


While the consumption of external energy (i.e., feeding) is essential to life, this action induces a temporary disturbance of homeostasis in an animal. A primary example of this effect is found in the regulation of glycemia. In the fasted state, stored energy is released to maintain physiological glycemic levels. Liver glycogen is liberated to glucose, glycerol and (glucogenic) amino acids are used to build new glucose molecules (i.e., gluconeogenesis), and fatty acids are oxidized to fuel long-term energetic demands. This regulation is driven primarily by the counterregulatory hormones epinephrine, growth hormone, cortisol, and glucagon. Conversely, feeding induces a rapid influx of diverse nutrients, including glucose, that disrupt homeostasis. Consistently, a host of hormonal and neural systems under the coordination of insulin are engaged in the transition from fasting to prandial states to reduce this disruption. The ultimate action of these systems is to appropriately store the newly acquired energy and to return to the homeostatic norm. Thus, at first glance it is tempting to assume that glucagon is solely antagonistic regarding the anabolic effects of insulin. We have been intrigued by the role of glucagon in the prandial transition and have attempted to delineate its role as beneficial or inhibitory to glycemic control. The following review highlights this long-known yet poorly understood hormone.
The Discovery of Glucagon and Insulin
In 1921 Banting and Best (1) identified insulin, a life-saving therapeutic for millions of individuals with diabetes, which set a new course for our understanding of glucose metabolism. Two years later Kimball and Murlin (2) described the second hormone, glucagon, which appeared to oppose insulin and elevate blood glucose (3). Subsequent work by Burger, Brandt, and Kramer (4–6) identified the liver as the primary target of glucagon-stimulated hyperglycemia. Finally, in 1948 Sutherland and de Duve (7) published the first evidence that glucagon was produced from the pancreatic α-cells, closing the loop between its initial discovery as a pancreatic hormone and its primary target tissue, the liver.
Since this early codiscovery, the contrasting roles of insulin and glucagon have been studied in detail, often with an emphasis on the pathophysiological role of unopposed glucagon action in diabetes (8–12). However, emerging preclinical studies have highlighted potential insulin-sensitizing effects of glucagon receptor (GCGR) agonism, both alone and in combination with other incretin signals (i.e., glucagon-like peptide 1 [GLP-1] and glucose-dependent insulinotropic polypeptide [GIP]) (13–20). Consistently, clinical studies of a single-molecule GCGR/GLP-1R coagonist uncovered reduced glucose excursion during a mixed-meal challenge (21). Although individual receptor contributions to this effect were not specifically investigated, similar findings have also been reported for single-molecule GCGR/GLP-1R/GIPR triagonists (22). Hence, a new emphasis has emerged on understanding the mechanisms and applications of GCGR agonism, especially in metabolic diseases.
Glucagon Secretion
Five main cell types (i.e., α-, β-, δ-, γ-, and ε-cells) make up the endocrine pancreas and are clustered into island-like structures called islets of Langerhans (23). Like insulin, glucagon is produced by the endocrine pancreas and secreted in response to changing nutritional demands (23). Glucagon is encoded by the proglucagon gene, which also encodes GLP-1, GLP-2, oxyntomodulin, glicentin, and the metabolically inert cleavage products glucagon-reactive polypeptide and major proglucagon fragment (24). Pancreatic α-cells preferentially express prohormone convertase-2, which is essential in processing the proglucagon peptide to produce the 29-amino-acid (AA) native glucagon peptide (25–27). Glucagon is secreted from the α-cells, which make up 15–20% of total rodent islet cells (23) but 30–45% of the human islet (28). Thus, in human islets there is far greater interaction (i.e., more contact) between α- and β-cells than in rodent islets. These compositional differences in islet morphology suggest that glucagon plays a greater physiological role in humans than in rodents.
Glucagon secretion is influenced by nutritional state and is best known in the context of fasting and hypoglycemia (29,30). α-Cells preferentially express the low-Km glucose transporter 1 (GLUT1) (31) and ATP-sensitive potassium (KATP) channels (32). Glucose-dependent increases in cellular ATP levels close KATP channels, depolarizing the cell and inhibiting glucagon secretion (33,34). Intriguingly, the regulation of glucagon secretion is not restricted to glucose alone.
Free fatty acids (FFA) may stimulate glucagon secretion. However, this regulation appears to be dependent on the FFA characteristics and if the FFA source was exogenous or endogenous (30). AAs, excluding the branched-chain AAs, stimulate glucagon secretion in dogs (35). This was consistent with the observation that high-protein meals (36–38), arginine (39,40), and alanine (41,42) stimulate glucagon secretion in humans. Importantly, the stimulatory effects of these AAs on glucagon secretion are far greater than those observed during hypoglycemia (20) yet are attenuated (43,44) or abolished (44) in the presence of hyperglycemia. Reciprocally, glucagon increases ureagenesis in hepatocytes to regulate AA metabolism (45). Insulin-resistant and steatotic individuals exhibit hyperaminoacidemia, leading to hyperglucagonemia and disruption of the liver–α-cell axis in humans (45). Likewise, inhibition of hepatic GCGR signaling results in increased circulating AAs and α-cell hyperplasia of both endogenous mouse islets and human islet transplants (46). Importantly, α-cell hyperplasia can be mimicked by culturing islets in high concentrations of AAs, especially l-glutamine (46). By extension, lipid-induced disruption of hepatic glucagon sensitivity has been postulated to contribute to impaired AA homeostasis, hyperglucagonemia, and eventually to type 2 diabetes (T2D) (47).
Glucagon secretion is also regulated via endocrine/paracrine factors, including insulin, amylin, zinc, GABA, GLP-1, GIP, and somatostatin. α-Cells express both insulin receptors (INSR) and GABA receptors (48,49). Consistently, insulin and GABA from neighboring β-cells both inhibit glucagon secretion (50–52). However, work in rat islets supports that the key inhibitory factor from β-cells may be zinc bound to the insulin protein (53). Similarly, somatostatin of the δ-cells inhibits glucagon secretion (54). Only a minority (∼20%) of mouse, rat, and human α-cells express GLP-1R (55,56). Thus, inhibition via GLP-1 (57–59) is likely secondary to GLP-1R–stimulated release of zinc-insulin, GABA, and amylin. Conversely, in healthy individuals GIP stimulates glucagon secretion in a glucose-dependent manner (i.e., during hypoglycemia) (60,61). Reciprocally, glucagon acts in a paracrine manner to increase insulin secretion through activation of both β-cell GCGR and GLP-1R (19).
Finally, glucagon secretion is directly mediated by the autonomic nervous system. Via their effects on insulin secretion, vagal stimulation (parasympathetic) inhibits (62), whereas splanchnic (sympathetic) stimulation increases, glucagon secretion (63–66). Together, these findings clearly support the idea that glucagon secretion is regulated in response to multiple stimuli and systems. Among them is a potential cosecretion with insulin in the early prandial state. Together these observations support a more complex role for glucagon beyond simple counterregulation of insulin in glucose homeostasis.
GCGR Tissue Distribution and Hepatic Signaling
GCGR is a member of the class B family of G protein–coupled receptors (67). Gcgr mRNA is primarily expressed in the liver, with low-level expression in the kidney, adipose tissue, pancreas, spleen, lymphoblasts, brain, gastrointestinal tract, and adrenal gland (68). Hepatic Gcgr expression and subsequent metabolic actions are restricted to the periportal area (69), where they overlap with INSR (Insr) expression (70). Hepatic GCGR signaling stimulates two intracellular cascades (Fig. 1), a cAMP stimulatory G protein, Gs, and a Gq protein that signals via Ca2+ (29,30). Canonical Gs signaling activates adenylate cyclase to produce cAMP. This second messenger stimulates both protein kinase A (PKA) and Rap guanine nucleotide exchange factor 3 (RAPGEF3; also known as EPAC1). EPAC1 activation stimulates the small GTPase Rap1 and the AMP-dependent protein kinase (AMPK) (71). Concomitantly, PKA phosphorylates the cAMP response element-binding protein (CREB) and stimulates protein phosphatase 2B-dependent dephosphorylation of the CREB-regulated transcription coactivator 2 (Crtc2) (72). CREB/CRTC2 signaling is associated with gluconeogenic and glycogenolytic gene expression (e.g., glucose-6-phosphatase [G6pc], phosphoenolpyruvate kinase [Pck1], and peroxisome proliferator-activated receptor γ coactivator 1-α [Ppargc1a]) (30). GCGR-stimulated Ca2+ signaling occurs downstream of Gq activation and is associated with hepatic glycogen phosphorylase activation, bile acid homeostasis, and liver regeneration (73).
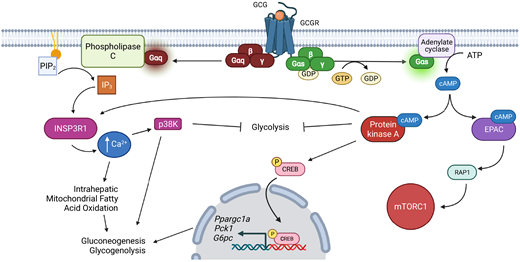
Overview of GCGR signaling pathways in the regulation of hepatic glucose homeostasis. Figure created with BioRender.com.
Overview of GCGR signaling pathways in the regulation of hepatic glucose homeostasis. Figure created with BioRender.com.
Termination of signaling is equally important to metabolic regulation. GCGR signaling is terminated by internalization of the ligand–receptor complex and occurs primarily via clathrin- and arrestin-facilitated endocytosis. Intriguingly, sustained GCGR signaling has been described after internalization, suggesting a second wave of signaling from this receptor (30). However, the biological relevance of this intracellular signaling has yet to be fully elucidated. Intracellular GCGR palmitoylation and ubiquitination have been observed and may also contribute to signal termination (30). Intriguingly, glucagon stimulates both GCGR internalization and deubiquitination, facilitating rapid recycling of the receptor (74).
Metabolic Actions of Hepatic GCGR Signaling
As introduced above, the best-known actions of GCGR signaling involve its counterregulatory effect on insulin action. In the context of glucose metabolism, GCGR signaling stimulates hepatic glycogenolysis and gluconeogenesis (GNG) with concomitant inhibition of glycogen synthesis (29). GCGR signaling rapidly increases hepatic glycogenolysis via a signaling cascade involving the canonical cAMP–PKA pathway. This signaling activates glycogen phosphorylase kinase and subsequent activation of glycogen phosphorylase. GCGR signaling (via PKA) likewise inhibits glycogen synthase, preventing hepatic glycogen synthesis (75).
GCGR regulation of hepatic GNG occurs via both transcriptional induction and allosteric modulation of GNG enzymes. PKA-dependent phosphorylation of phosphofructokinase 2 and pyruvate kinase shifts metabolic flux from glycolysis to GNG. GCGR signaling stimulates CREBSer133 phosphorylation coupled with dephosphorylation and nuclear translocation of its coactivator, Creb-regulated transcription coactivator 2 (Crtc2). These actions not only stimulate the induction of target GNG genes G6pc, Pck1, Ppargc1a and hepatocyte nuclear factor 4 (Hnf4a) but also regulate GNG-associated transcription factors FOXO1 and PGC-1-α via modulation of their acetylation states (30). Additionally, GCGR-stimulated Ca2+ signaling activates glycogenolysis and GNG via p38 kinase (76). Consistent with these signaling events, exogenous glucagon elevates glycemia (77). Moreover, genetic Gcgr deficiency and neutralizing antibodies targeting glucagon are sufficient to reduce glycemia (78–80). In contrast, the antidiabetic effects of Gcgr knockout in streptozotocin (STZ)-treated mice are lost when STZ is administered prior to Gcgr ablation (81). These rodent data must be interpreted with some caution, as GCGR antagonists clearly lower glycemia in individuals with T1D (82). Together, these findings highlight the complex and context-dependent relationship between glucagon and insulin in glucose homeostasis.
In addition to its effects on glucose metabolism, mounting evidence suggests hepatic glucagon is a potent regulator of energy balance, lipid homeostasis, and fat mass mobilization (30). In the context of energy balance, glucagon both stimulates energy expenditure and suppresses food intake, as highlighted by the negative energy balance observed in glucagonoma patients (83). This stimulation of energy expenditure and thermogenesis is conserved across a range of species (29). However, the conservation of this system in humans is still controversial, with reports observing both increased and unchanged energy expenditure (84,85). Energy expenditure regulation in mice is dependent upon hepatic GCGR signaling and is mechanistically associated with hepatic FXR activity and endocrine FGF21 action (14,15,86). Glucose futile cycling may also contribute to the upregulation of energy expenditure following GCGR agonism (87,88). Intriguingly, glucagon administration also decreases hunger and food intake in both rats (89) and human subjects (90,91). Consistently, GCGR agonism in diet-induced obese mice suppressed food intake; however, this effect was preserved in mice lacking hepatic Gcgr expression, suggesting that the liver is not the tissue of origin for this regulation (14).
Glucagon also regulates multiple components of lipid metabolism (29). Gcgr is expressed by rodent adipocytes (92). Consistently, glucagon mediates rodent white adipose tissue lipolysis (93). Conversely, evidence of Gcgr expression in human adipocytes is lacking (94), as is that for glucagon-induced lipolysis at physiological levels in patients (95). In rodents, glucagon-mediated white adipose tissue lipolysis (96,97) via hormone-sensitive lipase results in the liberation of nonesterified fatty acids (NEFA) (98). The majority of these NEFAs are catabolized. However, in the liver, NEFAs may be alternatively converted to ketone bodies to provide energy during times of glucose deficiency (99,100). Consistent with this shift to lipid energy substrates, glucagon exposure inhibits hepatic lipogenesis while stimulating FA transport and oxidation (101). Inhibition of hepatic lipogenesis occurs via two potential mechanisms: 1) CREB-mediated induction of insulin-induced gene 2 (Insig2) and sequestration of the lipogenic sterol regulatory element binding protein (SREBP) transcription factor (102) and 2) Ca2+-dependent activation of p38 kinase and subsequent inhibition of SREBP (76). GCGR agonism is also a potent regulator of bile acid metabolism, stimulating robust changes in the expression of bile acid enzymes and the composition of circulating bile acids (14). As introduced above, emerging data support that hepatic GCGR signaling is a crucial regulator of AA metabolism. GCGR agonism stimulates hepatic AA uptake and urea production and subsequently induces hypoaminoacidemia (103). Together these pieces of evidence point to glucagon as a potent regulator of AA and lipid homeostasis, energy balance, and fat mass mobilization.
Insulin, Insulin Action, and Hepatic INSR Signaling
Insulin is a powerful anabolic factor, stimulating growth and energy accrual throughout the organism. This pleiotropic hormone is essential to glucose metabolism and crucial to lipid and AA metabolism. Insulin action in the liver stimulates lipogenesis and glycogen synthesis while concomitantly inhibiting glycogenolysis, GNG, and liver fatty acid oxidation (104).
Insulin signals via the INSR, a member of the receptor tyrosine kinase family, and, to a lesser extent, the insulin-like growth factor 1 receptor. These receptors are endogenously inhibited by the recently discovered Inceptor protein in mouse β-cells (105). Insr is expressed in the central nervous system and a wide range of peripheral tissues. Unlike Gcgr, hepatic Insr expression is found in both periportal and perivenous zones (70). The role of this essential hormone and INSR signaling (summarized in Fig. 2) has been extensively covered, including the following review (104). Therefore, this Perspective will focus on hepatic signaling and biological functions arising from INSR activation. INSR signaling is initiated when insulin binds to the receptor, derepressing the receptor’s intrinsic kinase activity. INSR then phosphorylates intracellular substrates, including members of the insulin/insulin-like growth factor 1 receptor substrate (IRS) protein family, Gab-1, DOK1, Cbl, SH2B2 (APS), SHP2, and isoforms of Shc (104). Canonical insulin regulation of hepatic glucose and lipid metabolism involves subsequent IRS-dependent activation of phosphatidylinositol-3-kinase, 3′-phosphoinositide–dependent kinase 1 (PDK1), and AKT/PKB (104). AKT is a central node of hepatic insulin signaling and is crucial for both glucose and lipid metabolism. This serine/threonine kinase is activated by phosphorylation on two residues, Thr308 and Ser473. Thr308 phosphorylation occurs in a PDK1-dependent manner and is essential for AKT kinase activity. Ser473 is phosphorylated by the rapamycin-insensitive mTOR complex (mTORC2) and is permissive for full kinase activity (104). Importantly, the mechanisms of mTORC2 regulation remain uncertain. AKT activation leads to subsequent phosphorylation of forkhead box–containing protein, O subfamily (FOXO). FOXO proteins (especially members 1 and 6) are transcription factors that induce GNG. AKT-dependent phosphorylation triggers nuclear exclusion and, thus, is inhibitory to this action (106).

Overview of INSR signaling pathways in the regulation of hepatic glucose homeostasis. MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; PLCγ, phospholipase Cγ. Figure created with BioRender.com.
Overview of INSR signaling pathways in the regulation of hepatic glucose homeostasis. MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; PLCγ, phospholipase Cγ. Figure created with BioRender.com.
Insulin also regulates hepatic Ca2+ signaling. INSR activation stimulates phospholipase Cγ, generating inositol-1,4,5-triphosphate (InsP3). Increased InsP3 levels stimulate InsP3 ligand–gated Ca2+ channels of the endoplasmic reticulum and thus increase intracellular Ca2+ levels. Increased hepatic Ca2+ levels further stimulate INSR-dependent activation of the mitogen-activated protein kinase signaling cascade and activation of transcription factors (e.g., MYC, FOS, and JUN) in this mitogenic pathway (107).
Diabetes, whether type 1 (T1D) or type 2 (T2D), is defined by hyperglycemia and is ultimately the result of insufficient insulin action. In the case of T1D, this deficiency is caused by destruction of the pancreatic β-cell and therefore a lack of the insulin hormone. In T2D, insulin resistance accumulates to a point where β-cell compensatory hypersecretion is insufficient to counteract the resistance (108). In the liver, this insufficiency is manifested as a failure to suppress hepatic glucose output (i.e., GNG and glycogenolysis). Intriguingly, in T2D this resistance is often incomplete, resulting in a preservation of insulin-stimulated lipogenesis (108). Consistent with its counterregulatory role, both fasting and postprandial plasma glucagon levels are elevated in diabetes (109). However, these observations have been made in individuals with established cases of diabetes, and thus the causality of hyperglucagonemia is difficult to assign.
Overlapping Hepatic GCGR and INSR Actions
As a counterregulatory hormone with a role in maintaining fasting blood glucose, it is tempting to assume that glucagon opposes all actions of insulin. Consistent with this hypothesis, circulating glucagon levels are elevated in all known instances of T1D or T2D, including animal models of the disease (77). Likewise, preclinical GCGR ablation or pharmacological GCGR inhibition (including neutralizing antibodies against glucagon) in individuals with diabetes is sufficient to reduce glycemia and HbA1c. However, many of these strategies have been slowed due to adverse effects on liver transaminases, liver fat, and dyslipidemia (30).
Conversely, the increased concentrations and action of glucagon in the fasting state are well suited to potentiate subsequent insulin-mediated glucose control. To this point, glucagon acts in a paracrine manner to increase insulin secretion through activation of both β-cell GCGR and GLP-1R (19). Likewise, postprandial elevations of glucagon and GLP-1 contribute to the improved postprandial glucose profile observed in Roux-en-Y gastric bypass patients (110) and rodent models of this powerful intervention (111). Importantly, these physiological conditions are all characterized by their heightened insulin sensitivity. Regarding glucagon enhancement of insulin action, the use of the bionic pancreas (glucagon and insulin) must be mentioned (112). This technology was hypothesized to prevent life-threatening hypoglycemic episodes in people with diabetes. Beyond reducing hypoglycemic episodes, the bihormonal (glucagon and insulin) pump reduced average glycemia while requiring a similar total daily insulin dose in adolescents (112). Likewise, 13-h glucagon infusion increased both glucose appearance and disappearance in patients, suggesting that its regulation of human glucose metabolism is not restricted to increasing hepatic glucose output (113). Together, these observations support the hypothesis that glucagon, released during fasting and the prandial response, acts to prime metabolic tissues for the subsequent nutrient challenge of feeding. Moreover, it positions cooperative actions of glucagon and insulin as crucial to this physiology.
INSR and GCGR signaling also converge at the hepatocyte. Our group described the unexpected enhancement of insulin action in db/db mice following chronic (7-day) treatment with the long-acting GCGR agonist IUB288 (86). This initial observation was followed by more detailed investigation of acute (i.e., 60-min) GCGR agonism and its beneficial effect on insulin sensitivity (114). This work identified enhanced insulin-dependent signaling in the phosphorylation of AKTSer473 in mice treated with IUB288 60 min prior to insulin and was exclusive of PDK1-dependent phosphorylation (Thr308) (114). This single, acute IUB288 treatment increased insulin sensitivity, as defined by increased glucose infusion rate and improved insulin-stimulated suppression of hepatic glucose output during hyperinsulinemic-euglycemic clamps (114). These observations suggest GCGR and INSR signaling intersect via a TORC2-dependent phosphorylation of AKTSer473. Our observation was quickly followed by work by Besse-Patin et al. (115). This elegant study confirmed glucagon-enhanced AKTSer473 phosphorylation and identified glucagon-dependent induction of Ppargc1a as a transcriptional regulator of relative levels of hepatocyte IRS1:IRS2 ratios (115). This shift toward IRS2 favors insulin-dependent suppression of hepatic glucose output (115) and is consistent with our observations in hyperinsulinemic-euglycemic clamps (114). Congruous with our study and interpretation, Besse-Patin et al. concluded that glucagon (via PGC-1-α) primes the liver for subsequent insulin action.
However, an importation caveat to these studies is that the observations of Besse-Patin et al. were made 4 h after glucagon treatment. Subsequent observations in cultured hepatocytes suggest GCGR signaling transiently stimulates protein synthesis via an mTORC1-dependent action (116). This effect was also observed to be convergent with insulin signaling and dependent on EPAC activity (116). Additionally, work by Perry et al. (117) identified enhanced glucose tolerance and insulin sensitivity in rats infused with glucagon for 3.5 weeks. This work supported a role for inositol triphosphate receptor 1 (INSP3R1)-mediated calcium signaling downstream of GCGR activation. In this model, the benefits of GCGR signaling on glucose metabolism are related to hepatic mitochondrial oxidation (117). In summary, emerging data support a beneficial role for GCGR signaling in hepatic insulin glucose metabolism. While the precise mechanisms have yet to be elucidated, data support roles for mTORC1, mTORC2, and PCG1a-IRS2 as potential points for cross talk with hepatic insulin signaling (Fig. 3). INSP3R1 may also represent a mechanism by which hepatic GCGR signaling benefits glucose metabolism secondary to its regulation of mitochondrial oxidation.

Potential and reported cross talk in hepatic glucagon (GCG) and INSR signaling. PI3K, phosphatidylinositol 3-kinase. Figure created with BioRender.com.
Potential and reported cross talk in hepatic glucagon (GCG) and INSR signaling. PI3K, phosphatidylinositol 3-kinase. Figure created with BioRender.com.
GCGR and INSR Cross Talk in Emerging Therapeutics
As introduced above, GCGR ablation/antagonism is beneficial for glucose metabolism (78,79). Of note, treating mice with the INSR antagonist S961 induces severe insulin resistance, hyperglycemia, and ketonemia, yet the GCGR-blocking antibody REGN1193 was sufficient to normalize blood glucose and β-hydroxybutyrate levels in these mice (118). Subsequent clinical investigation uncovered reductions in fasting plasma glucose and HbA1c in REGN1193-treated T2D patients (119). Similar benefits in mice have been reported for the monoclonal antibody and competitive GCGR antagonist REMD 2.59 (120). Moreover, GCGR antagonism, when combined with GLP-1R agonism, stimulates cell regeneration in STZ-treated mice (121). However, enthusiasm for GCGR antagonism is offset by observations of dose-dependent increases in hepatic aminotransferases (122) and induction of profound dyslipidemia (79). Conversely, the benefits of GCGR agonism on energy expenditure, hepatic steatosis, and lipid homeostasis are of great therapeutic interest. Intriguingly, coupling of the antidiabetic properties of GLP-1R agonism with GCGR agonism profoundly enhances the therapeutic action of both receptors (17,18,123). The mechanisms underlying these benefits are still the focus of intense investigation. GLP-1/GCGR dual agonism drives weight loss in a synergistic manner. This weight loss is likely due to GCGR stimulation of energy expenditure and GLP-1R inhibition of gastric emptying (124), the latter also contributing to slower glucose uptake into the circulation. It is also likely that these compounds increase glucose-stimulated insulin secretion via activation of GCGR and GLP-1R at the β-cell while concomitantly enhancing insulin action via GCGR agonism at the liver. Based on this hypothesis, coupling GCGR agonism with other known insulin secretagogues should have similar effects. This hypothesis is supported by the observation in mice that tolbutamide enhanced glucagon-stimulated decreases in glycemia (19). It should be noted that while GLP-1/GCGR dual agonism drives weight loss and improves glucose homeostasis in both preclinical and clinical studies (21,125), clinical application of these molecules has targeted treatment of nonalcoholic steatohepatitis and nonalcoholic fatty liver disease (e.g., cotadutide) (125).
In summary, the glucagon peptide was discovered a century ago, yet our understanding of its metabolic actions is still evolving. The original view that GCGR signaling is antagonistic to insulin action is certainly true in some contexts yet is clearly incomplete. Studies currently underway will continue to refine the role of this long-known hormone and its therapeutic utility in metabolic diseases.
Article Information
Acknowledgments. I thank Dr. Teayoun Kim, Dr. Shelly Nason, and Jessica Antipenko (Comprehensive Diabetes Center and Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL) for helpful discussion.
Funding. The project described in this work was supported by National Institutes of Health grant 1R01DK112934 (K.M.H.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Prior Presentation. Parts of this work were presented at the 82nd Scientific Sessions of the American Diabetes Association, New Orleans, LA, 3–7 June 2022.