
Abstract
The fms-like tyrosine kinase 3 (FLT3) inhibitor gilteritinib is indicated for relapsed or refractory (R/R) FLT3-mutated acute myeloid leukemia (AML), based on its observed superior response and survival outcomes compared with salvage chemotherapy (SC). Frontline use of FLT3 tyrosine kinase inhibitors (TKIs) midostaurin and sorafenib may contribute to cross-resistance to single-agent gilteritinib in the R/R AML setting but has not been well characterized. To clarify the potential clinical impact of prior TKI use, we retrospectively compared clinical outcomes in patients with R/R FLT3-mutated AML in the CHRYSALIS and ADMIRAL trials who received prior midostaurin or sorafenib against those without prior FLT3 TKI exposure. Similarly high rates of composite complete remission (CRc) were observed in patients who received a FLT3 TKI before gilteritinib (CHRYSALIS, 42%; ADMIRAL, 52%) and those without prior FLT3 TKI therapy (CHRYSALIS, 43%; ADMIRAL, 55%). Among patients who received a prior FLT3 TKI in ADMIRAL, a higher CRc rate (52%) and trend toward longer median overall survival was observed in the gilteritinib arm versus the SC arm (CRc = 20%; overall survival, 5.1 months; HR = 0.602; 95% CI: 0.299, 1.210). Remission duration was shorter with prior FLT3 TKI exposure. These findings support gilteritinib for FLT3-mutated R/R AML after prior sorafenib or midostaurin.
Similar content being viewed by others

Introduction
Tyrosine kinase inhibitors (TKIs) that target the fms-like tyrosine kinase 3 (FLT3) receptor have demonstrated activity in patients with acute myeloid leukemia (AML) harboring activating mutations in FLT3 [1], namely, FLT3 internal tandem duplications (FLT3-ITD) in the juxtamembrane domain and FLT3 tyrosine kinase domain (FLT3-TKD) point mutations in the activation loop [2]. The effect of activating FLT3 mutations on clinical outcomes varies based upon the presence of certain recurrent chromosomal translocations, co-mutations such as NPM1, FLT3 mutation type, and FLT3-ITD to wild-type FLT3 allelic ratio [3, 4]. Patients with FLT3-ITD mutations—particularly those with a high FLT3-ITD mutant to wild-type allelic ratio at initial diagnosis—have short remission duration and poor survival [4]. In contrast, FLT3-TKD mutations may share aggressive clinical features with FLT3-ITD mutations, but have inconsistent prognostic effects [5]. Although prognostically less important, secondary FLT3-TKD mutations can confer resistance to certain FLT3 TKIs [6]. Notably, TKIs vary in their selectivity for the FLT3 receptor [7] and, to a certain extent, FLT3-TKD point mutations vary in their sensitivity to TKIs [6, 8]. Additionally, therapy with FLT3 inhibitors in patients with relapsed or refractory (R/R) AML who harbor FLT3-ITD mutations has been shown to promote drug-resistant clonal populations that contain secondary, on-target mutations in FLT3 that confer resistance to multiple TKIs [9, 10]. Because R/R AML is demonstrably polyclonal in nature, FLT3 TKIs may elicit clonal pressure to select for drug-resistant tumor cell populations with additional mutations that promote leukemic growth independent of the activation state of the FLT3 kinase [11,12,13], as well as for clones that lack FLT3 mutations entirely [11, 12].
First-generation FLT3 TKIs, midostaurin and sorafenib, display relatively limited selectivity for FLT3 and have relatively low potency in human plasma, which is thought to underlie their modest antileukemic efficacy when used as single agents in patients with R/R FLT3-mutated AML [14, 15]. However, the likelihood of response is improved by midostaurin and relapse rates decrease when either agent is combined with frontline 7 + 3 induction therapy in patients with newly diagnosed FLT3-mutated AML [16, 17]. Midostaurin in combination with chemotherapy was approved for this indication based on findings from the phase 3 RATIFY trial based upon an improvement in overall survival (OS) compared with placebo [17, 18]. Sorafenib is not approved for AML but is commonly used as maintenance therapy following allogeneic transplant, as supported by two randomized studies that show reductions in relapse rates and favorable effects on posttransplant relapse-free survival [19, 20]. Despite the observed benefit with first-generation FLT3 TKIs in the frontline setting, relapse is still common, especially in patients who are unable to undergo allogeneic transplantation [21].
The second-generation FLT3 TKIs, gilteritinib and quizartinib, have greater selectivity for the FLT3 receptor and, when administered continuously, appear to show fewer toxicities related to off-target effects compared with first-generation FLT3 TKIs [22]. Both agents have clinical activity that paired with demonstrated survival benefits compared to standard salvage chemotherapy (SC) when administered as single agents in patients with FLT3-mutated R/R AML [23,24,25]. However, while quizartinib has demonstrated activity against FLT3-ITD mutations, it is largely ineffective against FLT3-TKD mutations, which can emerge over time as a resistance mechanism [6, 10]. In contrast, gilteritinib has demonstrated activity against both FLT3-ITD and FLT3-TKD mutations [26, 27]. Despite this, secondary resistance to gilteritinib can develop from either on-target FLT3 mutations in a gatekeeper FLT3 residue (F691L), as well as off-target mechanisms, such as emergence of NRAS or related mutations that activate mitogen-activated protein kinase (MAPK) signaling downstream of FLT3 [12, 26]. Recently published evidence has demonstrated these and other mechanisms of resistance in a subset of patients with newly diagnosed FLT3-mutated AML who received midostaurin in combination with induction chemotherapy [11].
Gilteritinib was approved as single-agent therapy for patients with FLT3-mutated R/R AML based on findings from the phase 3 ADMIRAL trial (NCT02421939), which evaluated the safety and efficacy of a daily dose of 120-mg gilteritinib against SC in this patient population [24, 28]. The 120-mg/day dose (with the possibility of escalation up to 200 mg/day in case of no response) was identified as the recommended dose for the ADMIRAL study based on findings from the phase 1/2 dose-escalation/expansion CHRYSALIS trial (NCT02014558) of 20–450-mg gilteritinib in a FLT3-mutation–enriched R/R AML patient population [25]. In the ADMIRAL trial, patients assigned to 120-mg gilteritinib had significantly longer median OS than those assigned to SC (9.3 months vs 5.6 months, respectively; hazard ratio (HR) for death, 0.64; 95% confidence interval [CI]: 0.49, 0.83; P < 0.001); higher rates of complete remission (CR) with full or partial hematologic recovery were also observed in the gilteritinib arm (34.0% vs 15.3%, respectively) [24].
Because clonal evolution in AML has recently been shown to contribute to the development of resistance to initial FLT3 TKI therapy [11, 12] and there are limited data to guide clinical use of gilteritinib in an era where patients are commonly treated with frontline TKIs, we performed a retrospective analysis of the CHRYSALIS and ADMIRAL trials to evaluate response and survival in patients with FLT3-mutated, R/R AML who received or did not receive prior TKI therapy with midostaurin or sorafenib before treatment with gilteritinib.
Material and methods
CHRYSALIS and ADMIRAL study designs
CHRYSALIS was a multicenter phase 1/2 dose-escalation/expansion trial (start date: October 9, 2013; primary completion date: August 4, 2017) in which patients were enrolled across seven dose-escalation cohorts to receive 20-, 40-, 80-, 120-, 200-, 300-, and 450-mg doses of once-daily oral gilteritinib in 28-day cycles [25]. On the basis of emerging toxicity, pharmacokinetic and pharmacodynamic profile, and antileukemic response, the 120- and 200-mg dose-escalation cohorts were further expanded to include FLT3-mutated patients only [25]. A full description of the CHRYSALIS study design has been previously published by Perl and colleagues [25].
ADMIRAL was a global phase 3 trial (start date: October 20, 2015; primary completion date: September 17, 2021) of gilteritinib versus SC in patients with R/R FLT3-mutated AML [24]. Patients were randomized 2:1 to receive 120-mg gilteritinib or preselected high- or low-intensity SC [24]. Patients in the SC arm assigned to high-intensity SC received one to two cycles of treatment [24]. Treatment with gilteritinib or low-intensity chemotherapy was administered in 28-day cycles until disease progression or another discontinuation criterion was met [24]. Dose escalation to 200 mg/day was permitted for patients in the gilteritinib arm who did not have protocol-defined remission after the first treatment cycle. Complete details of the ADMIRAL study design and treatment are outlined in the primary publication [24]. The study protocols for the CHRYSALIS and ADMIRAL trials were approved by site-specific independent ethics committees or institutional review boards. All patients in the CHRYSALIS and ADMIRAL trials provided written informed consent at the time of enrollment.
CHRYSALIS and ADMIRAL patient populations
Adult patients with R/R FLT3-mutated AML who received 120-mg or 200-mg gilteritinib in the phase 1/2 CHRYSALIS trial and those who received 120-mg gilteritinib or SC in the ADMIRAL trial were included in this analysis. The subgroup of R/R AML patients who received 120- or 200-mg gilteritinib in the CHRYSALIS trial had locally confirmed FLT3 mutations and had received one or more lines of prior AML therapy [25]. Patients in the ADMIRAL trial had either relapsed after initial induction therapy or were refractory to initial induction therapy and were required to have central laboratory confirmed FLT3-ITD mutations or FLT3-TKD D835/I836 point mutations at study entry[24]; enrollment based on local FLT3 mutation testing was permitted in cases of rapidly proliferative disease. Complete inclusion and exclusion criteria for patients in both trials are outlined in the respective primary publications [24, 25].
Assessments
Treatment response was assessed using modified International Working Group criteria [29]. Complete definitions of treatment response parameters are presented in the Supplement (Table S1). The composite CR (CRc) rate was defined as the sum of patients who achieved CR, CR with incomplete hematologic recovery (CRi), and CR with incomplete platelet recovery (CRp). In the CHRYSALIS trial, FLT3 mutation status was determined based on local testing. In the ADMIRAL trial, FLT3 mutation status was assessed at enrollment (baseline) by a central laboratory using a polymerase chain reaction–based assay (LeukoStrat CDx) according to published methods; FLT3 mutation status based on local testing was permitted in cases of rapidly proliferative disease [24, 30].
Statistical analyses
Descriptive statistics were used to assess continuous variables. Categorical data were reported as frequencies and percentages. The Kaplan-Meier method and the Greenwood formula were used to estimate OS. Hazard ratio and supporting CIs were used to determine differences in OS between groups. As the statistical analysis plan did not include provisions for multiplicity correction with respect to evaluation of secondary outcomes or subgroup analyses, these results were reported as point estimates with 95% CIs. Statistical analyses were performed with SAS v9.3 or higher software.
Results
Baseline characteristics
Overall, 33 of 145 (22.8%) patients received 120- or 200-mg gilteritinib in the CHRYSALIS trial and 33 of 247 (13.4%) patients who received 120-mg gilteritinib in the ADMIRAL trial had received prior TKI therapy (Table 1). In the SC arm of the ADMIRAL trial, 15 patients had received prior TKI therapy. All patients who received prior TKIs in the 120- or 200-mg gilteritinib dose groups of the CHRYSALIS trial had received sorafenib. In the gilteritinib arm of the ADMIRAL trial, 58% (n = 19/33) of prior TKI-treated patients had received sorafenib and 42% (n = 14/33) had received midostaurin. Among prior TKI-treated patients in the SC arm of the ADMIRAL trial, 60% (n = 9/15) had received midostaurin and 40% (n = 6/15) had received sorafenib. For prior TKI-treated patients in the CHRYSALIS trial, the median time since last TKI therapy was 33 days (interquartile range [IQR], 16–149) for all patients who received 120-mg or 200-mg gilteritinib. For prior TKI-treated patients in the gilteritinib arm of the ADMIRAL trial, the median time since last TKI therapy was 34 days (IQR, 11–92). Demographic and baseline disease characteristics in prior TKI and no prior TKI subgroups within the CHRYSALIS and ADMIRAL trials were similar (Table 1). Among patients who received 120- or 200-mg gilteritinib in the CHRYSALIS trial, most (69%; n = 100/145) had received two or more lines of prior AML therapy. Baseline co-mutations in NPM1 occurred slightly more frequently among patients in the SC arm of the ADMIRAL trial who had received prior TKI therapy (64%) compared with corresponding patients in the gilteritinib arm (47%) and patients who had not received prior TKI therapy (gilteritinib, 48%; SC, 45%). Baseline co-mutations in RAS/MAPK pathway genes (ie, BRAF, CBL, KRAS, NRAS, or PTPN11) were observed in seven patients (15%) who had received prior TKI therapy (gilteritinib, n = 5; SC, n = 2) and in 18 patients (6%) without prior TKI exposure (gilteritinib, n = 13; SC, n = 5).
Survival outcomes
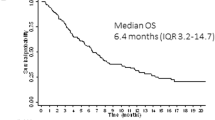
Median OS was similar in prior TKI-treated and no prior TKI subgroups (7.2 months and 7.5 months, respectively) following treatment with 120- or 200-mg gilteritinib in the CHRYSALIS trial (Fig. 1). Median OS by FLT3 mutation type is shown in Table S2.

CI confidence interval, OS overall survival, R/R relapsed or refractory, TKI tyrosine kinase inhibitor.
Among patients in the gilteritinib arm of the ADMIRAL trial, median OS duration was 9.5 months for those who did not receive prior TKIs compared with 8.7 months for those who received prior TKI therapy (Fig. 2). Among patients who received prior TKI therapy, a trend toward longer median overall survival (OS; 8.7 months) was observed in the gilteritinib arm than corresponding patients in the SC arm (median OS, 5.1 months; HR = 0.602; 95% CI: 0.299, 1.210). Among patients who did not receive prior TKI therapy, those in the gilteritinib arm had longer median OS (9.5 months) compared with those in the SC arm (6.1 months) (HR = 0.637; 95% CI: 0.482, 0.841). Median OS by FLT3 mutation type in patients treated with gilteritinib in the ADMIRAL trial (Table S2) did not show any trend. Among patients who had primary refractory AML, median OS was similar among those who received prior TKI therapy (10.6 months) and those who did not (10.2 months) (Table S3). Among patients with relapsed AML, median OS was 6.5 months in those treated with prior TKIs and 8.9 months in those who did not receive prior TKI therapy.

AML acute myeloid leukemia, CI confidence interval, OS overall survival, R/R relapsed or refractory, TKI tyrosine kinase inhibitor.
In the combined analysis of patients who received 120-mg gilteritinib in both trials, patients who did not receive prior TKI therapy had longer median OS duration than those who received prior TKI therapy (Fig. 3). Median EFS was similar for prior TKI and no prior TKI patients in the CHRYSALIS trial (3.6 months and 4.1 months, respectively). Likewise, in patients treated with 120-mg gilteritinib in the ADMIRAL trial, median EFS was the same (2.8 months) for both patients who received and did not receive prior TKI therapy.

AML acute myeloid leukemia, CI confidence interval, OS overall survival, R/R relapsed or refractory, TKI tyrosine kinase inhibitor.
Response outcomes
Overall, more than 40% of patients who received 120- or 200-mg gilteritinib in the CHRYSALIS and ADMIRAL trial after treatment with prior TKIs achieved CRc. Rates of CRc among gilteritinib-treated patients were not markedly different between prior TKI and no prior TKI subgroups (Table 2). In the CHRYSALIS trial, the overall CRc rate (includes pre- and posttransplant CRc) was 42% after prior treatment with sorafenib, with all patients achieving CRc prior to transplantation. Median durations of CR and CRc were shorter in the prior TKI group (9.1 and 1.9 months, respectively) than in the no prior TKI group (15.8 and 6.3 months, respectively). The median duration of CR in patients previously treated with sorafenib was 9.1 months. The cumulative incidence of relapse at 12 months after achievement of CR in patients previously treated with sorafenib was 100%; the cumulative incidence of relapse at 12 months after achievement of CRc in these patients was 79.2%. In patients treated with prior sorafenib, most relapses occurred within the first 4 months after CRc; in patients not previously treated with sorafenib, most relapses occurred within the first 10 months after CRc (data not shown).
In the gilteritinib arm of the ADMIRAL trial, the CRc rate was 57% in patients treated with prior midostaurin, with 21% achieving CR, 29% achieving CRi, and 7% achieving CRp. In ADMIRAL patients who received gilteritinib after prior sorafenib, the overall CRc rate was 47% with equal proportions of patients achieving CR, CRi, and CRp (all 16%) (Table 2). Notably, overall CRc rates in the gilteritinib arm after prior midostaurin (57%) or sorafenib (47%) were higher than CRc rates in corresponding prior TKI-treated subgroups in the SC arm (33% and 0%, respectively). The pretransplant CRc rates in the gilteritinib arm after prior midostaurin or prior sorafenib were 50% and 42%, respectively. Rates of CRc in prior TKI-treated patients were 47% for patients in relapse at baseline and 57% in patients who were refractory at baseline (Table S3); similarly high rates of CRc were observed among patients without prior TKI exposure within relapsed (60%) and refractory (46%) subgroups. As observed in the CHRYSALIS trial, median durations of CR and CRc in the gilteritinib arm of the ADMIRAL trial were shorter in patients who had received prior TKIs than in patients who had not. Patients in the gilteritinib arm who were previously treated with sorafenib had a median duration of CR of 12.9 months and those who were previously treated with midostaurin had a median duration of CR of 3.7 months. In the gilteritinib arm, all relapses occurred within the first 4 months after CRc in patients treated with prior midostaurin; in patients treated with prior sorafenib, most relapses occurred within the first 5 months. Among patients in the gilteritinib arm who did not receive prior TKI therapy, most relapses occurred within the first 12 months after achievement of CRc (data not shown). The cumulative incidence of relapse after achieving CRc in patients who received or did not receive prior TKI therapy before treatment with gilteritinib in both trials is shown in Figure S1.
In both trials, responses with gilteritinib therapy were observed in prior TKI-treated patients with baseline FLT3-ITD or FLT3-TKD mutations as well as in patients harboring both mutation types (Table 3). There was no observed trend in treatment response by FLT3 mutation type across prior TKI or no prior TKI subgroups. Combined response outcomes from both trials in patients treated with 120-mg gilteritinib show similarly high proportions of patients achieving CRc in both prior TKI therapy (52%) and no prior TKI (53%) groups (Table 4).
Transplantation and posttransplant gilteritinib maintenance therapy
In the CHRYSALIS trial, 30 patients who received 120- or 200-mg gilteritinib underwent hematopoietic stem cell transplantation (HSCT) and 12 received posttransplant gilteritinib maintenance therapy for a median of 564 days (range, 15–959). Of the 28 patients who underwent transplantation after CRc, 12 (43%) resumed gilteritinib after HSCT. Six patients with prior sorafenib exposure underwent HSCT, all of whom achieved CRc before HSCT (all CRi), but none received posttransplant gilteritinib maintenance therapy. Among patients in the gilteritinib arm of the ADMIRAL trial who received prior TKI therapy, five underwent HSCT during the trial, and four of these five patients resumed gilteritinib after HSCT. Durations of posttransplant gilteritinib therapy for patients with prior midostaurin exposure (n = 2) were 1 and 95 days, and for patients with prior sorafenib exposure (n = 2) the durations of posttransplant gilteritinib maintenance therapy were 33 and 135 days. Two of the four prior TKI-treated patients who received posttransplant gilteritinib had achieved pretransplant CRc (CRi, n = 2 [prior midostaurin, n = 1; prior sorafenib, n = 1]). Among ADMIRAL patients who had not received prior TKI, 59 underwent HSCT. Of the 38 gilteritinib-arm patients without prior TKI exposure who were transplanted after CRc, 24 (63%) received posttransplant gilteritinib maintenance therapy for a median of 643.5 days (range, 2–1505).
Discussion
The multi-kinase oral FLT3 TKIs, sorafenib, and midostaurin, are both efficacious in the frontline setting when used in combination with chemotherapy [16, 17, 31] and sorafenib is also beneficial in the post-transplant setting [19, 20] in patients with newly diagnosed FLT3-mutated AML. Although not approved for AML, sorafenib was one of the first and most widely used multi-kinase FLT3 TKIs [1]. Midostaurin is a multi-kinase FLT3 TKI approved in combination with high-intensity chemotherapy for patients with newly diagnosed FLT3-mutated AML [18]. The availability of more selective FLT3-targeted TKI therapies has further expanded treatment options with the approval of gilteritinib for patients with R/R FLT3-mutated AML [28]. This is an important clinical advance because outcomes in R/R AML with SC regimens have generally been poor [32]. Because FLT3 TKIs are increasingly being used as frontline therapy for FLT3-mutated AML, it is important to understand the degree to which prior TKI therapy alters the likelihood of response or survival benefit conferred by gilteritinib in R/R FLT3-mutated AML. Understanding the impact of prior FLT3 TKI therapy on the ability to respond to a subsequent FLT3 TKI might help guide treatment selection in the R/R AML setting.
This analysis of patients from two trials of gilteritinib in the FLT3-mutated R/R AML setting demonstrated that a high proportion of patients who received prior midostaurin or sorafenib still achieved remission with gilteritinib. High CRc rates with 120- or 200-mg gilteritinib were observed in heavily pre-treated R/R AML patients in the CHRYSALIS trial (42%) and in R/R AML patients who received 120-mg gilteritinib after a single line of prior induction therapy in the ADMIRAL trial (52%). In both trials, protocol-defined remissions were achieved across patients with FLT3-ITD, FLT3-TKD, or both mutations. Patients in the gilteritinib arm of the ADMIRAL trial who received prior TKIs had higher response rates than corresponding patients in the SC arm. High response rates with gilteritinib after prior TKI therapy were observed in both relapsed and refractory subgroups; remission duration was shorter in patients who received prior TKIs compared with those who did not. In both the CHRYSALIS and ADMIRAL trials, patients who received prior TKIs before treatment with gilteritinib had shorter remission duration and higher relapse rate compared with patients who did not. Although variability in remission quality from prior therapy likely underlies these differences in remission duration, other factors could have contributed to this observation. For example, per protocol in the CHRYSALIS trial, remission duration was censored prior to HSCT in all patients who proceeded to transplant after CRc. This affected 47% (n = 28/60) of CRc responses from the CHRYSALIS trial included in this analysis. Remission durations were also censored prior to transplant for patients in ADMIRAL who did not resume gilteritinib as posttransplant maintenance therapy, although this was relatively uncommon. Nonetheless, censoring of pretransplant remission duration affected a sizeable fraction of patients with prior TKI exposure who achieved CRc in either study (26%; n = 8/31). Overall, gilteritinib maintenance therapy was associated with longer survival during ADMIRAL and was administered to the majority of patients in this trial without prior TKI exposure who underwent HSCT after CRc. However, the proportion of patients in ADMIRAL with prior TKI who underwent HSCT after CRc and received gilteritinib maintenance was small, which also potentially contributed to observed differences in remission duration.
In an evaluation of the impact of sequential FLT3 TKI therapy in patients with FLT3-mutated AML, Yilmaz and colleagues reported that the rate of CRc declined from 77% for FLT3 TKI therapy in the frontline setting to 31% for TKI therapy administered in the R/R AML setting; the CRc rate further declined to 25% after the third TKI [33]. In a second cohort of patients who received their first TKI in the R/R AML setting, the rate of CRc was 45% after the first TKI and declined to 21% after the second TKI and 12% after the third TKI [33]. Although variations in patient and treatment characteristics render comparison of findings from our analysis to those reported by Yilmaz et al. [33] challenging, it is notable that a considerable proportion (>40%) of patients in our analysis still responded to single-agent gilteritinib after prior TKI therapy. Our observations of shortened OS in prior TKI-treated patients who received gilteritinib in the CHRYSALIS trial concurred with findings reported by Yilmaz and colleagues for patients receiving a second or third FLT3 TKI [33]. However, among patients in the gilteritinib arm of the ADMIRAL trial, median OS was similar among patients who received or did not receive prior TKIs and remained longer than the median OS for corresponding patients in the SC arm. The difference in survival trends related to prior TKI therapy between the CHRYSALIS and ADMIRAL trials may stem from the fact that most patients in the CHRYSALIS trial represented a heavily pretreated population, whereas patients in the ADMIRAL trial had received only one prior line of AML therapy.
Mutations associated with treatment resistance vary between type I and type II FLT3 TKIs [34]. Acquired off-target mutations in RAS/MAPK pathway genes are most commonly associated with treatment resistance to type I FLT3 inhibitors such as midostaurin and gilteritinib [12, 34]. Acquired FLT3-TKD mutations at codon D835 are the most common resistance mutations identified in patients treated with type II FLT3 TKIs sorafenib or quizartinib [6, 34]. In the current analysis, a greater proportion of prior-TKI–treated patients had previously received sorafenib (70%; n = 58/83) than midostaurin (30%; n = 25/83). As gilteritinib is effective against FLT3 D835 mutations, the potential acquisition of these mutations likely did not have a negative impact on treatment response. In the current analysis, we observed a CRc rate of 57% (CR, 21%; CRi, 29%; CRp, 7%), among patients who received prior midostaurin before gilteritinib in the ADMIRAL trial. As the use of midostaurin in the frontline setting becomes more prevalent, further investigation of response to gilteritinib as a second FLT3 TKI in the relapsed setting is warranted.
As is commonly seen in secondary analyses, our study was not sufficiently powered to detect significant differences between prior TKI and no prior TKI subgroups, and no adjustments for multiple comparisons were made. Because the numbers of patients who received prior TKI therapy in both trials was small, the results of this study should be interpreted with caution. Variability in prior treatment characteristics and the number of prior lines of AML therapy in the CHRYSALIS and ADMIRAL trials may have also had an impact on the observed outcomes. Furthermore, the presence of a high FLT3-ITD allelic ratio at baseline and persistence of measurable residual disease after gilteritinib therapy may have also had an impact on response duration and OS. We did not assess the impact of FLT3-ITD allelic ratio in gilteritinib-arm patients previously exposed or not exposed to a FLT3 TKI due to the small number of patients in the ADMIRAL trial with prior FLT3 TKI exposure (n = 33) and lack of available samples from the CHRYSALIS study. In addition, we did not evaluate the impact of baseline RAS/MAPK pathway mutations on response and survival outcomes in the gilteritinib arm because the small sample size precluded meaningful statistical comparisons between prior TKI–exposed (n = 5) and non-exposed (n = 13) patients in this co-mutation subgroup. However, some insights can be gleaned from a recent retrospective study of gilteritinib in patients with FLT3-mutated R/R AML previously treated with a FLT3 TKI (n = 113). Patients harboring RAS/MAPK pathway mutations (n = 19) had a lower rate of CRc (38%) and shorter median OS (4.9 months) than patients without these mutations (n = 62) (CRc = 59%; median OS: 7.8 months, HR = 2.4; 95% CI: 1.1, 5.4; P < 0.01) [35].
Findings from this analysis show that patients with FLT3-mutated R/R AML who received prior treatment with sorafenib or midostaurin do achieve high remission rates with single-agent gilteritinib. As the use of FLT3 TKIs such as midostaurin becomes more prevalent in the frontline setting, physicians may still consider using gilteritinib as a subsequent FLT3-targeted therapy in the R/R AML setting. Further studies in a larger patient population will help validate these findings and determine the molecular profile(s) of patients for whom gilteritinib as a second FLT3 TKI therapy at relapse fails to improve outcomes relative to alternate regimens.
Data availability
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx
References
Mosquera Orgueira A, Bao Perez L, Mosquera Torre A, Peleteiro Raindo A, Cid Lopez M, Diaz Arias JA, et al. FLT3 inhibitors in the treatment of acute myeloid leukemia: current status and future perspectives. Minerva Med. 2020;111:427–42.
Nguyen B, Williams AB, Young DJ, Ma H, Li L, Levis M, et al. FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget. 2017;8:10931–44.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35.
Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111:2527–37.
Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia. 2015;29:2390–2.
Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114:2984–92.
Tarver TC, Hill JE, Rahmat L, Perl AE, Bahceci E, Mori K, et al. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Blood Adv. 2020;4:514–24.
Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119:5133–43.
Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3.
Schmalbrock LK, Dolnik A, Cocciardi S, Strang E, Theis F, Jahn N, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood. 2021;137:3093–104.
McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Disco. 2019;9:1050–63.
Peretz CAC, McGary LHF, Kumar T, Jackson H, Jacob J, Durruthy-Durruthy R, et al. Single-cell DNA sequencing reveals complex mechanisms of resistance to quizartinib. Blood Adv. 2021;5:1437–41.
Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–45.
Pratz KW, Cho E, Levis MJ, Karp JE, Gore SD, McDevitt M, et al. A pharmacodynamic study of sorafenib in patients with relapsed and refractory acute leukemias. Leukemia. 2010;24:1437–44.
Röllig C, Serve H, Huttmann A, Noppeney R, Muller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–9.
Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl J Med. 2017;377:454–64.
Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017;129:3403–6.
Burchert A, Bug G, Fritz LV, Finke J, Stelljes M, Röllig C, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3-internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38:2993–3002.
Xuan L, Wang Y, Huang F, Fan Z, Xu Y, Sun J, et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: an open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020;21:1201–12.
Döhner K, Thiede C, Jahn N, Panina E, Gambietz A, Larson RA, et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood. 2020;135:371–80.
Larrosa-Garcia M, Baer MR. FLT3 Inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Ther. 2017;16:991–1001.
Cortes JE, Khaled S, Martinelli G, Perl AE, Ganguly S, Russell N, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20:984–97.
Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl J Med. 2019;381:1728–40.
Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18:1061–75.
Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129:257–60.
Mori M, Kaneko N, Ueno Y, Yamada M, Tanaka R, Saito R, et al. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest N. Drugs. 2017;35:556–65.
Dhillon S. Gilteritinib: first global approval. Drugs. 2019;79:331–9.
Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for diagnosis, standardization of response criteria, treatment outcomes, and reporting standards for therapeutic trials in acute myeloid leukemia. J Clin Oncol. 2003;21:4642–9.
Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD, et al. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5:96–102.
Larson RA, Mandrekar SJ, Huebner LJ, Sanford BL, Laumann K, Geyer S, et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: the Alliance CALGB 10603/RATIFY trial. Leukemia. 2021;35:2539–51.
Roboz GJ, Rosenblat T, Arellano M, Gobbi M, Altman JK, Montesinos P, et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J Clin Oncol. 2014;32:1919–26.
Yilmaz M, Alfayez M, DiNardo CD, Borthakur G, Kadia TM, Konopleva MY, et al. Outcomes with sequential FLT3-inhibitor-based therapies in patients with AML. J Hematol Oncol. 2020;13:132.
Alotaibi AS, Yilmaz M, Kanagal-Shamanna R, Loghavi S, Kadia TM, DiNardo CD, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3 inhibitors. Blood Cancer Disco. 2021;2:125–34.
Numan Y, Abdel Rahman Z, Grenet J, Boisclair S, Bewersdorf JP, Collins C, et al. Gilteritinib clinical activity in relapsed/refractory FLT3 mutated AML previously treated with FLT3 inhibitors. Am J Hematol. 2022;97:322–8.
Acknowledgements
This study was funded by Astellas Pharma, Inc. Medical writing/editorial support was provided by Kalpana Vijayan, PhD, Elizabeth Hermans, PhD, and Cheryl Casterline, MA, from Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, and funded by the study sponsor.
Author information
Authors and Affiliations
Contributions
AE Perl, N Podoltsev, C Smith, M Levis, S Strickland, N Hosono, N Hasabou, and JK Altman acquired the study data; AE Perl, P Montesinos, N Podoltsev, G Martinelli, C Smith, M Levis, S Strickland, C Röllig, N Hosono, N Hasabou, Q Lu, R Tiu, and J Altman contributed to the analysis of the study data; all authors contributed to the interpretation of study data, critically reviewed the manuscript, and provided final approval.
Corresponding author
Ethics declarations
Competing interests
AE Perl reports grants, personal fees and non-financial support from Astellas, during the conduct of the study; grants, personal fees, and non-financial support from FujiFilm; grants, personal fees, and non-financial support from Daiichi Sankyo; grants and personal fees from Abbvie and Actinium Pharmaceuticals; personal fees from Agios, Loxo, LLS/Beat AML, and Forma; non-financial support from Arog; personal fees and non-financial support from New Link Genetics, Novartis, Takeda, and Jazz; and grants from Bayer and Biomed Valley Discoveries, outside the submitted work. N Hosono reports no relevant conflicts of interest to disclose. P Montesinos reports research support from Pfizer, Abbvie, and Daiichi Sankyo; consultancy from Celgene, Pfizer, and Abbvie; and speakers bureau from Astellas, Novartis, and Janssen. N Podoltsev reports consultancy fees from Pfizer, Celgene, Agios Pharmaceuticals, Blueprint Medicines, Incyte, Novartis, Bristol-Myers Squibb, CTI Biopharma, PharmaEssentia, Constellation Pharmaceuticals, Cogent BioSciences, and AbbVie; and institutional research funding from Pfizer, Celgene, CTI Biopharma, Boehringer Ingelheim, Astellas, Daiichi Sankyo, Sunesis Pharmaceuticals, Jazz Pharmaceuticals, Astex Pharmaceuticals, Genentech, AI Therapeutics, Samus Therapeutics, Arog Therapeutics, and Kartos Therapeutics. G Martinelli reports grant funding and consultancy fees from Amgen, Ariad, Incyte, Pfizer, Roche, Celgene, Janssen, AbbVie, and Novartis. N Panoskaltsis reports no relevant conflicts of interest to disclose. C Recher reports grant funding and personal fees from Celgene, Sunesis, Amgen, and Novartis and personal fees from Incyte, Jazz Pharmaceuticals, AbbVie, Astellas, Macrogenics, and Otsuka. C Smith reports research funding from Abbvie, Revolution Medicines, Celgene, FujiFilm; consulting fees from Astellas, Daichi Sanyko, and Genentech to attend an advisory board meeting; and reports being a stockolder at Ligacept, LLC. MJ Levis reports grants and personal fees from Astellas and FujiFilm and personal fees from Daiichi Sankyo, Amgen, and Menarini. S Strickland reports consulting or advisory fees from Abbvie, Astellas Pharma, Jazz Pharmaceuticals, Kite, a Gilead company, Novartis, and Pfizer and research funding at an institutional level from Abbvie, Astellas Pharma, Inc, Celator/Jazz, Celgene, Daiichi Sankyo, Karyopharm Therapeutics, Menarini, Novartis, and Sunesis Pharmaceuticals. C Röllig has received grants from AbbVie, Novartis, and Pfizer; consulting fees from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Janssen, Jazz, Novartis, Pfizer, and Roche; and honoraria for speaker engagements from AbbVie, Amgen, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Janssen, Jazz, Novartis, Pfizer, and Roche. WC Chou reports no relevant conflicts of interest to disclose. H Lee has received honoraria for speaker engagements from AbbVie Korea and Astellas Korea, participated in and advisory board for AbbVie and Astellas, and is President of The Korean Society of Hematology. H Yokoyama reports having received honoraria from Astellas for speaking engagements. Q Lu, N Hasabou, M Groß-Langenhoff are employees of Astellas. R Tiu is a former employee of Astellas. JK Altman reports advisory or consulting fees from AbbVie, Amgen, Astellas, Daiichi Sankyo, Kura Oncology, Syros, and Theradex; institutional research funding for trials conducted by ALX Oncology, Amgen, Aptos, Astellas, Aprea, BioSight, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, FujiFilm, Immunogen, Kartos, Kura Oncology, and Loxo; reimbursement for travel from BioSight; and serves on a data monitoring committee for GlycoMimetics.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Perl, A.E., Hosono, N., Montesinos, P. et al. Clinical outcomes in patients with relapsed/refractory FLT3-mutated acute myeloid leukemia treated with gilteritinib who received prior midostaurin or sorafenib. Blood Cancer J. 12, 84 (2022). https://doi.org/10.1038/s41408-022-00677-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00677-7
This article is cited by
-
The GSK3β/Mcl-1 axis is regulated by both FLT3-ITD and Axl and determines the apoptosis induction abilities of FLT3-ITD inhibitors
Cell Death Discovery (2023)
-
FLT3 inhibitors as MRD-guided salvage treatment for molecular failure in FLT3 mutated AML
Leukemia (2023)
-
Gilteritinib activity in refractory or relapsed FLT3-mutated acute myeloid leukemia patients previously treated by intensive chemotherapy and midostaurin: a study from the French AML Intergroup ALFA/FILO
Leukemia (2023)
