
Abstract
Purpose
Intravenous vitamin C (IVC) is used in a variety of disorders with limited supporting pharmacokinetic data. Herein we report a pharmacokinetic study in healthy volunteers and cancer participants with IVC doses in the range of 1–100 g.
Methods
A pharmacokinetic study was conducted in 21 healthy volunteers and 12 oncology participants. Healthy participants received IVC infusions of 1–100 g; oncology participants received IVC infusions of 25–100 g. Serial blood and complete urine samples were collected pre-infusion and for 24 h post-infusion. Pharmacokinetic parameters were computed using noncompartmental methods. Adverse events were monitored during the study.
Results
In both cohorts, IVC exhibited first-order kinetics at doses up to 75 g. At 100 g, maximum concentration (Cmax) plateaued in both groups, whereas area under the concentration–time curve (AUC) only plateaued in the healthy group. IVC was primarily excreted through urine. No saturation of clearance was observed; however, the mean 24-h total IVC excretion in urine for all doses was lower in oncology participants (89% of dose) than in healthy participants at 100 g (99%). No significant adverse events were observed; thus, maximum tolerated dose (MTD) was not reached.
Conclusion
IVC followed first-order pharmacokinetics up to 75 g and at up to 100 g had complete renal clearance in 24 h. IVC up to 100 g elicited no adverse effects or significant physiological/biochemical changes and appears to be safe. These data can be used to rectify existing misinformation and to guide future clinical trials.
Registration
ClinicalTrials.gov identifier number NCT01833351.
Similar content being viewed by others

Intravenous vitamin C is widely used, yet basic pharmacokinetic information is not complete. |
A pharmacokinetic trial has been done in healthy people for the first time and additional data collected in cancer patients and found that with some precautions intravenous vitamin C is very safe. |
The PK study found that intravenous vitamin C, at doses up to 100 g, is distributed into extracellular water, is excreted by the kidneys, and exhibits predictable first order pharmacokinetics. |
1 Introduction
Parenteral-administered ascorbic acid or intravenous vitamin C (IVC) dates back many decades [1] and is known to be a pharmacologic agent when administered in this manner [2,3,4]. Orally administered vitamin C blood levels are tightly controlled by gastrointestinal tract absorption while IVC bypasses this tight control, resulting in markedly higher systemic concentrations. These higher concentrations produce well-documented effects not seen with oral administration, specifically the formation of ascorbyl radical and hydrogen peroxide, making it a pro-oxidative therapy [5,6,7]. While IVC clinical trials have been conducted in a variety of disorders such as cancer, sepsis, infection, and chelation [8,9,10,11,12,13,14,15,16], there are only limited pharmacokinetic data available to advise appropriate trial design [8, 9, 17,18,19,20,21,22,23]. Pharmacokinetic-based optimization of the IVC dosing strategy is essential to effectively design future IVC clinical trials. For example, the lack of pharmacokinetic data has hampered trial designs using IVC in acute and chronic infections.
This pharmacokinetic study is the first to look at escalating doses of IVC of 1–100 g in healthy participants and 25–100 g in participants with disease. In light of the increasing interest in the use of IVC, particularly in viral- and bacterial-infected patients [12,13,14, 16], the primary objectives of the study were to characterize the pharmacokinetic profile of IVC, to identify the maximum tolerated dose (MTD) to guide future trials, and to confirm or refute safety data collected in previous clinical trials [8, 9, 17,18,19,20,21,22,23].
2 Methods
2.1 Ethics
The study was conducted in compliance with the FDA-approved study protocol (IND number 65,805). The protocol and consent form were approved by the University of Kansas Health System, Institutional Review Board (IRB) and the Kansas Cancer Center Protocol Review and Management Committee. Trial oversight was provided by University of Kansas Health System, Kansas Cancer Center Data and Safety Monitoring Board with independent event adjudication. The Clinical Trials website (https://clinicaltrials.gov) study number was NCT01833351.
2.2 Study Population
The trial was conducted in two parts with Part 1 enrolling healthy adult participants and Part 2 enrolling adult oncology participants no longer eligible for cytoreductive therapy. Recruitment dates were from 2011 to 2015. The trial was a single-site study conducted at the University of Kansas Medical Center. The inclusion criteria and exclusion criteria are listed in Supplementary Table 1 (see electronic supplementary material [ESM]). As a result of the inclusion/exclusion criteria, none of the participants had impaired renal function. For Part 2, oncology participants had histologically or cytologically diagnosed malignancy, were unwilling or ineligible for further radiation or chemotherapy at the time of enrollment, and study participation was approved by the treating oncologist.
Participants were required to remain in the research center for approximately 10 hours the day of the infusion, discharged to home, and required to return the following morning for the 24-hour sample draw. All study procedures were completed at 24 hours.
2.3 Dosing and Administration
The design was a classic pharmacokinetic study with escalating doses from 1 to 100 g as an open-label trial designed to assess the safety and pharmacokinetics of pharmacologic ascorbate (IVC). The study drug was obtained from Mylan (Bioniche) Pharmaceutical under the name ASCORBIC ACID Injection, USP, 500 mg/mL in 50-mL sterile dispensing vials and buffered to a neutral pH. The study drug was housed in the Investigational Pharmacy at the University of Kansas Medical Center, where it was stored between 2 and 8 °C (36–46 °F) until the time of mixing, which was just prior to infusion.
Healthy participants in Part 1 were enrolled at six dose levels given as single administration infusions at 1, 5, 10, 25, 50, and 75 g, while the seventh dose level at 100 grams was administered with four infusions separated by 1-week intervals. Part 2 oncology participants received one of the following doses: 25, 50, 75, or 100 g, administered with four infusions at each dose level separated by 1-week intervals. Two of the four administrations were for pharmacokinetic analysis and two for safety analysis.
The study drug was prepared by the Investigational Pharmacy at the University of Kansas Health System and mixed in 1000 mL of carrier fluid for each dose level. The infusions were administered by infusion pump at a rate to deliver the total infusion in 2 hours for each dose administered.
Osmolarity of doses is shown in Supplemental Table 2 (see ESM). At higher doses, the osmolarity was elevated and contributed to a high osmotic load.
The study was accomplished by obtaining the following evaluations:
-
(1)
Toxicity grading for healthy participants using the Center for Biologics toxicity grading scale. Dose-limiting toxicity was defined as two or more participants experiencing the same GRADE ONE (mild) event on this scale (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/toxicity-grading-scale-healthy-adult-and-adolescent-volunteers-enrolled-preventive-vaccine-clinical); or any GRADE TWO or higher event with toxicity grading for oncology participants evaluated by the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.0 scored from 1 through 5 with 2–5 considered adverse events for the purposes of the study (https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm). Evaluation was conducted via physical examination, patient report, phlebotomy and urinalysis pre- and post-infusion for basic metabolic panel, bicarbonate (pH surrogate marker), complete blood count, osmolality, and electrocardiogram (ECG).
-
(2)
Pharmacokinetic evaluation of IVC first in healthy adults followed by evaluation in a cohort of oncology participants.
Three healthy participants were enrolled at each dose level. The protocol stipulated that if one of three participants accrued to a dose level developed Grade 1 toxicity, three more participants were to be accrued to that dose level. If no further toxicities were seen, the protocol recommended advancement to the next dose level. If two participants had toxicities identified at a dose level, the trial was to be stopped at that level. MTD was defined as the dose level BELOW the dose that encountered two dose-limiting toxicities, but not >100 g. Once MTD or 100 g was reached in the healthy participants, those enrolled received four doses separated by 1-week intervals with dose 1 and 4 scheduled as pharmacokinetic evaluation doses and dose 2 and 3 scheduled as safety evaluation doses.
The oncology cohort had a similar structure, enrolling three participants per dose level and evaluating adverse events as noted above. In contrast to the healthy participants, the oncology participants were enrolled at 25%, 50%, 75%, and 100% of MTD or 100 grams (whichever came first) or 25, 50, 75, and 100 grams. All enrolled oncology participants received four doses at each dose level separated by 1-week intervals with dose 1 and 4 scheduled as pharmacokinetic evaluation doses and doses 2 and 3 scheduled as safety evaluation doses. Data was collected over a 24-h period for all participants in both Part 1 and Part 2.
2.4 Sampling Schedule
Venous access was obtained by indwelling catheter placed at the outset of the infusion day, with baseline assessment obtained prior to starting the infusion. Infusion start was designated as Time 0 with outlined collections performed (see Supplemental Table 3 in the ESM). Safety data collection occurred at each infusion, although phlebotomy and urine samples were obtained at only dose 1 and dose 4 for pharmacokinetic analysis.
A baseline sample and total urine was collected, and volumes recorded for the intervals defined in Supplemental Table 3. Samples from each collection were analyzed for excreted ascorbic acid.
2.5 Analytical Methods
Sample acquisition and stability of samples was considered central to adequate pharmacokinetic evaluation, as degradation of vitamin C begins immediately upon sampling. Vitamin C plasma sample preparation has been described in detail previously [24, 25]. In brief, blood was collected in heparinized tubes at the prescribed schedule and plasma was immediately prepared by refrigerated centrifugation. Plasma was immediately aliquoted and frozen at −25 °C and then, within 72 h, transferred to a −80 °C C freezer until analyzed.
Plasma vitamin C levels were measured by using high-performance liquid chromatography (HPLC) with coulometric electrochemical detection with the sample processing and HPLC analysis performed as described [24,25,26].
Urine samples were collected, with 5-mL samples filtered through a micropore filter to remove proteins and impurities and stored at −80 ºC until analyzed by HPLC [25].
2.6 Pharmacokinetic Analysis
Pharmacokinetic parameters were determined by noncompartmental analysis using Phoenix WinNonlin, v. 8.3.3. (Certara). The pharmacokinetic parameters included maximum concentration (Cmax), time to Cmax (Tmax), area under the concentration–time curve (AUC), terminal half-life (t½), apparent clearance (CL), and apparent volume of distribution (VD). Participant body weights and serum creatinine levels were obtained for each participant’s first IVC at a given dose and were used to normalize doses and to calculate glomerular filtration rate (GFR). Kinetics of urinary excretion of vitamin C also were determined, and are reported as maximum rate of excretion and total excretion of vitamin C.
2.7 Statistical Analysis Methods
Baseline participant characteristics were summarized for both healthy and oncology participants. Mean ± standard deviation (SD) was presented for continuous variables like age, and count with percentages was presented for categorical variables such as race, gender, and tumor types. The change of plasma parameters between pre- and post-IVC infusion was analyzed using paired t-test. The plasma ascorbate concentrations after IV infusion were plotted for healthy participants and oncology participants, respectively. All analyses were conducted using SAS 9.4 (SAS, Cary, NC, USA).
3 Results
3.1 Participants Demographics
Participants in the healthy cohort were younger (34.9 ± 14.2 years) compared with the oncology cohort (54.8 ± 10.4 years). Gender enrollment in the healthy cohort had fewer females (7) compared with males (14), while the oncology cohort enrolled more females (7) than males (5). The enrolled healthy participants included Caucasian non-Hispanic (18), Hispanic (2), and Asian (1) ethnicities while the oncology participants were all Caucasian non-Hispanic (12). See Table 1.
3.2 Pharmacokinetics
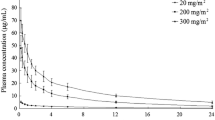
Serial plasma sampling and complete urine collections for 24 h, beginning at the start of vitamin C infusion, were performed in healthy and oncology participants to characterize the systemic exposure and the urinary excretion of vitamin C. Vitamin C concentrations in all samples were determined, noncompartmental analysis was performed, and when body weight and serum creatinine values were available, normalized doses and GFR were calculated. Compiled results are presented in Tables 2 and 3. Pre-dose plasma vitamin C concentrations were 1.3 ± 0.9 mg/dL in healthy participants and 2.2 ± 3.0 mg/dL in oncology participants, both within the normal concentration range previously reported [2, 23, 27, 28]. The plasma ascorbate concentration profiles following the IVC infusions are shown in Fig. 1.

Plasma ascorbate concentrations after IV infusion. IVC was given at the indicated doses, blood samples were drawn at various time points within 24 h, and ascorbate concentrations were detected via HPLC. Data shows mean ± SEM for at least 3 participants per dose. A Healthy participants. B Oncology participants. HPLC high-performance liquid chromatography, IVC intravenous vitamin C, SEM standard error of the mean
For healthy participants, both Cmax and AUC0–24 h increased linearly with dose up to 75 g, while both parameters appeared to plateau between 75- and 100-g doses (Table 2, Fig. 2A). The linear relationships between mean Cmax and AUC0–24 h values to dose over the range of 1–75 g resulted in r2 of 0.9892 and 0.9937, respectively. This linearity demonstrates that IVC follows first-order pharmacokinetics up to at least the 75-g dose, and this is further supported by the consistency of t½, VD, and clearance throughout this range (Table 2). AUC0–∞ values are also presented, and it is noteworthy that the mean extrapolated portions of those curves represent <1% of the total values.

Plasma Cmax and AUC for IVC in healthy and oncology participants. Doses for healthy participants were 1 g, 5 g, 10 g, 25 g, 75 g, and 100 g; and for oncological participants were 25 g, 75 g, and 100 g. A Cmax; B AUC (0–24 h). AUC (0–24 h) area under the concentration–time curve from 0 to 24 h, Cmax maximum concentration, IVC intravenous vitamin C
For the pharmacokinetic study in oncology participants, three participants were treated at the 25-, 50-, 75-, and 100-g dose levels. Sampling of one participant at the 75-g dose had missing data points and was not included in the analysis. Although some intra-individual differences in pharmacokinetic values were seen between the two infusions, most of those differences were <15% of the mean value for the parameter, and there was no consistent trend in these differences between the first and second pharmacokinetic study days. The pharmacokinetic parameters obtained for the oncology participants were similar to those seen with the healthy participants. Dose proportionality was observed for Cmax from 1 to 75 g, and plateaued between 75 and 100 g, similar to that in the healthy participants (shown in red in Fig. 2A, r2 = 0.9221). AUC0–24 h increased linearly across all doses used (r2 = 0.9519, Fig. 2B). Half-life (t½) in the oncological group was 2.4 ± 0.3 h, and in healthy participants was 1.9 ± 0.3 h (Table 2). The difference was not statistically significant (p = 0.065). All pharmacokinetic parameters determined for the oncology participants supported first-order kinetics for IVC infusion over this dose range, and none of the mean values differed significantly from those calculated for the healthy participants.
Dose normalization and scaling of VD and CL to body weight were performed and the compiled results are presented in Table 3. Dose normalization did not significantly affect inter-individual variability in pharmacokinetic values. Scaling of VD and calculation of individual GFR values were far more informative. As shown, VD for all doses were in the range of 0.12–0.25 L/kg, with a mean value of 0.20 L/kg, consistent with distribution into extracellular spaces but without extensive tissue uptake. CL ranged from 2.8 to 7.9 L/h across all groups, with mean values of 5.6 L/h and 5.1 L/h for the healthy participants and the oncology participants, respectively. CL/GFR for both participant groups at all doses from 10 to 100 g approached unity. This establishes renal filtration as the dominant clearance pathway. At the 1- and 10-g doses, this ratio was markedly lower, indicating significant renal reabsorption of filtered ascorbate at concentrations closer to physiological, rather than pharmacological.
Systemic pharmacokinetics for both study groups demonstrated >95% clearance of vitamin C during the 24-h sampling period. Urine excretion of vitamin C accounted for 99% of the dose for healthy participants and 89% for oncology participants (Table 4, Fig. 3). The percent recovery of the vitamin C doses in the urine is significantly lower in oncology participants than in healthy participants (Fig. 2; p = 0.0037). The reasons for this are not clear. Given the slopes of the curves of Cmax and AUC, and no difference in 24-h blood ascorbate concentration, the difference in the excreted ascorbate amount is presumably because of tissue utilization of ascorbate in oncology participants.

Urinary excretion of intravenous vitamin C (IVC). 24-h urinary excretion of IVC shown as percentage of recovery of the received doses. All doses combined. *p = 0.0037 by t test
Both the maximum rate of urinary excretion and the area under the urinary excretion curve show linear dose proportionality in both groups, establishing that urinary excretion of vitamin C even at the 100-g infusion dose is first order. Urinary excretion of vitamin C from the two groups also establishes that this is the primary path for clearance. Maximum rate of urinary excretion, area under the urinary excretion curve, and the amount of vitamin C recovered in the urine all are consistent with this complete clearance within the 24-h period, and that at least 89% of the administered dose is excreted as vitamin C (Table 3).
MTD was not identified. No significant adverse events were identified at the top dose of 100 g in either Part 1 or Part 2.
3.3 Laboratory Analysis
Laboratory analyses were carried out with plasma samples pre- and post-IVC infusion for all participants, assessing renal function (blood urea nitrogen [BUN] and creatinine), liver function (ALT, AST, alkaline phosphatase), glucose, calcium, hemogram, blood pH surrogate markers (bicarbonate), as well as other metabolic and coagulation parameters (Table 5). Complete blood counts were not changed in either group at any dose, including absolute counts and percentages of lymphocytes, neutrophils, basophils, eosinophils, monocytes, red blood cells, and platelet counts. Hematocrit was not changed, as well as mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), and mean corpuscular volume (MCV), at all doses in both groups. Hemoglobin levels were also not changed. As shown in Table 5, no clinically significant changes were seen in any measured parameter.
Electrocardiograms (ECGs) were also obtained pre- and post-IVC infusion for evaluation of cardiac status, with no noted changes elicited by IVC.
3.4 Safety and Tolerability
The first line of adverse event review was provided by the principal investigator (JD) during study drug infusions, and as values were made available to include physical examination, clinical laboratory test results, vital sign measurements, and ECG assessment. For purposes of ongoing evaluation, all data were reviewed by the Kansas Cancer Center Protocol Review and Management Committee (PRMC) and the Data Safety Monitoring Board (DSMB) appointed by the University of Kansas Health System, Institutional Review Board, and the FDA as part of investigational new drug review reporting. Committee reviews were independent of investigator input. Hematologic, metabolic, and constitutional adverse events were made available to PRMC, DSMB, and IRB Human Subjects Committee (HSC) on schedule prescribed by the approved protocol.
See Table 6 for adverse event reporting and monitoring schedule. Two treatment-emergent adverse events were reported during the conduct of the trial in two participants. One incident was related to pain at the infusion site, which was deemed unlikely to be study-drug related; a separate incident of Grade 1 chills was deemed to have a reasonable possibility of being study-drug related. The episode of chills, seen at the 100-g dose in an oncology participant, was not associated with any change in vital signs including temperature, blood pressure, pulse rate, or respiratory rate. Laboratory analysis was unremarkable for this participant. This incident was reviewed and considered Grade 1; by protocol definition, the event was not significant to trigger further enrollment at this dose level.
ECG tracings at baseline and after study drug infusion were read by the study cardiologist (RF). There were no changes in ECG tracings, including Q wave analysis, attributable to the study drug.
4 Discussion
There is increasing interest in clinical applications of intravenous vitamin C (IVC) in a variety of diseases including cancer, sepsis, and infections. Clinical trials conducted to date, however, suffer from insufficient pharmacokinetic information, resulting in different dosing regimens and varied outcomes [13, 14]. This report serves to provide data to encourage future trial design to include more robust dosing without fear of adverse events. Increased dosing of IVC is associated with significant positive pharmacologic effects and outcomes [8, 9, 22].
This is a classic pharmacokinetic study using escalating doses of IVC, unique in that it characterizes pharmacokinetics first in healthy participants followed by an oncology population no longer eligible for cytoreductive therapy. Characterizing the pharmacokinetics of IVC is critical to understanding the feasibility of this approach to preventing or treating human disease. Prior reported IVC pharmacokinetic studies have been in specific disease populations using blood concentration data obtained during clinical trials to assess safety and possible efficacy [8, 9, 17,18,19,20,21,22,23]. These studies, however, utilized far shorter sampling periods, hindering the calculation of t½ and complete AUC. Our work provides a more rigorous characterization of IVC pharmacokinetics, advancing our understanding in three ways. First, by covering a full 24-h period from infusion start we have characterized near quantitative clearance of ascorbate and supported a more accurate determination of primary pharmacokinetic parameters. Second, by complete collection and analysis of urine we have documented and quantified renal excretion as the single dominant pathway for ascorbate clearance, and that it is not saturated even at these high doses. Third, we have demonstrated that for IVC doses of 10–100 g, CL is approximately equal to GFR.
Key parameters to assess IVC as a therapeutic intervention include understanding dosing to provide sufficient exposure to exert a therapeutic effect, the demonstration of distribution that allows the agent to reach its therapeutic targets, and the determination of elimination kinetics to advise dosing schedules. Furthermore, defining the constancy of pharmacokinetic parameters is essential to establishing a predictable dose–exposure relationship. All these goals were achieved in the current study. Hoffer et al. [17] reported an IVC study in cancer patients with similar doses and dosing to this study. Based on their data they proposed equations for predicting Cmax and AUC based on dose and body weight. We used those equations with data from this study and calculated the ratios between our observed results and those predicted by the equations. Results were mixed. For our healthy participants, the mean ratio (± SD) of all individual observed Cmax and AUC(0-24 h) values divided by the calculated values were 0.92 ± 0.27 and 1.08 ± 0.28, respectively. For the oncology participants in this study those ratios were 1.04 ± 0.44 and 1.54 ± 0.55. With our study participants, the Hoffer et al. equations gave acceptable predictions for Cmax, but the results for AUC were less accurate. Further testing of the validity of these equations in different populations and determining whether efficacy of IVC more closely corresponds to Cmax or AUC will be required to assess the clinical value of these dose–exposure predictions.
Of note, MTD was not defined, as the study dictated the stopping point as either MTD or 100-g dose, whichever came first. Future trials may be necessary to define MTD using doses higher than 100 g.
Our previous work [4,5,6, 8, 9] and that of other groups [2, 3, 17,18,19,20,21,22,23, 28] clearly demonstrate the need to bypass the tightly controlled gut absorption of vitamin C to achieve pharmacological concentrations systemically, and this is achieved only by parenteral administration. Baseline vitamin C concentrations are approximately 2 mg/dL [23], and administration of 2 g and 10 g by bolus injection increased initial vitamin C plasma concentrations to 3.5 and 20 mg/dL, respectively.
From our investigation of the cytotoxic effects of vitamin C in pancreatic cell cultures, the optimal effective concentration in vitro to effect pancreatic cancer death is 88 mg/dL (5 mM) [8]. We extrapolate this to in-vivo environments to suggest that even a 10-g bolus may not be sufficient to generate cytotoxic effects. Indeed, our current reported in-vivo data demonstrate that a Cmax >88 mg/dL was achieved by a 25-g infusion, and that the 100-g infusion produced a Cmax >4.6 times higher than the optimal in-vitro concentration.
Our data also established a VD of about 0.2 L/kg, consistent with free distribution of vitamin C into extracellular water, but also suggested that extensive tissue binding does not occur. This apparent distribution will provide vitamin C exposure in the extracellular space, where it may drive Fenton chemistry with production of hydrogen peroxide to become a pro-oxidant agent. This is prevented in blood space secondary to peroxidases, catalase, and other biochemical reactions [5,6,7].
The results reported here expand our understanding of the pharmacokinetics of vitamin C administered by infusion. Our data show that in both healthy and oncology participants, the pharmacokinetics of vitamin C remain first-order at doses of up to 75–100 g in a 2-h infusion. Moreover, even at the 100-g dose, there is virtually quantitative clearance of vitamin C from the body within a 24-h period following infusion. These findings demonstrate that even at these high infusion doses, neither the distribution nor the elimination of vitamin C become saturated or show any significant deviation from first-order kinetics. This validation of first-order pharmacokinetics for intravenous vitamin C over this wide dose range supports accurate prediction of dose–exposure relationships for this pharmacological intervention.
The treatment-appropriate IVC dose is determined by sampling the plasma for peak levels immediately after the infusion is complete, as we have shown in this trial. This ensures adequate vitamin C is present to trigger Fenton chemistry (hydrogen peroxide formation) in the extracellular space. Inadequate dosing is unlikely to result in the prooxidative burst necessary for antineoplastic, antiviral, or antibiotic effects. Infusion rate also contributes to peak plasma concentration and an infusion rate of 0.5–1.0 g/min is recommended [8, 9]. Therefore, it is imperative to check plasma vitamin C to confirm adequate IVC levels before assigning treatment dosing.
Harmful effects have been mistakenly attributed to vitamin C, including hypoglycemia, rebound scurvy, infertility, mutagenesis, and destruction of vitamin B12, but vitamin C does not produce these effects [27]. We have convincingly shown there are no changes in cardiac function by ECG, bicarbonate (pH surrogate marker), glucose levels, bleeding times, liver enzymes, Hb/Hct, platelet counts, or white blood cell count (WBC). There is no evidence that plasma vitamin C levels at 24 h were lower than baseline to suggest rebound scurvy might result. This reported study shows that renal injury does not occur even at the highest dose of 100 g. The transient rise in creatinine at the 100-g dose may be related to the IVC but more likely is related to the very high osmolarity of the infusate [29,30,31].
As noted, IVC distribution is in body water. Of concern is the obesity epidemic that is associated with alterations in body composition with expanded extracellular water [32,33,34]. Expanded body water has implications for dosing in obese individuals. To address this concern for future trialists and clinicians, we have provided g/kg doses at each dose level as a guide with the caveat to check plasma levels.
A note of caution for future trialists and treating clinicians. Never obtain point of care or glucometer fingerstick blood glucose readings after IVC infusion. The glucometer will misidentify vitamin C as glucose and result in an erroneously elevated glucose reading. Hypoglycemic medications such as insulin should never be administered in this setting as there is not elevated blood glucose. If hypoglycemic medications are administered this could result in significant hypoglycemia with untoward effects. If it is critical to assess glucose levels post IVC, a routine laboratory blood draw must be obtained as this is the only accurate assessment of blood glucose levels in this setting [35, 36].
A potential limitation of this study is the paucity of ethnic diversity, especially for African Americans. There is no clinical evidence that Africans or African Americans will have different IVC pharmacokinetic characteristics compared with other ethnic groups. A non-synonymous single nucleotide polymorphism in the vitamin C transporter SVCT1 (SLC23A1) may decrease its activity [37]. This polymorphism is rare but has increased allelic frequency in people of African origin [37]. However, it is unlikely that modified SVCT1 activity will affect pharmacokinetic parameters of vitamin C at the pharmacologic concentrations produced with IV administration.
Overall, the infusion at all doses is safe.
5 Conclusions
IVC is a pharmacologic agent, compared with oral vitamin C, with pro-oxidative properties that present only at higher doses. Emerging from these data is confirmation of safety even at highest doses. This pharmacokinetic study will help define whether dosage and administration schedules are appropriate in future clinical trials and will allow analysis of efficacy and ongoing safety of IVC interventions.
References
Drisko JA. Chapter 8 Orthomolecular approaches for the use of intravenous vitamin C. In Vissers M, Chen Q (editors) Cancer and vitamin C: a new Odyssey for an old vitamin—biochemistry and function, vol II. pp 153–165. New York: CRC Press; 2020. https://doi.org/10.1201/9781003017493-8
Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, Park JB, Lazarev A, Graumlich JF, King J, Cantilena LR. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93(8):3704–9. https://doi.org/10.1073/pnas.93.8.3704.
Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, Wesley RA, Levine M. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140(7):533–7. https://doi.org/10.7326/0003-4819-140-7-200404060-00010.
Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102(38):13604–9. https://doi.org/10.1073/pnas.0506390102.
Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, Choyke PL, Pooput C, Kirk KL, Buettner GR, Levine M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104(21):8749–54. https://doi.org/10.1073/pnas.0702854104.
Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105(32):11105–9. https://doi.org/10.1073/pnas.0804226105.
Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology, and the treatment of cancer. Biochim Biophys Acta. 2012;1826(2):443–57. https://doi.org/10.1016/j.bbcan.2012.06.003.
Polireddy K, Dong R, Reed G, Yu J, Chen P, Williamson S, Violet PC, Pessetto Z, Godwin AK, Fan F, Levine M, Drisko JA, Chen Q. High dose parenteral ascorbate inhibited pancreatic cancer growth and metastasis: mechanisms and a phase I/IIa study. Sci Rep. 2017;7(1):17188. https://doi.org/10.1038/s41598-017-17568-8.
Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med. 2014;6(222):222ra18. https://doi.org/10.1126/scitranslmed.3007154
Ma Y, Chen P, Drisko JA, Khabele D, Godwin AK, Chen Q. Pharmacological ascorbate induces “BRCAness” and enhances the effects of Poly(ADP-Ribose) polymerase inhibitors against BRCA1/2 wild-type ovarian cancer. Oncol Lett. 2020;19(4):2629–38. https://doi.org/10.3892/ol.2020.11364.
Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS ONE. 2010;5(7): e11414. https://doi.org/10.1371/journal.pone.0011414.
Liu F, Zhu Y, Zhang J, Li Y, Peng Z. Intravenous high-dose vitamin C for the treatment of severe COVID-19: study protocol for a multicentre randomised controlled trial. BMJ Open. 2020;10: e039519. https://doi.org/10.1136/bmjopen-2020-039519.
Kashiouris MG, L’Heureux M, Cable CA, Fisher BJ, Leichtle SW, Fowler AA. The emerging role of vitamin C as a treatment for sepsis. Nutrients. 2020;12(2):292. https://doi.org/10.3390/nu12020292.
Marik PE, Khangoora V, Rivera R, Hooper MH, Catravas J. Hydrocortisone, vitamin C, and thiamine for the treatment of severe sepsis and septic shock: a retrospective before-after study. Chest. 2017;151(6):1229–38. https://doi.org/10.1016/j.chest.2016.11.036.
Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Drisko JA, Lee KL. Design of the trial to assess chelation therapy (TACT). Am Heart J. 2012;163(1):7–12. https://doi.org/10.1016/j.ahj.2011.10.002.
Zhang J, Rao X, Li Y, Zhu Y, Liu F, Guo G, Luo G, Meng Z, De Backer D, Xiang H, Peng Z. Pilot trial of high-dose vitamin C in critically ill COVID-19 patients. Ann Intensive Care. 2021;11(1):5. https://doi.org/10.1186/s13613-020-00792-3.
Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, Rousseau C, Robitaille L, Miller WH Jr. Phase I clinical trial of iv ascorbic acid in advanced malignancy. Ann Oncol. 2008;19(11):1969–74. https://doi.org/10.1371/journal.pone.0120228.
Hoffer LJ, Robitaille L, Zakarian R, Melnychuk D, Kavan P, Agulnik J, Cohen V, Small D, Miller WH Jr. High-dose intravenous vitamin C combined with cytotoxic chemotherapy in patients with advanced cancer: a phase I-II clinical trial. PLoS ONE. 2015;10(4): e0120228. https://doi.org/10.1371/journal.pone.0120228.
Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;72(1):139–46. https://doi.org/10.1007/s00280-013-2179-9.
Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ. Elimination of ascorbic acid after high-dose infusion in prostate cancer patients: a pharmacokinetic evaluation. Basic Clin Pharmacol Toxicol. 2015;116(4):343–8. https://doi.org/10.1111/bcpt.12323.
Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, Pillai MV, Newberg AB, Deshmukh S, Levine M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS ONE. 2012;7(1): e29794. https://doi.org/10.1371/journal.pone.0029794.
Welsh, J.L., Wagner, B.A., van’t Erve, T.J, Zehr PS, Berg DJ, Halfdanarson TR, Yee NS, Bodeker KL, Du J, Roberts II LJ, Drisko J, Levine M, Buettner GR, Cullen JJ. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol. 2013;71:765–775. https://doi.org/10.1007/s00280-013-2070-8
de Grooth HJ, Manubulu-Choo WP, Zandvliet AS, Spoelstra-de Man AME, Girbes AR, Swart EL, Oudemans-van Straaten HM. Vitamin C pharmacokinetics in critically ill patients: a randomized trial of four IV regimens. Chest. 2018;153(6):1368–77. https://doi.org/10.1016/j.chest.2018.02.025.
Washko PW, Hartzell WO, Levine M. Ascorbic acid analysis using high-performance liquid chromatography with coulometric electrochemical detection. Anal Biochem. 1989;181:276–82. https://doi.org/10.1016/0003-2697(89)90243-1.
Washko PW, Welch RW, Dhariwal KR, Wang Y, Levine M. Ascorbic acid and dehydroascorbic acid analyses in biological samples. Anal Biochem. 1992;204(1):1–14. https://doi.org/10.1016/0003-2697(92)90131-p.
Espey MG, Chen Q, Sun AY, Kim HS, Padayatty S, Wang Y, Tu H, Margolis S, Levine M. Ascorbate methodologies and considerations. In Das DK (editor) Handbook of the methods for studying redox signaling. New York: Mary Ann Liebert, pp 24–31; 2009.
Levine M, Rumsey SC, Daruwala R, Park JB, Wang Y. Criteria and recommendations for vitamin C intake. JAMA. 1999;281(15):1415–23. https://doi.org/10.1001/jama.281.15.1415.
Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. PNAS. 2001;98(17):9842–6. https://doi.org/10.1073/pnas.171318198.
Bankir L, Plischke M, Bouby N, Haas M. Urine osmolarity and risk of dialysis initiation in a CKD cohort. Ann Nutr Metab. 2015;66(3):14–7. https://doi.org/10.1159/000381240.
Feig PU, McCurdy DK. The hypertonic state. N Engl J Med. 1977;297(26):1444–54. https://doi.org/10.1056/NEJM197712292972608.
Johnson R, Rodriguez-Iturbe B, Roncal-Jimenez C, et al. Hyperosmolarity drives hypertension and CKD—water and salt revisited. Nat Rev Nephrol. 2014;10:415–20. https://doi.org/10.1038/nrneph.2014.76.
Das SK. Body composition measurement in severe obesity. Curr Opin Clin Nutr Metab Care. 2005;8(6):602–6. https://doi.org/10.1097/01.mco.0000171122.60665.5f.
Sartorio A, Malavolti M, Agosti F, Marinone PG, Caiti O, Battistini N, Bedogni G. Body water distribution in severe obesity and its assessment from eight-polar bioelectrical impedance analysis. Eur J Clin Nutr. 2005;59(2):155–60. https://doi.org/10.1038/sj.ejcn.1602049.
Waki M, Kral JG, Mazariegos M, Wang J, Pierson RN Jr, Heymsfield SB. Relative expansion of extracellular fluid in obese vs. nonobese women. Am J Physiol. 1991;261(2 Pt 1):E199–203. https://doi.org/10.1152/ajpendo.1991.261.2.E199
Ma Y, Sullivan GG, Schrick E, Choi IY, He Z, Lierman J, Lee P, Drisko JA, Chen Q. A convenient method for measuring blood ascorbate concentrations in patients receiving high-dose intravenous ascorbate. J Am Coll Nutr. 2013;32(3):187–93. https://doi.org/10.1080/07315724.2013.791167
Howell AP, Parrett JL, Malcom DR. Impact of high-dose intravenous vitamin C for treatment of sepsis on point-of-care blood glucose readings. J Diabetes Sci Technol. 2021;15(2):309–316. https://doi.org/10.1177/1932296819889638
Corpe CP, Lee JH, Kwon O, Eck P, Narayanan J, Kirk KL, Levine M. 6-Bromo-6-deoxy-L-ascorbic acid: an ascorbate analog specific for Na+-dependent vitamin C transporter but not glucose transporter pathways. J Biol Chem. 2005;280(7):5211–20. https://doi.org/10.1074/jbc.M412925200.
Acknowledgments
The authors are indebted to the excellent care provided to the clinical trial participants by Jean Sunega, BS, CCRP and Elizabeth Schrick, RN. We thank Mylan Pharmaceutical (formally Bioniche Pharma and currently Viatris) for providing the study drug.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Funding for the study was provided by private foundations through both Jefferson Medical Endowment and the University of Kansas Medical Center Endowment. Pharmacokinetics support was provided by the Clinical Pharmacology Shared Resource of the University of Kansas Cancer Center (P30 CA168524). ML was supported by the Intramural Research Program, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health. Study drug was provided at no cost to the study by Mylan Pharmaceutical (Viatris).
Conflicts of interest
Ping Chen, Greg Reed, Joyce Jiang, Yaohui Wang, Jean Sunega, Ruochen Dong, Yan Ma, Anna Esparham, Ryan Ferrell, Mark Levine, Jeanne Drisko, and Qi Chen declare that they have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval
The study was conducted in compliance with the FDA-approved study protocol (IND number 65,805). Trial oversight was provided by University of Kansas Health System, Kansas Cancer Center Data and Safety Monitoring Board with independent event adjudication. The Clinical Trials website https://clinicaltrials.gov study number was NCT01833351.
Consent to participate
The protocol and consent form were approved by the University of Kansas Health System, Institutional Review Board and the Kansas Cancer Center Protocol Review and Management Committee. All study participants were allowed to ask questions about study participation prior to signing the consent form. All signed consent forms are maintained in study files.
Consent for publication
Not applicable.
Availability of data and material
Requests for study data will be honored after written request to the corresponding author and pending approval by appropriate oversight bodies.
Code availability
Not applicable.
Author contributions
JD, QC, GR, PC, ML, and JJ participated in trial design, data analysis, and interpretation. All authors were involved in preparation, review, and approval of this article. PC, YW, RD, YM, RF, AE, and JS were involved in sample collection, preparation, and analysis. No other entity or persons, other than the authors, contributed to the writing, review, or approval of the publication. The funders of the study and the pharmaceutical company did not contribute to the design, conduct, or interpretation of data.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chen, P., Reed, G., Jiang, J. et al. Pharmacokinetic Evaluation of Intravenous Vitamin C: A Classic Pharmacokinetic Study. Clin Pharmacokinet 61, 1237–1249 (2022). https://doi.org/10.1007/s40262-022-01142-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01142-1