
Abstract
Pregnane X receptor (PXR) is highly expressed in the liver and plays a pivotal role in xenobiotic and endobiotic metabolism. We previously reported that PXR activation by its specific mouse agonist pregnenolone 16α-carbonitrile (PCN) significantly induces liver enlargement and lipid accumulation. However, the effect of long-term PCN treatment on PXR and mouse liver is still unknown. This study aimed to explore the influence of long-term administration of PCN on mouse liver and hepatic lipid homeostasis. Male C57BL/6 mice were injected intraperitoneally with PCN (100 mg/kg once a week) for 42 weeks. Serum and liver samples were collected for biochemical and histological analysis. PXR activation was investigated by Western blot. Ultra-high-performance liquid chromatography coupled with electrospray ionization high-resolution mass spectrometry (UHPLC-ESI-HRMS)-based lipidomics analysis was performed to explore the change in different lipid categories. The results showed that long-term treatment with PCN significantly promoted hepatomegaly without hepatocyte proliferation and enlargement. Long-term treatment with PCN did not upregulate PXR target proteins in mice, and there was no significant upregulation of CYP3A11, CYP2B10, UGT1A1, MRP2, or MRP4. Lipidomics analysis showed obvious hepatic lipid accumulation in the PCN-treated mice, and the most significant change was found in triglycerides (TGs). Additionally, long-term treatment with PCN had no risk for carcinogenesis. These findings demonstrated that long-term PCN treatment induces hepatomegaly and lipid accumulation without hepatocyte proliferation or enlargement.
Similar content being viewed by others

Introduction
Pregnane X receptor (PXR, NR1I2) is a ligand-activated transcription factor that is enriched in the liver and intestine [1]. PXR belongs to the nuclear receptor (NR) superfamily and plays an important role in various aspects, such as drug metabolism and endobiotic homeostasis. PXR can be activated by a wide range of compounds. In addition to pregnane, steroids, bile acids and other endobiotic chemicals [2], many xenobiotic compounds, such as pregnenolone 16α-carbonitrile (PCN, a typical mouse PXR agonist), and clinical drugs have been found to activate PXR, such as mifepristone, dexamethasone, schisandrol B, nifedipine [3] and corticosterone [4]. PXR contains a DNA-binding domain (DBD) at the N-terminus that facilitates binding to DNA responsive elements and a ligand-binding domain (LBD) at the C-terminus, which is responsible for ligand binding and interaction with coregulators [5]. After binding to its ligand, PXR forms a heterodimer with retinal X receptor (RXR), translocates from the cytoplasm to the nucleus and binds to the promoter region of its target genes that are involved in xenobiotic and endobiotic metabolism, endobiotic homeostasis and some liver diseases [6].
It is well known that the liver possesses the ability to maintain a constant mass and size (hepatostat) [7]. Previous studies have demonstrated that PXR activation induces liver enlargement by increasing hepatocyte size and proliferation [8,9,10]. Our previous study also reported that after 5 days of administration of PXR activators, such as PCN, mifepristone [11], dexamethasone [12], or schisandrol B [13], the liver/body weight ratio was significantly increased in mice, which may be due to proliferation and enlargement [9]. Additionally, lipid accumulation occurred in the mouse liver, especially the accumulation of triglycerides [14]. Other reports have shown that PXR activation stimulates growth factor-mediated hepatocyte proliferation by inhibiting forkhead box O3 (FOXO3) to accelerate cell cycle progression [10]. In addition, PXR activation does not promote hepatocarcinogenesis, in contrast to constitutive androstane receptor (CAR, NR1I3); rather, it attenuates CAR-mediated liver cancer [15]. However, the effects of long-term treatment with the mPXR agonist PCN on the activation of PXR, lipid homeostasis, liver size, and liver cancer are still unclear.
Therefore, the purpose of this study was to investigate the effects of long-term administration of PCN on liver size, lipid metabolism, and liver cancer. The findings may provide new insights for the long-term use of PXR agonists and may help promote the rational use of PXR agonists in the future.
Materials and methods
Chemicals and reagents
PCN with 98% purity (Cat. C3884-250) was purchased from APExBIO Technology (Houston, USA). Corn oil (Cat. C116025) was purchased from Aladdin Company (Shanghai, China). N-Nitrosodiethylamine (DEN) was purchased from Sigma (St. Louis, USA). Antibodies used in this study were anti-CYP3A4 (ABclonal Technology, Wuhan, China, Cat. A13484, available to detect CYP3A4 in human and CYP3A11 in rodents), anti-CYP2B6 (ABclonal Technology, Wuhan, China, Cat. A1463, available to detect CYP2B6 in human and CYP2B10 in rodents), anti-UGT1A1 (ABclonal technology, Wuhan, China, Cat. A1359), anti-MRP2 (ABclonal Technology, Wuhan, China, Cat. 8405), anti-MRP4 (ABclonal Technology, Wuhan, China, Cat. 2198), anti-CAR (ABclonal Technology, Wuhan, China, Cat. A17066), anti-FXR (Abcam, Cambridge, UK, Cat. Ab129089), anti-PPARα (Santa Cruz Biotechnology, Dallas, USA, Cat. sc-398394), anti-CDK4 (Sangon, Shanghai, China, Cat. D120396), anti-CCNA1 (Sangon, Shanghai, China, Cat. D220507), anti-CCND1 (Sangon, Shanghai, China, Cat. D120509), anti-CCNE1 (Sangon, Shanghai, China, Cat. D151593), anti-PCNA (Cell Signaling Technology, Danvers, USA, Cat. 13110), anti-CTNNB1 (BD Biosciences, San Jose, USA, Cat. 610153), anti-AFP (Santa Cruz Biotechnology, Dallas, USA, Cat. sc-130302) and anti-β-ACTIN (Cell Signaling Technology, Danvers, USA, Cat. 4970).
Animals and treatment
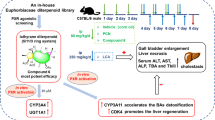
Male C57BL/6 mice (2 weeks old, 8–10 g) were purchased from Guangdong Medical Laboratory Animal Centre (Foshan, China). Mice were housed in a temperature- and light-controlled room (22–24 °C, 12 h light-dark cycle) and had free access to standard chow and water. The experiment protocol was approved by Institutional Animal Care and Use Committee of Sun Yat-sen University. The animals were randomly divided into 4 groups. PCN was dissolved in corn oil at the concentration of 10 mg/mL. The vehicle group and PCN group were injected intraperitoneally with corn oil or PCN (100 mg/kg) per week for 42 weeks. The DEN and DEN + PCN group mice were injected intraperitoneally with 5 mg/kg DEN for only once. All mice were sacrificed 7 days after the last treatment. The body and liver weight were measured to calculate the liver/body weight ratio. Serum and liver samples were collected and kept at −80 °C for further study. A part of the liver was immediately fixed in 4% paraformaldehyde for further histological assessment.
Biochemical and histological assessment
The levels of serum alanine aminotransferase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), albumin (ALB), total bile acid (TBA), and total bilirubin (TBIL) were measured using URIT-8021A (URIT Medical Electronic Co., Ltd, Guilin, China) following the manufacturer’s standard protocols.
According to our previously reported method [9], liver tissues were fixed in neutral buffered formalin and embedded in paraffin to obtain liver sections. Liver sections were then deparaffinized by different concentrations of xylene and ethyl alcohol and then stained with hematoxylin and eosin (H&E) solutions (Servicebio, Wuhan, China). For immunohistochemical (IHC) staining, paraffin-embedded sections were stained with primary antibodies against proliferating cell nuclear antigen (PCNA), β-catenin (CTNNB1), and α-fetoprotein (AFP) after heat-induced epitope retrieval in citrate buffer. Images were obtained by a DMI 3000B microscopy (Olympus, Tokyo, Japan).
Quantitative real-Time polymerase chain reaction (qRT–PCR) analysis
RNA isolation, cDNA acquisition, and qRT–PCR analysis were performed according to manufactory’s instructions using AG RNAex Pro reagent, Evo M-MLV RT Mix Kit, and SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, Hunan, China), respectively. The fold changes of mRNA levels were analyzed by ΔΔCt method.
Western blot
Total proteins were extracted from liver tissues as described previously [9, 16, 17]. BCA assay kit (Thermo Fisher Scientific, Waltham, USA) was used to determine protein concentration. Briefly, 40 μg of protein lysate from liver samples of different groups was used. Protein samples were separated by 10% SDS-PAGE gels and then transferred onto polyvinylidene difluoride membranes. Blots were blocked in 5% skim milk, which was followed by incubation with appropriate primary antibodies overnight at 4 °C. Next day, the membranes were incubated with secondary anti-rabbit or anti-mouse antibodies at room temperature for 60 min. The ECL Detection Kit (Millipore, Darmstadt, Germany) was employed to develop the blots. The intensity of the bands was quantified by Quantity One software (Bio-Rad, Hercules, USA).
UHPLC-ESI-HRMS-based lipidomic analysis
Liver lipidomics analysis was performed according to our previously reported procedures and methods [14, 18]. Briefly, methyl tert-butyl ether (MTBE) method was used to extract lipids from the liver tissues. About 30 mg liver samples were homogenized and the homogenate was dissolved in 1.2 mL of chilled methanol/MTBE/H2O (4:5:5, v/v/v). After being vortexed thoroughly and centrifuged, the organic phase was removed and evaporated to dryness under nitrogen, evaporated extracts were dissolved in MeOH/ isopropanol (1:1 v/v; 1 mL), and centrifuged to remove insoluble materials. A 2 μL aliquot of supernatant samples was injected into the UHPLC-ESI-HRMS (Thermo Fisher Scientific, Waltham, USA). The chromatographic separation and MS analysis were performed according to our previous report [12].
Lipid components identification and data analysis
Lipidomic data processing was performed using LipidSearch software (Thermo Scientific, San Jose, USA). The data matrix was further analyzed using SIMCA-P 13.0 (Umetrics, Kinnelon, USA).
Statistical analysis
All data were expressed as the means ± standard deviation (SD). Two-tailed Student’s t tests were used to compare the significant difference between two groups using GraphPad Prism 8.0 software (GraphPad Software, San Diego, USA) and SPSS 23.0 software (IBM Analytics, Armonk, USA). P value < 0.05 was regarded as significantly different.
Results
Long-term treatment with PCN induces significant liver enlargement
We previously reported that 5- or 10-day treatment with PCN (100 mg/kg every day) significantly promotes liver size [9]. To demonstrate the effect of long-term PCN treatment after 42 weeks on liver size, mice were injected with 0.1 mL/10 g corn oil or 100 mg/kg PCN once a week for 42 weeks (Fig. 1a). The results showed that long-term PCN treatment significantly increased the liver/body weight ratio, which was 22% higher than that of the vehicle group (Fig. 1b, c).

a Mice were treated with vehicle (corn oil) or PCN (100 mg/kg per week, i.p.) for 42 weeks. b Morphological photographs of representative mouse livers. c Liver/body weight ratios. d Serum ALT, AST, ALP, ALB, TBA and TBIL levels. e The hepatic mRNA levels of Il-6, Tnf-α and Ifn-γ. f Histopathological analysis of representative mouse liver samples following H&E staining. Data are presented as the means ± SD. (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 versus the vehicle group.
It is known that liver enlargement may be associated with inflammation or hepatotoxicity [19]. To confirm whether PCN-induced hepatomegaly was associated with liver injury, serum biochemical indices were measured. The serum ALP, ALB, TBA and TBIL levels of the PCN group had no obvious changes, while the ALT and AST levels were slightly upregulated but less than 2-fold (Fig. 1d). Similarly, H&E staining and the mRNA levels of several inflammatory factors, such as Tnf-α, Il-6, and Ifn-γ, showed no significant difference between the two groups (Fig. 1e, f). These results demonstrated that long-term PCN treatment induced hepatomegaly without liver injury.
Long-term PCN treatment does not upregulate proteins downstream of PXR
To investigate the effect of long-term PCN treatment on PXR activation, the hepatic protein expression levels of PXR downstream targets, such as CYP3A11, CYP2B10, UGT1A1, MRP2, and MRP4, were measured by Western blot analysis (Fig. 2a, b). Interestingly, there were no significant increases in these downstream targets of PXR. PXR expression was also measured to explore the impact of long-term PCN treatment on PXR itself (Fig. 2a, b). Similarly, the protein expression of PXR did not change. These data suggested that after long-term PCN stimulation, PXR downstream proteins were not upregulated.

a Western blot analysis of PXR target proteins and PXR. b Quantification of the protein levels of PXR target genes and PXR. c Western blot analysis of PPARα, CAR, and FXR. d Quantification of the protein levels of PPARα, CAR, and FXR. Data are presented as the means ± SD. (n = 3).
Given that there is crosstalk among NRs, we further detected the expression of other NRs that are related to hepatomegaly. The results showed that the expression of peroxisome proliferator-activated receptor α (PPARα), CAR, and farnesoid X receptor (FXR) showed no significant change (Fig. 2c, d).
Long-term treatment with PCN does not induce hepatocyte proliferation and enlargement
It was reported that PCN can induce hepatocyte proliferation [10]. To determine whether hepatocyte proliferation was provoked by long-term PCN treatment, the expression of PCNA was evaluated by immunohistochemistry (Fig. 3a). There were no visible PCNA-positive cells either in the vehicle group or the PCN group around the central vein and portal vein areas, indicating that long-term PCN treatment did not promote hepatocyte proliferation. Moreover, the levels of proliferation-related proteins were measured using Western blotting (Fig. 3b). The expression of cyclin A1 (CCNA1), CCND1, CCNE1, cyclin-dependent kinase 4 (CDK4) and PCNA was unchanged compared with that of the vehicle group (Fig. 3c). These data also indicated that hepatocyte proliferation did not contribute to long-term PCN treatment-induced liver enlargement.

a IHC staining for PCNA. b Protein expression levels of proliferation-related proteins. c Quantification of the expression of proliferation-related proteins. d IHC staining for CTNNB1 and e Quantification of CTNNB1 staining. Data are presented as the means ± SD. (n = 3).
Next, IHC staining of CTNNB1 was performed to investigate the change in cell size after long-term PCN treatment (Fig. 3d). The results showed no significant difference in cell size between the vehicle group and the PCN group (Fig. 3e), suggesting that hepatocyte enlargement was not the reason for hepatomegaly induced by long-term PCN treatment.
Taken together, long-term treatment with PCN did not promote hepatocyte enlargement or proliferation.
Long-term PCN treatment induces lipid accumulation
Lipidomic analysis was used to characterize the change in lipids. First, unsupervised principal component analysis (PCA) was performed on the lipidomic data of liver tissues from the vehicle and PCN groups. Then, orthogonal projections to latent structures discriminant analysis (OPLS-DA) models were developed to eliminate interferences other than sample classification and maximize the separation between the two groups (Fig. 4a). The data clustered well, and there was an obvious separation under positive and negative ion modes, indicating diverse lipid profiles between the vehicle and PCN-treated groups.

a Principal component analysis (PCA) and orthogonal partial least discriminant analysis (OPLS-DA) score plots of lipidomic profiles under positive and negative ion modes (Blue diamonds: the vehicle group; Red diamonds: the PCN group). b Heatmap of different lipids between the vehicle group and the PCN group. c Hepatic TG content in vehicle- and PCN-treated mice. d qRT–PCR analysis of TG-related genes in mouse livers after long-term treatment with PCN. Data are presented as the means ± SD. (n = 5–6). *P < 0.05, **P < 0.01, ****P < 0.0001 versus the vehicle group.
Several classes of lipid biomarkers were identified, including triglyceride (TG), phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS). According to the criteria of variable importance (VIP) values > 1.0, P < 0.05 and fold-change > 5.0, 48 biomarkers were selected, as shown in the heatmap. TGs were upregulated dramatically among these lipids, accounting for about half of all, including TG(22:5/18:2/22:6), TG(20:5/18:2/22:6), TG(20:1/18:1/18:2), TG(19:5/18:2/18:2), TG(18:4/17:1/18:1), TG(18:2/17:2/22:6), TG(18:2/17:1/22:6), TG(18:2/17:1/18:2), TG(18:2/13:1/18:2), TG(18:1/20:4/22:6), TG(18:1/18:2/22:6), TG(18:1/18:2/20:4), TG(18:1/18:2/18:2), TG(16:1/18:2/22:6), TG(16:1/18:2/18:3), TG(16:1/18:2/18:2), TG(16:1/17:1/18:2), TG(16:1/16:2/18:2), TG(16:1/16:1/18:2), TG(16:0/18:2/20:4), TG(16:0/18:2/18:2), TG(15:2/18:2/18:2), TG(15:1/18:2/18:2), TG(15:0/18:2/22:6) and TG(12:0/18:2/22:6). Additionally, several PC, PE and PS were downregulated in the PCN group (Fig. 4b). Hepatic TG content was also measured, and the results suggested that long-term PCN treatment significantly increased hepatic TG to 2-fold of the vehicle group from 0.08 to 0.18 mmol/g protein (Fig. 4c), which was consistent with the lipidomic analysis results above.
The mRNA expression of genes involved in hepatic TG homeostasis was measured. As shown in Fig. 4d, the mRNA levels of genes encoding hepatic fatty acid binding protein 1 (Fabp1), Cd36 and acyl-CoA synthetase long-chain family member 1 (Acsl1) were elevated to 4.04-, 4.03-, and 11.22-fold of the vehicle group. In addition, the relative levels of mRNA encoding hormone-sensitive lipase (Hsl or Lipe) and adipose triglyceride lipase (Pnpla2) were upregulated to 2.47- and 3.97-fold of the vehicle group (Fig. 4d). Taken together, TG uptake and synthesis were enhanced, whereas the metabolism of TGs was suppressed, which eventually contributed to the accumulation of TGs.
In conclusion, lipidomics and gene analysis suggested that long-term treatment with PCN had a significant influence on hepatic lipid homeostasis in mice. Significant TG accumulation in the liver may eventually lead to liver enlargement.
Long-term PCN treatment does not induce or aggravate tumors
The carcinogenesis risk in mouse liver after long-term PCN treatment was further studied after a single DEN at the beginning of the administration (Fig. 5a). The results indicated that both the DEN and DEN + PCN groups displayed visible tumor foci. Additionally, the vehicle and PCN groups showed no observable tumor foci (Fig. 5b). The tumors of the DEN + PCN group were smaller and more sporadic than those of the DEN group.

a Mice were treated with vehicle (corn oil) or PCN (100 mg/kg per week, i.p.) for 42 weeks after a single injection of saline or DEN (5 mg/kg). b Morphological photographs of representative mouse livers. c The relative mRNA levels of Krt8 and Afp. d IHC staining for AFP. Data are presented as the means ± SD. (n = 3). *P < 0.05 versus the vehicle group.
To investigate whether PXR can induce liver cancer, the mRNA levels of cytokeratin 8 (Krt8), a liver preneoplastic tumor marker [20], and Afp, a liver tumor marker [21], were measured in all four groups. The results showed that the mRNA levels of Krt8 and Afp were not upregulated in the PCN group compared with the vehicle group, suggesting that no preneoplastic or neoplastic foci existed in the livers of long-term PCN-treated mice. Moreover, Krt8 and Afp levels were upregulated in both the DEN and DEN + PCN groups, but there was no significant difference between the two groups, indicating that long-term PCN treatment did not increase the risk of DEN-induced carcinogenesis (Fig. 5c). IHC staining of AFP was further performed to confirm the above result. Similar to the mRNA levels, long-term use of PCN did not upregulate the expression of AFP in either cancerous or normal mice (Fig. 5d). All these data demonstrated the safety of the long-term use of the mPXR agonist PCN.
Discussion
Unlike other organs, the liver always maintains constant weight and size to maintain body homeostasis. PXR activation has been reported to induce physiological liver enlargement with hepatocyte proliferation and enlargement after 5- or 10-day administration [8, 9]. However, the effect of long-term PCN treatment on the liver is still unclear. The current study showed that long-term PCN treatment for 42 weeks induced hepatomegaly (liver/body weight ratio from 4.3% to 5.2%, 1.2-fold). However, the liver/body weight ratio was significantly lower than that of 5-day PCN treatment in our previous study (from 4.6% to 6.7%, 1.5-fold [9]). Abe et al. reported that the liver/body weight ratio gradually decreased after the withdrawal of PCN. There was no statistical significance against the vehicle group at day 6 after PCN withdrawal [22]. In the current study, liver samples were collected 7 days after the last dose. Although the liver/body weight ratio was still higher in the PCN group, discontinuation of PCN may have resulted in a substantial reduction in liver enlargement compared to that of the 5-day treatment. Hepatocyte hypertrophy and proliferation were not observed after long-term PCN treatment. A possible reason for the hepatomegaly is that hepatocyte proliferation and enlargement occurred in the early stage during PCN treatment simultaneously. Over time, the liver gradually adapts to repeated exogenous stimulation, and thus, there was no hepatocyte hypertrophy or proliferation after long-term treatment with PCN.
As a pivotal xenobiotic receptor, PXR functions in a ligand-binding-dependent manner. Many reports and our previous studies have shown that administration of PCN can significantly upregulate the expression of PXR target genes [9, 23]. However, in the current study, PXR inactivation after 42 weeks of PCN treatment was validated by the protein levels of CYP3A11, CYP2B10, UGT1A1, MRP2, and MRP4. In addition, the expression of PXR itself was not changed. The results above indicated that long-term PCN treatment does not upregulate PXR downstream proteins, and the reasons for this phenomenon may be various. It has been illustrated that the liver has a self-regulating mechanism to adapt to changes when facing external stimuli, and long-term agonist administration may change the receptor status. The multidimensional regulatory mechanism of PXR has been revealed, including the genetic and epigenetic regulation of PXR expression, transcriptional regulation, subcellular localization, ligand-dependent activation, and protein–protein interactions [24]. Notably, we collected liver samples 1 week after the last dose of PCN, which may result in the reversal of PXR activation after withdrawal, with a concomitant reversal of liver enlargement and cessation of hepatocyte proliferation. Thus, the results may have been different if liver samples were collected 24 h after the last dose. The reasons why PXR downstream proteins were not upregulated after long-term treatment with PCN still need further investigation. The transcriptional activity of PXR can also be modulated through crosstalk with many other NRs, including FXR, CAR, PPARα, liver X receptor (LXR), and androgen receptor (AR), many of which have been reported to induce hepatomegaly. Therefore, we further detected other NRs related to hepatomegaly. Although the results show that the expression of PPARα, CAR and FXR was not changed, their downstream proteins should be further detected to confirm whether these NRs were activated after long-term treatment with PCN.
Hepatomegaly is often attributed to changes in cellular contents, including excess glycogen, water retention, and lipid accumulation [25]. Lipids are crucial elements of cellular or subcellular membranes. Lipids can be divided into eight categories, including fatty acyls, glycerophospholipids, glycerolipids, polyketides, prenol lipids, saccharolipids, sphingolipids, and sterol lipids, each of which contains distinct classes and subclasses [26]. PXR maintains lipid homeostasis by regulating the enzymes and transporters involved in lipid synthesis and disposition [27]. We previously reported that 5-day PCN treatment induces lipid deposition, which is mostly attributed to TGs by regulating the genes related to TG uptake, biosynthesis, and metabolism [14]. Furthermore, dexamethasone, an agonist of PXR, also promotes liver enlargement by accumulating lipids [12]. In this study, long-term treatment with PCN affected lipid homeostasis in a similar way. Long-term PCN treatment affected the expression of genes associated with TG uptake, synthesis, and metabolism. The hepatic accumulation of TGs in lipid droplets represents the first step in the development of nonalcoholic fatty liver disease (NAFLD) [28]. However, several studies have recently defined TGs as having a protective rather than harmful role in the progression from steatosis to nonalcoholic steatohepatitis (NASH) [29, 30], which is consistent with our results regarding physiological liver enlargement. Thus, TG accumulation may contribute greatly to long-term PCN treatment-induced hepatomegaly.
TG’s original sources include de novo lipogenesis and intracellular stores. FABP1 and CD36 are important fatty acid transporters in the liver [31]. Our results showed significant upregulation of Fabp1 and CD36 expression, leading to increased TG uptake. In addition, the mRNA level of Acsl1, which can increase TG synthesis and β-oxidation [32], was most dramatically upregulated by 11.22-fold. HSL and PNPLA2 are key players in TG catabolism, providing fatty acids as energy substrates and metabolic intermediates [33]. Although the mRNA levels of Hsl and Pnpla2 were upregulated, the total effect of gene expression promoted TG accumulation.
Multiple lipids were significantly changed in addition to TGs. PC and PE are the two most abundant phospholipids in the plasma membranes of all mammalian cells [34]. An abnormal PC/PE ratio influences energy metabolism and is linked to NAFLD, as well as liver failure, liver regeneration, and the severity of alcoholic fatty liver disease [35]. In the current study, 5 PCs and 7 PEs were dramatically downregulated after long-term PCN treatment, indicating that the PC/PE ratio might change. The possibility of liver pathological changes caused by an abnormal PC/PE ratio should be considered during long-term PXR agonist treatment. PS is an anionic immunosuppressive phospholipid that is expressed on the inner leaflet of the cell membrane and is synthesized from PC or PE by exchanging the base head group with serine [36]. Thus, the decrease in PC and PE may result in the reduction of PS, which is consistent with the current study. Furthermore, PS is a key signal in the coagulation cascade, and extracellular exposed PS is also a pivotal factor in the recognition and clearance of apoptotic cells, indicating that its activity depends on the location [37]. In the current study, 4 PSs were significantly decreased after long-term PCN treatment. However, the change in the subcellular distribution of PS was unclear.
In this study, the Afp and Krt8 expression levels and AFP staining showed that long-term PCN treatment did not induce or aggravate liver cancer. Growing evidence strongly points to the significant ability of PXR in defense, detoxification, homeostasis maintenance and proliferation, which are antagonistic for cancer development [24]. PXR activation does not promote hepatocarcinogenesis, in contrast to constitutive androstane receptor (CAR, NR1I3), and rather attenuates CAR-mediated liver cancer [15]. Deepak et al. found that PXR is significantly downregulated in DEN-induced liver cancer and that PXR is not related to hepatic carcinogenesis or cancer progression [38]. PXR is capable of suppressing the growth, proliferation, and migration of cancer cells by inducing cellular cycle arrest. The antitumor bioregulation of PXR is achieved via functional interaction with the transcriptional regulation of p21, E2F, cullin1e3, MAD2L1 (mitotic spindle assembly checkpoint protein MAD2A), ANAPC2 (anaphase-promoting complex subunit 2) and other PXR-related pathways [39, 40]. In addition, PXR was reported to attenuate liver cancer formation by inhibiting the epithelial-mesenchymal transition (EMT) of liver cancer cells [15]. Taken together, these results suggest that PXR could be regarded as a promising drug target with a large safety margin for long-term treatment.
In summary, long-term treatment with PCN did not upregulate PXR downstream proteins but still promoted hepatomegaly and lipid accumulation without hepatocyte enlargement and proliferation. The accumulated TG may lead to long-term PCN treatment-induced hepatomegaly. In addition, long-term treatment with PCN did not induce liver cancer. Our study demonstrated the safety of long-term PCN use and suggested that PXR could be a promising and safe drug target for long-term treatment.
References
Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82.
Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends Pharmacol Sci. 2012;33:552–8.
Drocourt L, Pascussi JM, Assenat E, Fabre JM, Maurel P, Vilarem MJ. Calcium channel modulators of the dihydropyridine family are human pregnane X receptor activators and inducers of CYP3A, CYP2B, and CYP2C in human hepatocytes. Drug Metab Dispos. 2001;29:1325–31.
Bhadhprasit W, Sakuma T, Hatakeyama N, Fuwa M, Kitajima K, Nemoto N. Involvement of glucocorticoid receptor and pregnane X receptor in the regulation of mouse CYP3A44 female-predominant expression by glucocorticoid hormone. Drug Metab Dispos. 2007;35:1880–5.
Di Masi A, De Marinis E, Ascenzi P, Marino M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol Asp Med. 2009;30:297–343.
Wang Y, Ong S, Chai S, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8:803–17.
Michalopoulos GK. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology. 2017;65:1384–92.
Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos. 2001;29:1467–72.
Jiang Y, Feng D, Ma X, Fan S, Gao Y, Fu K, et al. Pregnane X receptor regulates liver size and liver cell fate by yes-associated protein activation in mice. Hepatology. 2019;69:343–58.
Shizu R, Abe T, Benoki S, Takahashi M, Kodama S, Miayata M, et al. PXR stimulates growth factor-mediated hepatocyte proliferation by cross-talk with the FOXO transcription factor. Biochem J. 2016;473:257–66.
Yao X, Jiao T, Jiang Y, Fan S, Zhao Y, Yang X, et al. PXR mediates mifepristone-induced hepatomegaly in mice. Acta Pharmacol Sin. 2021;43:146–56.
Jiao T, Yao X, Zhao Y, Zhou Y, Gao Y, Fan S, et al. Dexamethasone-induced liver enlargement is related to PXR/YAP activation and lipid accumulation but not hepatocyte proliferation. Drug Metab Dispos. 2020;48:830–9.
Zhao Y, Yao X, Jiao T, Tian J, Gao Y, Fan S, et al. Schisandrol B promotes liver enlargement via activation of PXR and YAP pathways in mice. Phytomedicine. 2021;84:153520.
Jiang Y, Yao X, Fan S, Gao Y, Zhang H, Huang M, et al. Lipidomic profiling reveals triacylglycerol accumulation in the liver during pregnane X receptor activation-induced hepatomegaly. J Pharm Biomed Anal. 2021;195:113851.
Shizu R, Ishimura M, Nobusawa S, Hosaka T, Sasaki T, Kakizaki S, et al. The influence of the long-term chemical activation of the nuclear receptor pregnane X receptor (PXR) on liver carcinogenesis in mice. Arch Toxicol. 2021;95:1089–102.
Gao Y, Fan S, Li H, Jiang Y, Yao X, Zhu S, et al. Constitutive androstane receptor induced-hepatomegaly and liver regeneration is partially via yes-associated protein activation. Acta Pharm Sin B. 2021;11:727–37.
Fan S, Gao Y, Qu A, Jiang Y, Li H, Xie G, et al. YAP-TEAD mediates PPAR alpha-induced hepatomegaly and liver regeneration in mice. Hepatology. 2022;75:74–88.
Zhang H, Wang Y, Guan L, Chen Y, Chen P, Sun J, et al. Lipidomics reveals carnitine palmitoyltransferase 1C protects cancer cells from lipotoxicity and senescence. J Pharm Anal. 2021;11:340–50.
Carthew P, Nolan BM, Edwards RE, Smith LL. The role of cell death and cell proliferation in the promotion of rat liver tumours by tamoxifen. Cancer Lett. 1996;106:163–9.
Kakehashi A, Kato A, Inoue M, Ishii N, Okazaki E, Wei M, et al. Cytokeratin 8/18 as a new marker of mouse liver preneoplastic lesions. Toxicol Appl Pharmacol. 2010;242:47–55.
Wong RJ, Ahmed A, Gish RG. Elevated alpha-fetoprotein: differential diagnosis - hepatocellular carcinoma and other disorders. Clin Liver Dis. 2015;19:309–23.
Abe T, Shizu R, Sasaki T, Shimizu Y, Hosaka T, Kodama S, et al. Functional interaction between pregnane X receptor and Yes-associated protein in xenobiotic-dependent liver hypertrophy and drug metabolism. J Pharmacol Exp Ther. 2019;371:590–601.
Fisher CD, Jackson JP, Lickteig AJ, Augustine LM, Cherrington NJ. Drug metabolizing enzyme induction pathways in experimental non-alcoholic steatohepatitis. Arch Toxicol. 2008;82:959–64.
Xing Y, Yan J, Niu Y. PXR: a center of transcriptional regulation in cancer. Acta Pharm Sin B 2020;10:197–206.
Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–76.
Holčapek M, Liebisch G, Ekroos K. Lipidomic analysis. Anal Chem. 2018;90:4249–57.
Wada T, Gao J, Xie W. PXR and CAR in energy metabolism. Trends Endocrinol Metab. 2009;20:273–9.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51.
Yamaguchi K, Yang L, Mccall S, Huang J, Yu X, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–74.
Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–51.
Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–108.
Li LO, Ellis JM, Paich HA, Wang S, Gong N, Altshuller G, et al. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem. 2009;284:27816–26.
Brejchova K, Radner FPW, Balas L, Paluchova V, Cajka T, Chodounska H, et al. Distinct roles of adipose triglyceride lipase and hormone-sensitive lipase in the catabolism of triacylglycerol estolides. Proc Natl Acad Sci USA. 2021; 118.
Van Der Veen JN, Kennelly JP, Wan S, Vance JE, Vance DE, Jacobs RL. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. 2017;1859:1558–72.
Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, et al. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006;3:321–31.
Kim HY, Huang BX, Spector AA. Phosphatidylserine in the brain: metabolism and function. Prog Lipid Res. 2014;56:1–18.
Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27.
Kotiya D, Jaiswal B, Ghose S, Kaul R, Datta K, Tyagi RK. Role of PXR in hepatic cancer: its influences on liver detoxification capacity and cancer progression. PLoS One. 2016;11:e0164087.
Ouyang N, Ke S, Eagleton N, Xie Y, Chen G, Laffins B, et al. Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells. Br J Cancer. 2010;102:1753–61.
Niu Y, Wang Z, Huang H, Zhong S, Cai W, Xie Y, et al. Activated pregnane X receptor inhibits cervical cancer cell proliferation and tumorigenicity by inducing G2/M cell-cycle arrest. Cancer Lett. 2014;347:88–97.
Acknowledgements
The work was supported by the National Natural Science Foundation of China [Grant number: 82025034, 81973392], the National Key Research and Development Program (Grant number: 2017YFE0109900), the Shenzhen Science and Technology Program (KQTD20190929174023858), the 111 project (Grant number: B16047), and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program [Grant number: 2017BT01Y093].
Author information
Authors and Affiliations
Contributions
MH and HCB participated in research design. YFZ, JY, SCF, YG, and YMJ performed experiments. YFZ, YG, and HCB wrote or revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, Yf., Gao, Y., Yang, J. et al. Long-term treatment with the mPXR agonist PCN promotes hepatomegaly and lipid accumulation without hepatocyte proliferation in mice. Acta Pharmacol Sin 44, 169–177 (2023). https://doi.org/10.1038/s41401-022-00925-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00925-3
Keywords
This article is cited by
-
Adverse outcome pathway for pregnane X receptor-induced hypercholesterolemia
Archives of Toxicology (2023)
