
Abstract
Glioblastoma (GBM) derived from the “stem cell” rich subventricular zone (SVZ) may constitute a therapy-refractory subgroup of tumors associated with poor prognosis. Risk stratification for these cases is necessary but is curtailed by error prone imaging-based evaluation. Therefore, we aimed to establish a robust DNA methylome-based classification of SVZ GBM and subsequently decipher underlying molecular characteristics. MRI assessment of SVZ association was performed in a retrospective training set of IDH-wildtype GBM patients (n = 54) uniformly treated with postoperative chemoradiotherapy. DNA isolated from FFPE samples was subject to methylome and copy number variation (CNV) analysis using Illumina Platform and cnAnalysis450k package. Deep next-generation sequencing (NGS) of a panel of 130 GBM-related genes was conducted (Agilent SureSelect/Illumina). Methylome, transcriptome, CNV, MRI, and mutational profiles of SVZ GBM were further evaluated in a confirmatory cohort of 132 patients (TCGA/TCIA). A 15 CpG SVZ methylation signature (SVZM) was discovered based on clustering and random forest analysis. One third of CpG in the SVZM were associated with MAB21L2/LRBA. There was a 14.8% (n = 8) discordance between SVZM vs. MRI classification. Re-analysis of these patients favored SVZM classification with a hazard ratio (HR) for OS of 2.48 [95% CI 1.35–4.58], p = 0.004 vs. 1.83 [1.0–3.35], p = 0.049 for MRI classification. In the validation cohort, consensus MRI based assignment was achieved in 62% of patients with an intraclass correlation (ICC) of 0.51 and non-significant HR for OS (2.03 [0.81–5.09], p = 0.133). In contrast, SVZM identified two prognostically distinct subgroups (HR 3.08 [1.24–7.66], p = 0.016). CNV alterations revealed loss of chromosome 10 in SVZM– and gains on chromosome 19 in SVZM– tumors. SVZM– tumors were also enriched for differentially mutated genes (p < 0.001). In summary, SVZM classification provides a novel means for stratifying GBM patients with poor prognosis and deciphering molecular mechanisms governing aggressive tumor phenotypes.
Similar content being viewed by others
Introduction
Glioblastoma (GBM), the most common primary brain tumor, is characterized by an infiltrative growth pattern and inherent refractoriness to therapy. This inevitably leads to local therapy failure and dismal prognosis, with an overall survival (OS) of approximately 15 months after surgery and chemoradiotherapy [16], and up to 20.5 months with the addition of tumor-treating fields (TTFields) [46]. The resistance of GBM to therapy has been attributed to a variety of factors, such as inter- as well as intra-tumoral heterogeneity, tumor origin, stem cell-like characteristics and tumor-stroma communication at the tumor vessel/immune response [1, 16, 19]. Predictive biomarkers such as promoter methylation status of the DNA repair enzyme O6-methylguanine DNA methyltransferase (MGMT) was causally linked to enhanced vulnerability of GBM cells to DNA damage inducing agents such as temozolomide chemotherapy [20, 53]. Hence, identification of molecular subtypes has advanced patient selection for ongoing prospective trials, such as the NCT Neuro Master Match (N2M2), a basket trial wherein poor prognostic MGMT hypomethylated tumors are molecularly stratified to different targeted therapies administered concurrently with radiotherapy [52].
A growing body of data indicates a prognostic impact for GBM localized to the subventricular zone (SVZ), a 3–5 mm-thick region located adjacent to the lateral ventricles that harbors neural stem cells (NSCs) throughout adult life [4, 5, 24, 25, 36]. These data examined the hypothesis that tumors originating from this stem cell-rich niche may conserve NSC characteristics. Indeed, genetic models have impressively demonstrated that the introduction of a few aberrations selectively targeted to a few hundred NSCs present in the murine SVZ region are sufficient to form tumors with human GBM characteristics [28, 31]. Moreover, ablation of this low-cycling cell population in established tumors was associated with improved prognosis and outcomes [57]. Accordingly, the association of GBM in SVZ areas was attributed to a stem cell-like phenotype with aggressive clinical behavior and poor survival [3, 4]. Classification of SVZ GBM in clinical studies largely relies on non-invasive magnetic resonance imaging (MRI). High interobserver variability and a lack of objective criteria for defining “central” tumors originating from the SVZ region versus secondary infiltration of “peripheral” tumors to this region limits implementation of a robust imaging-based clinical classifier. Accordingly, part of the poor prognosis in MRI-based SVZ GBM may be related to selection for a more invasive and migratory GBM subtype secondarily infiltrating this area [5]. As such, GBM in contact with the SVZ defined by MRI are more likely to recur as multifocal disease [5, 30]. Therefore, reliable molecular classifiers of SVZ GBM are urgently needed [44] to assist physicians in efforts to circumvent the uncertainty of current MRI-based methods.
Pioneering studies have led to introduction of DNA methylome analysis into the clinical realm, revolutionizing tumor classification in neuropathology [9, 13, 14]. A particular strength of this method relates to the long-term preservation of the epigenetic fingerprint, as opposed to more dynamic transcriptome signatures, thereby better allowing for determination of the cell of origin. This study reports on the utilization of DNA methylome analysis for discovery of a novel molecular SVZ classification principle that could potentially improve current imaging-based stratification. The SVZ methylome (SVZM) classifier was subsequently employed for molecular characterization of these tumors on multiple chromosomal aberrations (CNV), mutational signatures (exome), and transcriptome levels.
Materials and methods
Patients and tissue sampling.
This study was approved by the Institutional Review Board and Ethics Committee (No. S-056/2015) and retrospectively evaluated 54 isocitrate dehydrogenase-1 (IDH1) wild-type GBMs with available tissue samples that completed a course of radiotherapy (RT) in the Department of Radiation Oncology at University Hospital Heidelberg from 2005–2013. Of these patients, radiographic assessment using magnetic resonance imaging (MRI) was conducted for stratification into two cohorts (SVZ– and SVZ+). The latter (also labeled as “central” GBMs) was defined by the contrast-enhanced lesion having infiltrated the borders of the lateral ventricle and SVZ (5 mm margin lateral to the lateral ventricles), which could be accompanied by subependymal spreading (tumor-related contrast enhancement spreading along the ventricle walls). The remainders were categorized as SVZ–, or “peripheral” GBMs. The SVZ– patients were matched with the SVZ+ patients according to tumor localization, performance status, age, availability of sufficient pre-radiotherapeutic tumor tissue samples, and follow-up data.
Of note, because these patients were treated as early as 2005, not all patients received concurrent and adjuvant temozolomide (TMZ), and a variety of RT dose/fractionation regimens were utilized. RT in all patients was delivered as three-dimensional conformal RT. Target volume delineation was based on T1-weighted MRI, and included the primary tumor region, resection cavity (if applicable), and T2-hyperintense areas. A clinical target volume (CTV) margin of up to 2 cm was added on the gross tumor volume (GTV) and resection cavity to account for microscopic tumor spread, respecting anatomic borders. The planning target volume (PTV) was made from the CTV+ a 5 mm margin for setup inaccuracies.
Tumor samples, obtained before commencing RT, were archived in the Department of Neuropathology at the University Hospital of Heidelberg’s Institute of Pathology. As part of contemporary diagnostic assessment based on molecular factors, all tissue samples were re-evaluated based on histopathological criteria, immunohistochemical staining, and molecular analyses according to the 2016 World Health Organization classification of central nervous system tumors [33]. O6-methylguanine DNA methyltransferase (MGMT) promoter status was determined as previously described [10].
DNA extraction and molecular analysis
Tumor DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) material. Representative tumor areas with high tumor content (> 80%) were utilized. DNA extraction was performed using the automated Maxwell system (Promega, Madison, WI, USA).
DNA methylation analysis was carried out in the Genomics and Proteomics Core Facility at the German Cancer Research Center (DKFZ, Heidelberg) according to the manufacturer’s instructions as previously described [21]. Genomic positions refer to the hg19 assembly.
GBMs have previously been reported to cluster in distinct subgroups: RTKI, RTKII, H3.3 K27, and H3.3 G34 mutated, IDH mutated, and mesenchymal (Suppl. Figure 2A and B, online resource) [13]. We additionally selected representative reference cases of each subgroup (n = 10 RTK1, n = 11 RTKII, n = 15 H3.3 G34 mut., n = 19 H3.3 K27 mut., n = 11 IDH mut., n = 16 mesenchymal) and compared DNA methylation patterns between SVZ – and SVZ + GBMs via unsupervised hierarchical clustering analyses of 30,000 probes showing the highest median absolute deviation (MAD) in beta values (Suppl. Figure 2A, online resource).
CNV data were extracted from methylation arrays utilizing the minfi package. Downstream processing (identification of segments, and automatic threshold selection for the identification of gains/losses) was performed with the cnAnalysis450k package, using references for normal tissue as described earlier [27].
For methylome analysis, idat files were processed with the minfi R package [8, 18]. Data were filtered for probes mapping to chromosome X and Y, as well as single nucleotide polymorphisms (SNPs) and repetitive sequences. Data were funnorm normalized, and M-values were used for analysis. Identification of differentially methylated CpGs between SVZ+ and SVZ– tumors is outlined in Suppl. Figure 1, online resource.
Lastly, for DNA sequencing, the customized SureSelect kit (Agilent), encompassing 130 brain tumor relevant genes, was used for target-enrichment and sequenced on a NextSeq 500 machine (Illumina) as previously described [40]. Mutational data were obtainable for 45 patients, but the DNA quantity was insufficient for the remaining samples.
Validation dataset
To validate the findings herein using an independent dataset, the GBM data collection (TCGA-GBM) from The Cancer Genome Atlas (TCGA) was utilized. Retrieved data from the TCGA data portal included level 2 Illumina 450 k methylation data, RNASeq data, whole exome sequencing data (level 3), and clinical data (follow_up_v1.0). RNASeq was rlog transformed prior to analysis. Silent mutations were filtered. Enrichment tests between groups were performed using Barnard’s tests per gene and any type of mutation vs no/silent mutation. IDH1 mutant samples (WXS data) were excluded from analyses. Matching MRI data for the TCGA-450 k cohort were retrieved from The Cancer Imaging Archive (TCIA) and classified as SVZ+ or SVZ– by two independent raters. Patient characteristics of the respective cohorts are shown in Suppl.-Tbl. 1, online resource.
Survival analysis
To examine the association of SVZ apposition (and correlation with molecular/genetic factors) with clinical outcomes, evaluation of survival was conducted. As a prerequisite to this analysis, all patients were followed up clinically and with MRI 4 weeks after RT, followed by 3 month intervals until recurrence/progression or death. Tumor localization and progression were assessed by an experienced radiologist based on the RANO criteria [51]. Overall survival (OS) was calculated using the Kaplan–Meier method as the time between the date of commencing RT until the date of death, or censored at last contact.
Statistical analyses
Statistics were carried out in R v3.6.1 R Core Team, [47] and utilized a two-sided α of 0.05 unless stated otherwise. For categorical tests, the chi-squared, Fisher’s, or Barnard tests were used. Survival was additionally assessed using Cox proportional hazard models or parametric survival regressions with the survival package [48]. Random forest analyses were conducted with the randomForest package [29]. The psych and irr packages were used to calculate intraclass correlations. Pathway activity estimation from expression data was performed with PROGENy [43].
Results
SVZ classification based on MRI
A retrospective cohort of 54 GBM patients classifiable by MRI for SVZ association were denoted as the training set. Among them, tumors of 30 (55.6%) patients were classified as SVZ– and 24 (44.4%) were SVZ + GBMs as discerned by MRI features. Table 1 demonstrates clinical characteristics of both cohorts.
The vast majority of patients (≥ 90%) underwent resection prior to RT, the median dose of which was 60 Gy (range: 40.1–60). The MRI classified SVZ– group was associated with better OS than the SVZ + patients (p = 0.05, LRT, Fig. 1A). All 54 patients were IDH1/2 wt as determined by panel sequencing.

Subventricular zone positive “central” (SVZ+) and subventricular zone negative “peripheral” (SVZ–) glioblastoma differ in their epigenomic signatures. a Imaging-based (MRI) classification of GBM patients with poor prognosis SVZ+ tumors (Kaplan–Meier, Cox model likelihood ratio test, LRT). b Identification of SVZ specific DNA methylome signature (SVZM) consisting of 15 CpGs. Left: random forest derived rank order of single CpGs according to their relevance to differentiate SVZ state are shown (left red dots, right CpG annotations and importance score). c SVZM Classification separates the training cohort into two main clusters (heatmap, hcl with Euclidean distance and complete linkage). Molecular (MGMT, G-CIMP) and MRI classifications are also provided. An inferior prognosis of GBM patients with SVZM+ tumors was found by Kaplan–Meier analysis of patient survival
Development of a DNA-methylation-based SVZ classifier
The MRI classification was utilized to identify the most relevant differentially methylated probes (DMP, 225 CpGs, defined with p < 0.001) using Illumina 450 K microarrays. Hierarchical clustering (HCL) for identification of the least stable classification error between the two modalities and the most relevant DMP among these 188 CpGs were selected by random forest analysis (details in Suppl. Figure 1, online resource). This analysis resulted in a 15 CpG DNA methylation signature distinguishing between SVZ+ and SVZ– tumors (SVZM, Fig. 1B, Suppl, Fig. 11, online resource, upper row). This separation was also seen on global epigenetic level (non-selected CpGs, Suppl. Figure 11, online resource, bottom row). Among the selected CpGs, 5 of the 15 DMP were associated with the LRBA gene. Heatmap and HCL of the SVZM classified training cohort combined with other relevant parameters (i.e., MRI evaluation, IDH1, CIMP, and MGMT status) are provided in Fig. 1C. SVZM clearly separated the training cohort into prognostic subgroups (p = 0.003, LRT). No enrichment of SVZ assigned tumors to previously discovered glioma methylation subtypes was found based on hierarchical cluster analysis of most variant probes (Suppl. Figure 2, online resource), using the v11b4 neuropathology classifier [13] revealed that all subtypes were present in SVZM+ and negative tumors (Suppl. Figure 2C, online resource). A previously observed trend between MRI classification and degree of resection (p = 0.08, chi-squared test) was weaker for SVZM (p = 0.26, Suppl. Figure 3A, online resource). In concordance, Goodman Kruskal’s lambda showed smaller values for SVZM, however the 95% CI included 0 (no association) for all performed comparisons (Suppl. Figure 3A, online resource). Finally, multivariable survival analysis of the extent of resection and SVZ classification method did not show a significant contribution of adding the extent of resection in either case (SVZM: p = 0.47 and MRI: p = 0.43, Suppl. Figure 3B, online resource).
Reassessment of misclassified tumors
As compared to MRI-based classification, eight (14.8%) tumors were reclassified following molecular SVZM assessment. Interestingly, SVZM was able to separate these patients into two distinct prognostic subgroups confirmed on survival analysis (p = 0.02 by Weibull distribution, Fig. 2A, updated survival data after extended follow-up). Representative MRI images of tumors with mismatching radiographic and molecular classifications illustrate current difficulties associated with discrimination of SVZ-driven central GBM from secondary infiltration of peripheral tumors into the SVZ. Most MRI SVZ-/SVZM + tumors showed FLAIR signal reaching the SVZ (Fig. 2B, Suppl. Figure 13, online resource). FLAIR signal for classification, however, is only useful if the SVZ region is not reached (Suppl. Figure 14, online resource).

Discordance between SVZM vs. MRI-based classifications. a Overall survival of all differently assigned patients indicating an inferior outcome in patients with SVZM+ but according to MRI SVZ negative tumors. Kaplan–Meier curves and parametric survival model (Weibull distribution, dashed line, LRT). b Tumor localization of representative patients with discordant classification highlights the difficulty to distinguish between secondary invasion to the SVZ region and tumors originating from this region solely by the imaging method. c A significantly increased hazard ratio (HR: 2.48, p < 0.004) for SVZM by univariable survival analysis (Cox model) versus other parameter including classifications based on MRI SVZ+ vs SVZ–; female vs male; performance status (KPS ≥ 80 vs < 80); multi- vs unifocal presentation; chemotherapy (TMZ: adjuvant/concurrent vs. incomplete treatment); radiation dose (≥ 60 Gy vs < 60 Gy), surgery: subtotal vs total and MGMT status
Univariable analysis for OS identified SVZM (hazard ratio HR 2.48, 95% CI [1.35–4.58] p = 0.004), MRI SVZ (HR 1.83 [1.00–3.35] p = 0.049), age (HR 1.04 [1.01–1.07] p = 0.013), performance status (HR 0.24 [0.12–0.48] p < 0.001), use of TMZ (HR 0.25 [0.13–0.5] p < 0.001), Radio-Chemotherapy (HR 0.19, [0.06–0.38], p < 0.001) and MGMT promoter methylation status (HR 2.04 [1.03–4.04] p = 0.041) as significant covariates (Fig. 2C). Comparative multivariable analysis for MRI-based versus SVZM with clinical (age, performance status) and other molecular/treatment associated covariates (MGMT promoter methylation, Radiochemotherapy) showed a significant independent contribution only for the SVZM-based classification with Cox-PH models (Suppl. Figure 4, online resource). After parametric models were applied to compensate for relatively low n, MGMT and Radiochemotherapy also showed a statistically significant contribution (Suppl. Figure 12, online resource).
Validation of the SVZM and molecular characterization
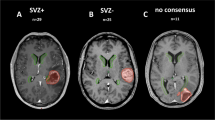
For validation of the SVZM by an independent cohort, TCGA methylome data of 132 GBM patients were utilized. An overview of the available levels of molecular-/imaging data is shown in Fig. 3A. Patients were assigned to SVZM + vs. SVZM– groups via HCL of the 15 DMP of the SVZM signature (Fig. 3B). Two main clusters could be identified, which showed significant differences in OS (p = 0.04, LRT). To further validate the proposed SVZM, available matched TCIA MRI imaging data was assessed by a single independent observer yielding an intraclass correlation (ICC) of 0.51 (Fig. 3C). Of note, for 15 out of 39 patients (38%), no consensus classification could be determined by MRI classification. Among the patients with a consensus MRI-based classification, SVZM performed better (SVZM HR 3.08 [CI 1.24–7.66], p = 0.016) compared to MRI (HR 2.03 [CI 0.81–5.09], p = 0.13) (Fig. 3C).

Performance of SVZM vs. MRI in the validation cohort. a Overview of multiple layer of data that were correlated with the SVZM state in training/validation cohorts. Methylome and copy number variation (CNV) analysis by 450 K microarrays, T1 contrast enhanced (CE) MRI, mutational profile by whole exome sequencing (WXS), deep “panel” NGS and RNAseq. b SVZ± assignment of the validation cohort by cluster analysis of the 15 CpG signature (maximum distance, ward.D) and prognostic evaluation (Kaplan–Meier, Cox model, and LRT). c Heterogeneity of SVZ classification by MRI in the validation cohort. Manual rating of patients to SVZ classes based on MRI shows discordance between the three observations for a fraction of patients (intraclass correlation, ICC). Comparative univariable survival analyses (bottom) for the 24 most consistently rated tumors by MRI vs. SVZM (Cox model)
Segmental CNV alterations revealed loss of chromosome 10 in SVZM– tumors and gains on chromosome 19 in SVZM– tumors (Fig. 4A). Segmental and gene/transcript level CNVs are shown in Suppl. Figure 6A and B, online resource. On whole exome sequencing data (WES, TCGA cohort), evaluation of variant calls from four pipelines revealed differentially enriched mutations mostly confined to SVZM– tumors (Fig. 4B), whereas the total number of non-silent mutations did not differ between the two classes (Suppl. Figure 7A, online resource). More specifically, frame shift deletions/insertions, in-frame deletions, mutations in splice regions, and splice sites were enriched in SVZM + tumors (Fig. 4D). EPHA1, DECAF12L2 and ADCY5 mutations were detected exclusively in SVZM + tumors, whereas CNTNAP2, AHNAK2 and ITIH6 mutations were among the most significantly enriched in SVZ– tumors (p < 0.01) (Fig. 4B). Deep panel sequencing of the Heidelberg cohort revealed no mutational enrichments as a function of SVZM classification; at 10% FDR for the exclusive presence of ARID1B1 and BRCA2 in SVZ+, and VHL in SVZM– tumors. Cross-comparison of mutational readouts between the Heidelberg and TCGA cohorts was limited by differences in methodology (e.g., sequencing depth and consequently VAF cut-off criteria applied to define mutations). Therefore, it was not surprising that repetitive elements difficult to detect with WES such as TERTp mutations were found in 46 (85%) samples of the Heidelberg cohort but not reported in the TCGA study (Fig. 4C). Accordingly, we failed to confirm an exclusive enrichment of mutations in SVZ+ tumors identified in the Heidelberg cohort with TCGA WES data (Suppl. Figure 7C, online resource).

Differential CNV and mutational profile of SVZ GBM. a Segmental CNV alterations indicate a relative loss of chromosome 10 in SVZM– and gains on chromosome 19 in SVZM– tumors in both training and validation cohorts. b Among differentially mutated genes identified by WXS in the validation cohort, a significantly lower number of mutations in SVZ+ compared to SVZ– tumors was found (left, p < 0.001 by Wilcoxon test). Most significantly enriched mutations as a function of SVZ state are shown as heatmap (right, Barnard’s test). Scale bar of the heatmap correspond to non-silent variants, identified as differential between SVZ± in ≥ 3 out of 4 mutation calling pipeline datasets. #calls indicate the number of pipelines identifying a mutation in the respective sample. c Differentially enriched mutations as a function of SVZ state identified by ultra-deep panel NGS of the training HD-Cohort. p value: Barnard’s test for associations between mutational enrichment in SVZM or SVZ-MRI classified groups, respectively. d Interactions between the type of mutation and SVZM status using a linear mixed model (random factor variant calling method) indicate significant association between SVZM+ frameshift (insertion/deletion), in frame deletions and splice region/sites mutations (black bars)
A consensus set of 439 CpGs was identified as being differentially methylated in both cohorts as a function of SVZM status (FDR < 0.05 by SAM with 500 permutations, Fig. 5A and Suppl. Figure 5A, online resource). DMPs were significantly hypomethylated in SVZM + vs. SVZM– tumors in both studied cohorts (p < 0.001). Global alterations on methylome and gene expression levels revealed an inverse relationship between DMPs and differentially expressed genes (DEG). The mean expression of DEGs was significantly increased in SVZ + tumors (Fig. 5B), showing a hypomethylation of 430 CpGs and higher expression of 3456 genes (total regulated genes: 3456 + 55, n = 55 being in average less expressed in SVZM + tumors, t test, FDR < 0.05 for selection of differential genes, Fig. 5C).

Molecular characterization of SVZ GBM via integrative omics. a A consensus set of 439 SVZM associated differentially methylated probes, intersect between training (HD) and validation (TCGA) cohort, was identified. A significant relative hypomethylation in SVZM+ compared to SVZM- tumors was found (right, mean methylation of all CpGs, test: linear model). b 3456 genes showed an inverse gene expression vs. CpG methylation pattern. c In line with CpG hypomethylation pattern, an enhanced mean gene-expression was found in SVZM+ tumors (selection: t-test, FDR < 0.05. test: linear model). d Rank ordered genes based on the number of differentially methylated CpG sites found in the 439-consensus signature. Of note, MAB21L2/LRBA from the SVZM random forest classifier (15 CpG RF set) are with 9 CpGs among the top ranked genes. e Intersection between different molecular layers showing an inverse relationship between methylation and expression for MAB21L2 (less expressed in SVZ+) and additional genes higher expressed. Expression and high-resolution CNV alterations show overlap for GRK5, NDST2, ZNF559-ZNF117, and ADGRE3. Methylation and CNV show hypomethylation and relative loss for ICAM5 and ONECUT3 (CNV: p < 0.05, Fisher test; methylation: FDR < 0.05, SAM; expression: FDR < 0.05, t test). f LRBA and MAB21L2 methylation (left, n = 132, linear model), expression (middle, n = 47, neg-binomial model, count data, log offset [total counts], one-sided p value, transformation for visualization) and correlation between methylation and expression (n = 47, Pearson). g PROGENy inferred enhanced pathway activity in SVZM+ tumors derived from RNAseq data, linear model analysis
With 9 CpGs, MAB21L2, LRBA, and ZNF177 were the most frequently abundant in the consensus 439 SVZ associated DMPs (Fig. 5D, Suppl. Figure 5C, online resource). The LRBA and MAB21L2 CpGs are located on chromosome 4 on overlapping positions, and ZNF177 is located on chromosome 19 (Suppl. Figure 5C+D, online resource). A paralog of ZNF177 on chromosome 19 (hypomethylated in SVZ +), ZNF559-ZNF177, showed a CN loss in SVZ+ and was less expressed, whereas ZNF177 did not show a significant difference in expression (Suppl. Figure 8, online resource). Five of the 9 LRBA/MAB21L2 annotated and differentially methylated CpGs were part of the SVZM signature (Suppl. Figure 5B, online resource). The next highest ranked (≥ 6CpGs) were ENPP4, SLC32A1 and FAM43A. NETO1, KCNH1 and BMP3 were present with > 3 CpGs. Additional overlaps between all DMPs and 15CpG RF signature were detected for CPSF1 and EHHADH (Suppl. Figure 5B, online resource).
Pair-wise co-alterations of SVZM differential genes (methylation [M], expression [E], copy number [CN]) are shown in Fig. 5E. In SVZ+ , co-regulation analysis between expression and methylation identified MAB21L2 (high E/low M), and for low E/high M MSC, RNASE11, RNASE12, PTGER2 and WNK4. Expression vs copy number analysis identified GRK5 and NDST2 with high E/CN gain, ZNF599-ZNF177 low E/CN loss, and ADGRE3 high E/CN loss. Methylation and copy number co-alteration showed low M/CN loss for ONECUT3 and ICAM5.
SVZ± population-based testing for differences in LRBA and MAB21L2 expression revealed decreased expression in SVZ+ for both genes. Moreover, a negative correlation between methylation pattern and gene-expression was found (Fig. 5F, – 0.76 for MAB21L2, -0.42 for LRBA, Pearson p < 0.01). Detailed methylation analysis of MAB21L2 and LRBA showed significant hypermethylation in 14 out of 16 MAB21L2 annotated CpGs (88%), and 19 CpGs located in the body region of LRBA (Fig. 5F, Suppl. Figure 5C, online resource). Potential sources of LRBA might be T-cells and/or microglia (Suppl. Figure 10 [50]), a direct link to tumor cells seems less likely (Suppl. Figure 9 [38, 56]).
Finally, we performed more global pathway activity estimation from expression data which revealed higher inferred activity for TNFα, NFκB, TGFβ, estrogen, p53, and hypoxia in SVZM + tumors (p < 0.05, Fig. 5G).
Discussion
This study reports on the discovery of a novel molecular classifier of SVZ-driven GBM based on DNA methylome analysis – the SVZM. Based on the growing implementation of DNA methylome analysis in neuropathology [13, 14, 24, 55], the existence of such classifiers could be of utmost relevance for designing prospective studies where SVZM complement current MRI-based classification for a more accurate and robust stratification of patients. In the training cohort, where patients with a clear consensus MRI-based SVZ association were selected to guide methylome classifier development, clustering based on the SVZM signature showed superior prognostic performance. Moreover, detailed re-analysis of MRI data based on SVZM assignment in SVZM vs. MRI discordant cases provided a plausible explanation for a possible erroneous MRI classification of peripheral tumors with secondary infiltration to the SVZ region or contact to the SVZ on T2 sequence data. This is also consistent with the improved performance of SVZM on multivariable analysis and its ability to discriminate prognostic subgroups among the discordantly classified patients. In addition to secondary infiltration as a source for the MRI classification error, heterogeneity in treatment and clinical variables as well as known prognostic subgroups such as enrichment for mutant IDH/G-CIMP in peripheral tumors might contribute to differential outcomes attributed to SVZ status [25]. Therefore, IDH-mut/G-CIMP positive patients were excluded in our study and clinically relevant parameters were well balanced in our training cohort. Moreover, for detection of the tumor cell of origin, preservation of the epigenetic fingerprint by DNA-methylome analysis may pose advantages over more dynamic molecular readouts such as transcriptome analysis. Together, MRI classification bias with potential enrichment for invasive tumors in the SVZ group (secondary infiltration of peripheral tumors), heterogeneity and unintended enrichment for prognostic subgroups as well as contamination of stroma cell signatures by analysis of tumor bulk might provide plausible explanations for previous failure to molecularly characterize SVZ GBM as a distinct biological subgroup [37]. Consequently, recent attempts to reduce the influence of the aforementioned variables (e.g., by excluding IDHmut tumors) reported successful characterization of SVZ GBM as a distinct gene-expression subtype with enrichment of cancer stem cell-like markers (e.g., CD133) and increased expression of genes associated with Notch and DNA-repair pathways [24, 45].
Male abnormal 21 (MAB21) homolog protein MAB21L2 (Mab-21 Like 2) and LRBA (lipopolysaccharide-LPS–responsive vesicle trafficking, beach- and anchor-containing) build a nested gene pair (embedding of one gene in another), which is a unique evolutionarily conserved feature reaching back to C. elegans [49]. MAB21 like protein family members are linked to cell fate determination, neuronal development, and increasingly functionally connected to the immune response. In addition to involvement in key immune pathways such as TGF signaling described for MAB21L2, their nucleotidyltransferase activity has been recently studied and compared with another prominent MAB21 family member, cGAS (cyclic GMP-AMP synthase, also known as MAB21 domain-containing protein 1–MB21D1), which is a pivotal cytosolic DNA sensor and activator of the innate immune system [15]. Intriguingly, 14/16 SVZM+ DMPs and 1/3 of the classifier was dominated by probes related to MAB21L1/LRBA, all demonstrating a hypermethylation pattern further correlated with decreased expression in SVZM+ tumors. Mab21L2 expression is considered for classification of medulloblastoma subtypes (is among the Nanostring signature genes [39]), differentially expressed in brain vs. bone metastases of breast cancer [26], and low expression of lnc-MAB21L2-1 correlated with resistant to neoadjuvant chemoradiotherapy in rectal cancer [17]. Silencing of MAB21L2, as a TGFβ transcriptional repressor, was shown to induce an immune-suppressive microenvironment in myelodysplastic syndrome (MDS) [41]. Accordingly, we found increased TGFβ pathway activity in SVZM+ tumors. MAB21L2 hypermethylation was associated with chemotherapy resistance in gastric cancer [34], could discriminate between different thyroid tumors [35], was among the top hypermethylated DMP in pathology-free regions of multiple sclerosis-affected brains [22], associated with neurogenesis [55], as well as linked to neural differentiation and human hippocampal neurogenesis in Alzheimer’s disease [6]. Additionally, LRBA was shown to be involved in trafficking key immune checkpoints (e.g., CTLA-4), is known to contribute to immune dysregulation [32], and is correlated with both disease mortality and recurrence in breast cancer [7]. The source cell of LRBA, however, remains elusive [50, 56]. Together with the epigenetic and transcriptional silencing of MAB21L2/LRBA in SVZM+ tumors found in our study, these data provide a plausible explanation for a prognostic and potentially functional relevance of these genes at the tumor immune microenvironment interface contributing to the observed inferior clinical outcome in SVZM+ GBM. This hypothesis is further supported by the presence of probes associated with IL-6 immune signaling and collagen-18, a precursor to the endogenous angiogenesis inhibitor endostatin [2], in the SVZM classifier and warrants further investigation.
Multiscale molecular characterization of SVZM+ tumors further revealed a global hypomethylation (98% of SVZ associated DMP) and increased gene expression in SVZM+ vs. SVZM– tumors as well as segmental CNV alterations and a significant enrichment for differentially mutated genes in SVZ– tumors. Differential regulation patterns as a function of SVZM status was found on two or more levels, such as in the ZNF599-ZNF177 locus (CNV, methylation and expression). Interestingly, ARID1B1 and BRCA2 mutations were found to be SVZM+ tumor exclusive by deep NGS of the Heidelberg cohort as well as EPHA1, DECAF12L2, and ADCY5 by WES in the TCGA cohort. Neither ARID1B1 nor BRCA2 were detected in the unselected original Heidelberg GBM cohort evaluated for this panel [40], suggesting that MR-based enrichment of the cohort for SVZ GBM was relevant for this discovery. Moreover, our gene panel constituted relevant mutations in adult as well as pediatric neuro-oncology. Therefore, the presence of mutations such as ARID1B1 in 3/21 (~ 14%) of SVZM+ tumors was a relatively surprising finding. ARID1B mutations are known to appear in ~ 10% of neuroblastoma patients and are correlated with poor clinical outcome. ARID1B mutations and alterations are also believed to serve as a driver of tumorigenesis in a small fraction of medulloblastoma and other solid tumors such as breast and ovarian cancers [42]. Exclusive association of BRCA2 mutations in ~ 14% of SVZM+ provide another interesting target for therapeutic targeting such as synthetic lethal interactions with DNA-damage repair inhibitors and radiotherapy as the cornerstone of postoperative therapy. Among the SVZM+ exclusive mutations identified in the TCGA cohort, the ephrin family member EPHA1 mutation may provide an interesting therapeutic target as it was recently attributed to an improved response to anti-PD-L1 immune checkpoint inhibition in lung cancer [11]. Furthermore, DECAF12L2 could be an attractive biomarker for further investigation as it is among the top 15 driver mutations of GBM (Suppl. Figure 7C, online resource).
Unfortunately, we were not able to validate our deep panel NGS finding with TCGA WES data. This might be due to the limited depth of WES and prefiltering criteria leading to selection of genes with VAF > 50% and other technical issues. For example, TERT promoter (pTERT) mutations were rarely found in the TCGA analysis; from 291 GBM patients with WES data 42 had whole genome NGS but only 25 samples had adequate coverage (read count > 10) of the TERT promoter for mutational analysis [12]. In contrast, by inclusion of intronic/non-coding regions to cover the TERT promoter with an average coverage of 550-fold, we found pTERT mutations in 85% (n = 46) of the Heidelberg GBM cohort. The depth of reads might also have been advantageous for detection of genes with large exons like BRCA1 and its association with SVZM+ in our training cohort. These limitations notwithstanding, these findings warrant further validation in well-powered prospective cohorts and may have ramifications for improved diagnostic and therapeutic tailoring of SVZ+ GBM.
References
Abdollahi A, Folkman J (2010) Evading tumor evasion: current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist Updat 13:16–28. https://doi.org/10.1016/j.drup.2009.12.001
Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W et al (2004) Endostatin’s antiangiogenic signaling network. Mol Cell 13:649–663. https://doi.org/10.1016/s1097-2765(04)00102-9
Adeberg S, Bostel T, Konig L, Welzel T, Debus J, Combs SE (2014) A comparison of long-term survivors and short-term survivors with glioblastoma, subventricular zone involvement: a predictive factor for survival? Radiat Oncol 9:95. https://doi.org/10.1186/1748-717X-9-95
Adeberg S, Harrabi SB, Bougatf N, Bernhardt D, Mohr A, Rieber J et al (2016) Do increased doses to stem-cell niches during radiation therapy improve glioblastoma survival? Stem cells international 2016:8793462. https://doi.org/10.1155/2016/8793462
Adeberg S, Konig L, Bostel T, Harrabi S, Welzel T, Debus J et al (2014) Glioblastoma recurrence patterns after radiation therapy with regard to the subventricular zone. Int J Radiat Oncol Biol Phys. https://doi.org/10.1016/j.ijrobp.2014.07.027
Altuna M, Urdanoz-Casado A, Sanchez-Ruiz de Gordoa J, Zelaya MV, Labarga A, Lepesant JMJ et al (2019) DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin Epigenetics 11:91. https://doi.org/10.1186/s13148-019-0672-7
Andres SA, Brock GN, Wittliff JL (2013) Interrogating differences in expression of targeted gene sets to predict breast cancer outcome. BMC Cancer 13:326. https://doi.org/10.1186/1471-2407-13-326
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD et al (2014) Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30:1363–1369. https://doi.org/10.1093/bioinformatics/btu049
Bady P, Kurscheid S, Delorenzi M, Gorlia T, van den Bent MJ, Hoang-Xuan K et al (2018) The DNA methylome of DDR genes and benefit from RT or TMZ in IDH mutant low-grade glioma treated in EORTC 22033. Acta Neuropathol 135:601–615. https://doi.org/10.1007/s00401-018-1810-6
Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C et al (2012) MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 124:547–560. https://doi.org/10.1007/s00401-012-1016-2
Bai H, Duan J, Li C, Xie W, Fang W, Xu Y et al (2020) EPHA mutation as a predictor of immunotherapeutic efficacy in lung adenocarcinoma. J Immunother Cancer. https://doi.org/10.1136/jitc-2020-001315
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. https://doi.org/10.1016/j.cell.2013.09.034
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M et al (2018) Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 136:181–210. https://doi.org/10.1007/s00401-018-1879-y
de Oliveira Mann CC, Kiefersauer R, Witte G, Hopfner KP (2016) Structural and biochemical characterization of the cell fate determining nucleotidyltransferase fold protein MAB21L1. Sci Rep 6:27498. https://doi.org/10.1038/srep27498
Debus J, Abdollahi A (2014) For the next trick: new discoveries in radiobiology applied to glioblastoma. Am Soc Clin Oncol Educ Book. https://doi.org/10.14694/EdBook_AM.2014.34.e95
Ferrando L, Cirmena G, Garuti A, Scabini S, Grillo F, Mastracci L et al (2020) Development of a long non-coding RNA signature for prediction of response to neoadjuvant chemoradiotherapy in locally advanced rectal adenocarcinoma. PLoS ONE 15:e0226595. https://doi.org/10.1371/journal.pone.0226595
Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ et al (2014) Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 15:503. https://doi.org/10.1186/s13059-014-0503-2
Geisenberger C, Mock A, Warta R, Rapp C, Schwager C, Korshunov A et al (2015) Molecular profiling of long-term survivors identifies a subgroup of glioblastoma characterized by chromosome 19/20 co-gain. Acta Neuropathol 130:419–434. https://doi.org/10.1007/s00401-015-1427-y
Hegi ME, Genbrugge E, Gorlia T, Stupp R, Gilbert MR, Chinot OL et al (2019) MGMT promoter methylation cutoff with safety margin for selecting glioblastoma patients into trials omitting temozolomide: a pooled analysis of four clinical trials. Clin Cancer Res 25:1809–1816. https://doi.org/10.1158/1078-0432.CCR-18-3181
Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R et al (2013) Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125:913–916. https://doi.org/10.1007/s00401-013-1126-5
Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD et al (2014) Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat Neurosci 17:121–130. https://doi.org/10.1038/nn.3588
Janin M, Ortiz-Barahona V, de Moura MC, Martinez-Cardus A, Llinas-Arias P, Soler M et al (2019) Epigenetic loss of RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a stress adaptive translational program. Acta Neuropathol 138:1053–1074. https://doi.org/10.1007/s00401-019-02062-4
Jungk C, Mock A, Exner J, Geisenberger C, Warta R, Capper D et al (2016) Spatial transcriptome analysis reveals Notch pathway-associated prognostic markers in IDH1 wild-type glioblastoma involving the subventricular zone. BMC Med 14:170. https://doi.org/10.1186/s12916-016-0710-7
Jungk C, Warta R, Mock A, Friauf S, Hug B, Capper D et al (2019) Location-dependent patient outcome and recurrence patterns in IDH1-Wildtype glioblastoma. Cancers (Basel). https://doi.org/10.3390/cancers11010122
Klein A, Olendrowitz C, Schmutzler R, Hampl J, Schlag PM, Maass N et al (2009) Identification of brain- and bone-specific breast cancer metastasis genes. Cancer Lett 276:212–220. https://doi.org/10.1016/j.canlet.2008.11.017
Knoll M, Debus J, Abdollahi A (2017) cnAnalysis450k: an R package for comparative analysis of 450k/EPIC Illumina methylation array derived copy number data. Bioinformatics 33:2266–2272. https://doi.org/10.1093/bioinformatics/btx156
Lee JH, Lee JE, Kahng JY, Kim SH, Park JS, Yoon SJ et al (2018) Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 560:243–247. https://doi.org/10.1038/s41586-018-0389-3
Liaw A, Wiener M (2002) Classification and regression by random forest. R News 2:18–22
Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S et al (2007) Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol 9:424–429. https://doi.org/10.1215/15228517-2007-023
Liu HK, Wang Y, Belz T, Bock D, Takacs A, Radlwimmer B et al (2010) The nuclear receptor tailless induces long-term neural stem cell expansion and brain tumor initiation. Genes Dev 24:683–695. https://doi.org/10.1101/gad.560310
Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C et al (2015) Autoimmune disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 349:436–440. https://doi.org/10.1126/science.aaa1663
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. https://doi.org/10.1007/s00401-016-1545-1
Maeda O, Matsuoka A, Furukawa K, Miyahara R, Hirooka Y, Ando Y (2019) Alterations in gene expression and DNA methylation profiles in gastric cancer cells obtained from ascitic fluids collected before and after chemotherapy. Mol Clin Oncol 11:91–98. https://doi.org/10.3892/mco.2019.1858
Mancikova V, Buj R, Castelblanco E, Inglada-Perez L, Diez A, de Cubas AA et al (2014) DNA methylation profiling of well-differentiated thyroid cancer uncovers markers of recurrence free survival. Int J Cancer 135:598–610. https://doi.org/10.1002/ijc.28703
Mistry AM (2019) Clinical correlates of subventricular zone-contacting glioblastomas: a meta-analysis. J Neurosurg Sci 63:581–587. https://doi.org/10.23736/S0390-5616.17.04274-6
Mistry AM, Wooten DJ, Davis LT, Mobley BC, Quaranta V, Ihrie RA (2019) Ventricular-subventricular zone contact by glioblastoma is not associated with molecular signatures in bulk tumor data. Sci Rep. https://doi.org/10.1038/s41598-018-37734-w
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ et al (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178(835–849):e821. https://doi.org/10.1016/j.cell.2019.06.024
Northcott PA, Shih DJ, Remke M, Cho YJ, Kool M, Hawkins C et al (2012) Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol 123:615–626. https://doi.org/10.1007/s00401-011-0899-7
Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D et al (2016) Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 131:903–910. https://doi.org/10.1007/s00401-015-1519-8
Sarhan D, Wang J, Sunil Arvindam U, Hallstrom C, Verneris MR, Grzywacz B et al (2020) Mesenchymal stromal cells shape the MDS microenvironment by inducing suppressive monocytes that dampen NK cell function. JCI Insight. https://doi.org/10.1172/jci.insight.130155
Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X et al (2013) Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45:12–17. https://doi.org/10.1038/ng.2493
Schubert M, Klinger B, Klunemann M, Sieber A, Uhlitz F, Sauer S et al (2018) Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat Commun 9:20. https://doi.org/10.1038/s41467-017-02391-6
Smith AW, Parashar B, Wernicke AG (2016) Subventricular zone-associated glioblastoma: a call for translational research to guide clinical decision making. Neurogenesis (Austin) 3:e1225548. https://doi.org/10.1080/23262133.2016.1225548
Steed TC, Treiber JM, Taha B, Engin HB, Carter H, Patel KS et al (2020) Glioblastomas located in proximity to the subventricular zone (SVZ) exhibited enrichment of gene expression profiles associated with the cancer stem cell state. J Neurooncol 148:455–462. https://doi.org/10.1007/s11060-020-03550-4
Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA et al (2015) Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA 314:2535–2543. https://doi.org/10.1001/jama.2015.16669
Team RC (2018) R: A language and environment for statistical computing. https://www.R-project.org/
Therneau TM (2015) A Package for Survival Analysis in S.
Tsang WH, Shek KF, Lee TY, Chow KL (2009) An evolutionarily conserved nested gene pair - Mab21 and Lrba/Nbea in metazoan. Genomics 94:177–187. https://doi.org/10.1016/j.ygeno.2009.05.009
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S et al (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22:1021–1035. https://doi.org/10.1038/s41593-019-0393-4
Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E et al (2010) Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol 28:1963–1972. https://doi.org/10.1200/JCO.2009.26.3541
Wick W, Dettmer S, Berberich A, Kessler T, Karapanagiotou-Schenkel I, Wick A et al (2019) N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro Oncol 21:95–105. https://doi.org/10.1093/neuonc/noy161
Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A et al (2014) MGMT testing–the challenges for biomarker-based glioma treatment. Nat Rev Neurol 10:372–385. https://doi.org/10.1038/nrneurol.2014.100
Wiestler B, Capper D, Sill M, Jones DT, Hovestadt V, Sturm D et al (2014) Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol 128:561–571. https://doi.org/10.1007/s00401-014-1315-x
Wong YM, Chow KL (2002) Expression of zebrafish mab21 genes marks the differentiating eye, midbrain and neural tube. Mech Dev 113:149–152. https://doi.org/10.1016/s0925-4773(02)00012-6
Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D et al (2019) Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci 22:1696–1708. https://doi.org/10.1038/s41593-019-0491-3
Zhu Z, Khan MA, Weiler M, Blaes J, Jestaedt L, Geibert M et al (2014) Targeting self-renewal in high-grade brain tumors leads to loss of brain tumor stem cells and prolonged survival. Cell Stem Cell 15:185–198. https://doi.org/10.1016/j.stem.2014.04.007
Funding
Open Access funding enabled and organized by Projekt DEAL. National Center for Tumor diseases, NCT-PRO-2015-21, Amir Abdollahi, Deutsche Forschungsgemeinschaft, UNITE SFB13-89, Deutsches Krebsforschungszentrum.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adeberg, S., Knoll, M., Koelsche, C. et al. DNA-methylome-assisted classification of patients with poor prognostic subventricular zone associated IDH-wildtype glioblastoma. Acta Neuropathol 144, 129–142 (2022). https://doi.org/10.1007/s00401-022-02443-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-022-02443-2