf. Comput. Sci. 44, 931–939 (2004).
Landgrebe, T. C. W. & Duin, R. P. W. A simplified volume under the ROC hypersurface. SAIEE Afr. Res. J. 98, 94–100 (2007).
Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 27, 861–874 (2006).
Paclík, P., Lai, C., Landgrebe, T. C. W. & Duin, R. P. W. ROC analysis and cost-sensitive optimization for hierarchical classifiers. in 2010 International Conference on Pattern Recognition 2977–2980 (2010). https://doi.org/10.1109/ICPR.2010.729
Empereur-Mot, C. et al. Predictiveness curves in virtual screening. J. Cheminform. 7, 52–68 (2015).
Pearlman, D. A. & Charifson, P. S. Improved scoring of ligand-protein interactions using OWFEG free energy grids. J. Med. Chem. 44, 502–511 (2001).
Srinivas, R., Klimovich, P. V. & Larson, E. C. Implicit-descriptor ligand-based virtual screening by means of collaborative filtering. J. Cheminform. 10, 56–75 (2018).
Shi, L. et al. The Microarray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 28, 827–838 (2010).
Ballabio, D., Grisoni, F. & Todeschini, R. Multivariate comparison of classification performance measures. Chemom. Intell. Lab. Syst. 174, 33–44 (2018).
Boughorbel, S., Jarray, F. & El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PLoS ONE 12, e0177678 (2017).
Bradley, D. Dealing with a data dilemma. Nat. Rev. Drug Discov. 7, 632–633 (2008).
Mendolia, I., Contino, S., Perricone, U., Ardizzone, E. & Pirrone, R. Convolutional architectures for virtual screening. BMC Bioinforma. 21, 310–323 (2020).
Bochevarov, A. D. et al. Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 113, 2110–2142 (2013).
Yanai, T., Tew, D. P. & Handy, N. C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004).
Irfan, A., Jin, R., Al-Sehemi, A. G. & Asiri, A. M. Quantum chemical study of the donor-bridge-acceptor triphenylamine based sensitizers. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 110, 60–66 (2013).
Song, J. & Xu, J. Density functional theory study on D-π-A-type organic dyes containing different electron-donors for dye-sensitized solar cells. Bull. Korean Chem. Soc. 34, 3211–3217 (2013).
Arunkumar, A., Shanavas, S., Acevedo, R. & Anbarasan, P. M. Acceptor tuning effect on TPA-based organic efficient sensitizers for optoelectronic applications—quantum chemical investigation. Struct. Chem. 31, 1029–1042 (2020).
Acknowledgements
The authors gratefully acknowledge the help of Dr. Sharon D. Bryant and her team at Inte:Ligand in this research. The help and guidance they provided were essential for its successful completion.
Author information
Authors and Affiliations
Contributions
H.S. conceptualized the idea of the research and supervised it. H.B. curated the data for the research. H.B. processed the data from the DSSCSB, generated the pharmacophore models, validated and analyzed the results. H.B. performed VS with the selected models, extracted the candidates and performed QM calculations on the final candidates which were selected by H.B. with assistance from H.S. All software manipulations were performed by H.B. H.B. was responsible for data visualization. H.B. drafted the manuscript, which was edited and reviewed by H.S. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41524_2022_823_MOESM1_ESM.pdf
Photovoltaphores: Pharmacophore models for identifying metal-free dyes for dye-sensitized solar cells - Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Binyamin, H., Senderowitz, H. Photovoltaphores: pharmacophore models for identifying metal-free dyes for dye-sensitized solar cells. npj Comput Mater 8, 142 (2022). https://doi.org/10.1038/s41524-022-00823-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00823-6
