
Enhancing Dissolution and Oral Bioavailability of Ursodeoxycholic Acid with a Spray-Dried pH-Modified Extended Release Formulation


Abstract
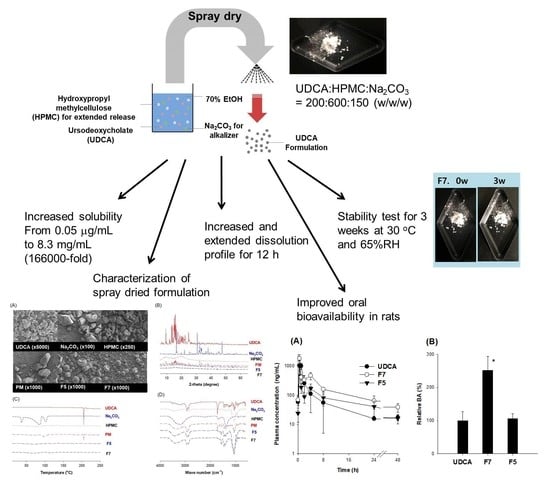
:
1. Introduction
2. Materials and Methods
2.1. Materials
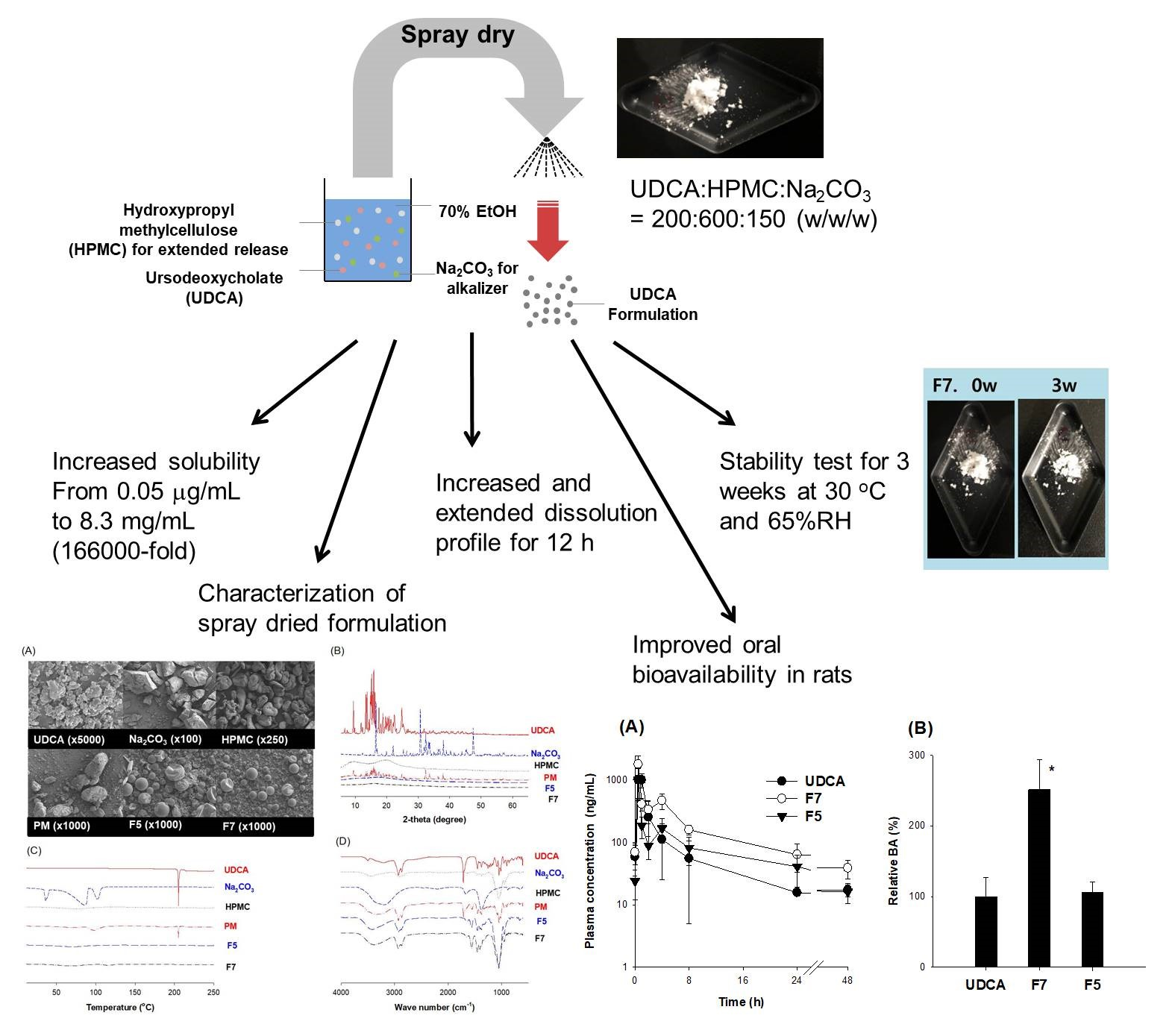
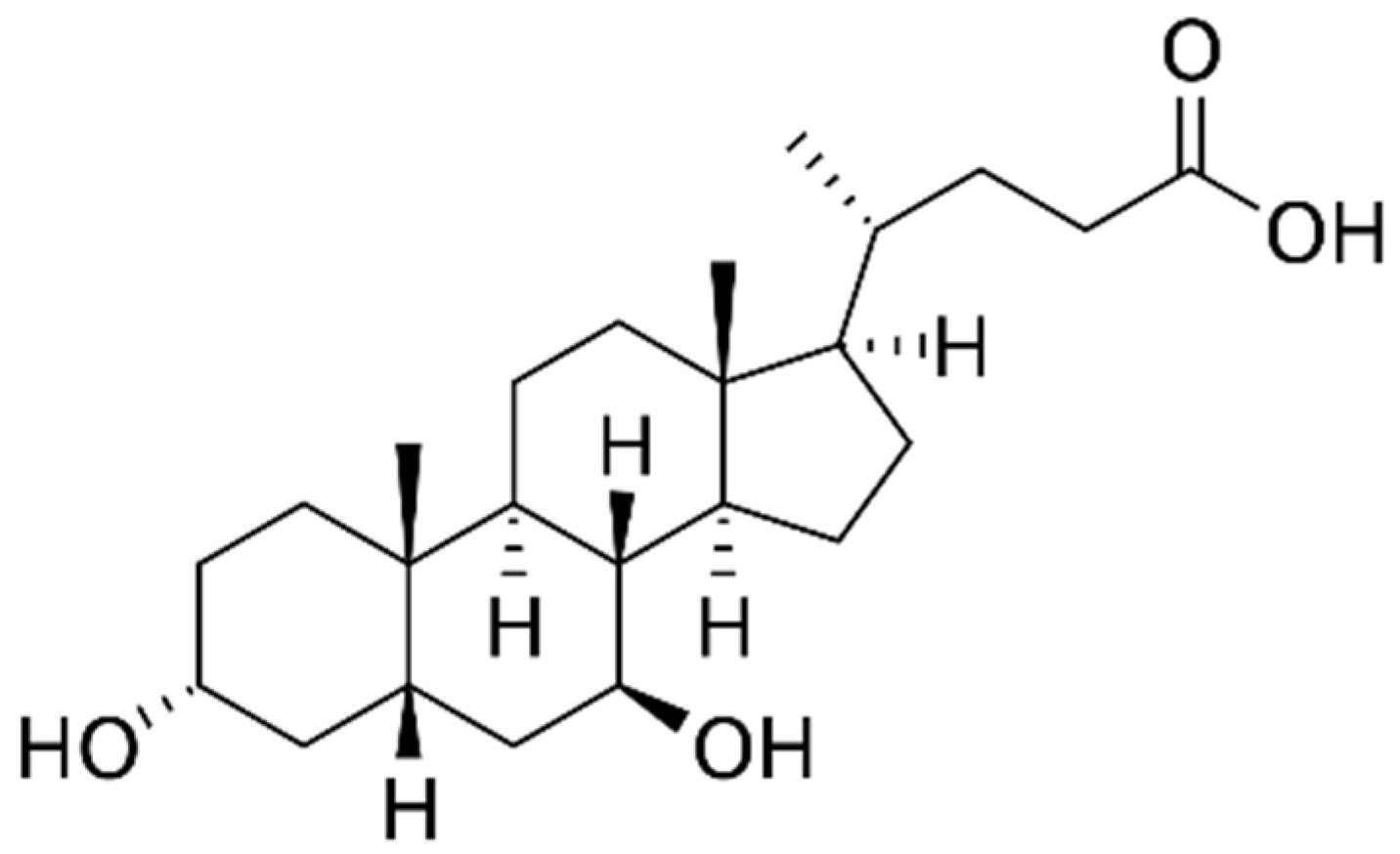
2.2. pH-Dependent Solubility of UDCA
2.3. pH-Dependent Permeability of UDCA
2.4. Optimization of UDCA Formulation
2.4.1. Optimization of HPMC
2.4.2. Optimization of Na2CO3
2.4.3. Preparation of UDCA Formulation
2.5. Characterization of UDCA Formulation
2.6. Pharmacokinetic Study
2.7. LC-MS/MS Analysis of UDCA
2.8. Data Analysis
3. Results
3.1. pH-Dependent Solubility and Permeability of UDCA
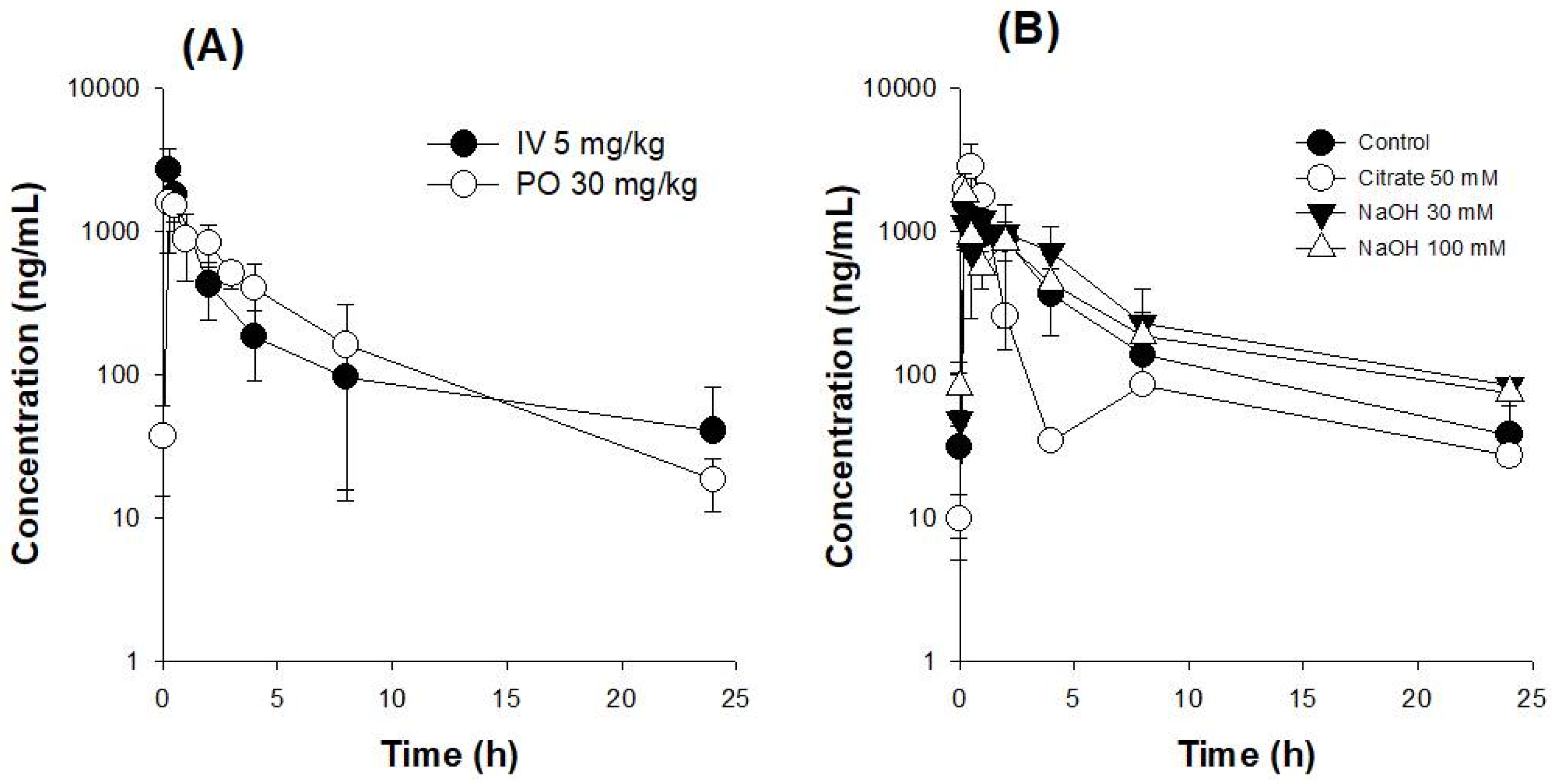
3.2. Oral Bioavailability (BA) of UDCA
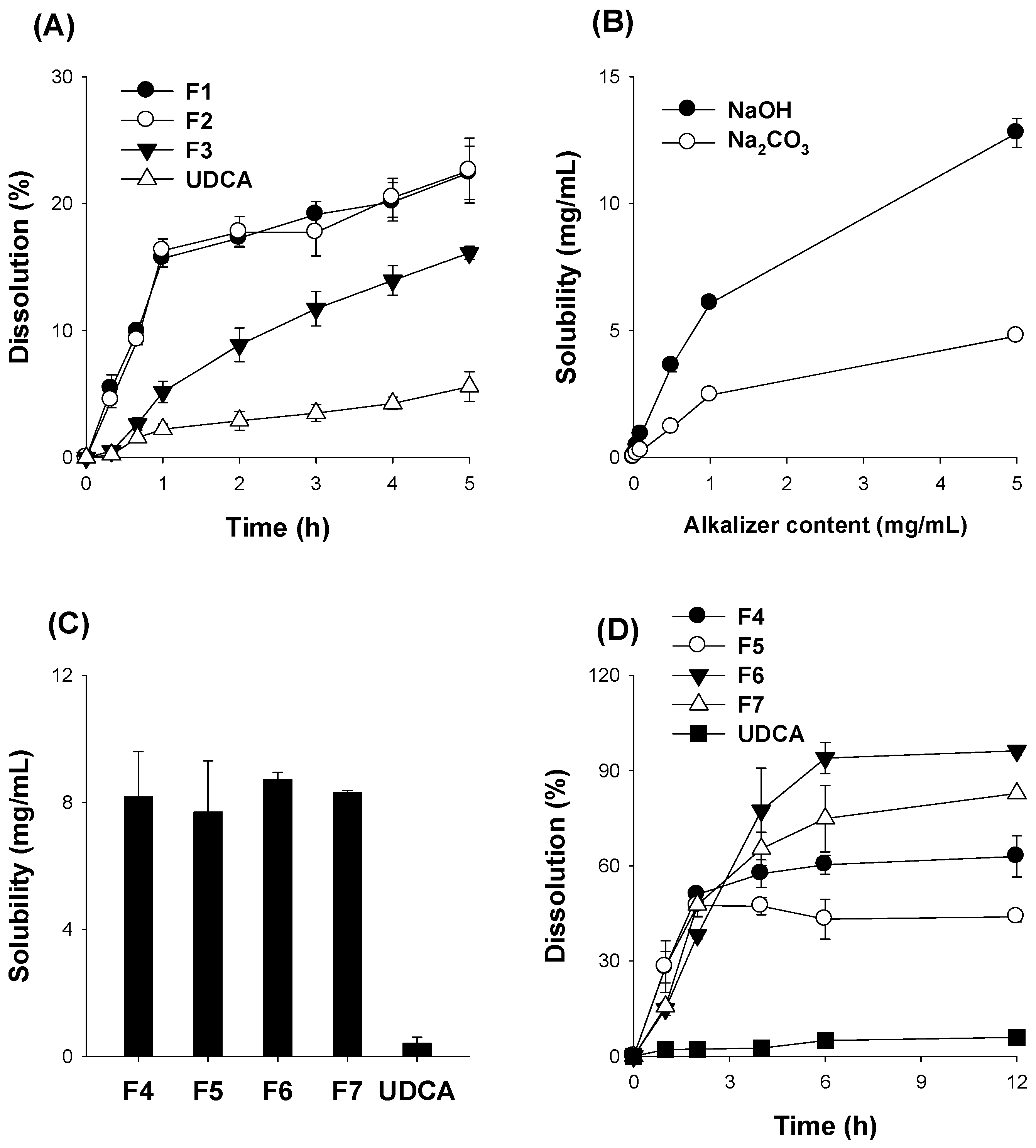
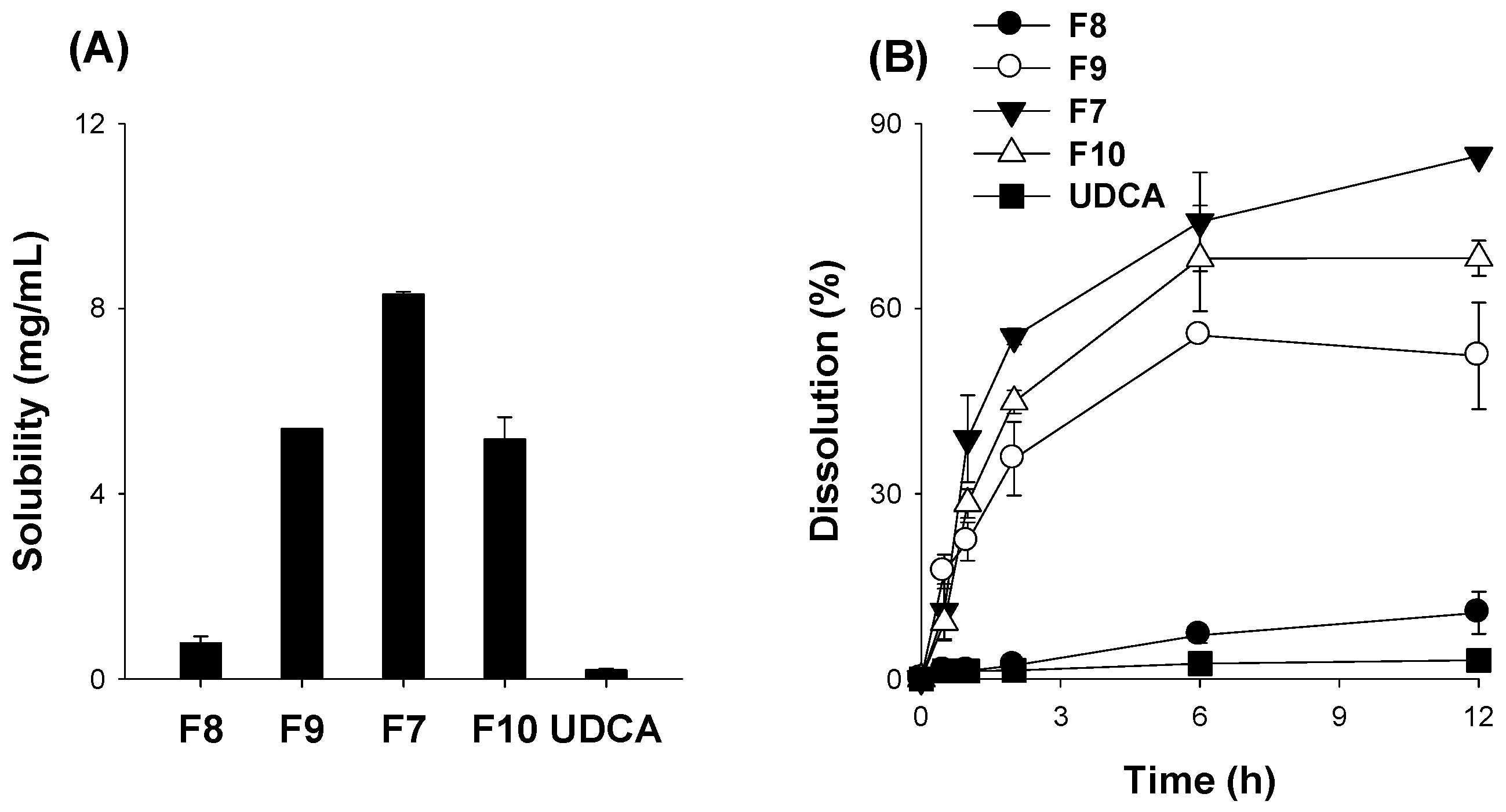
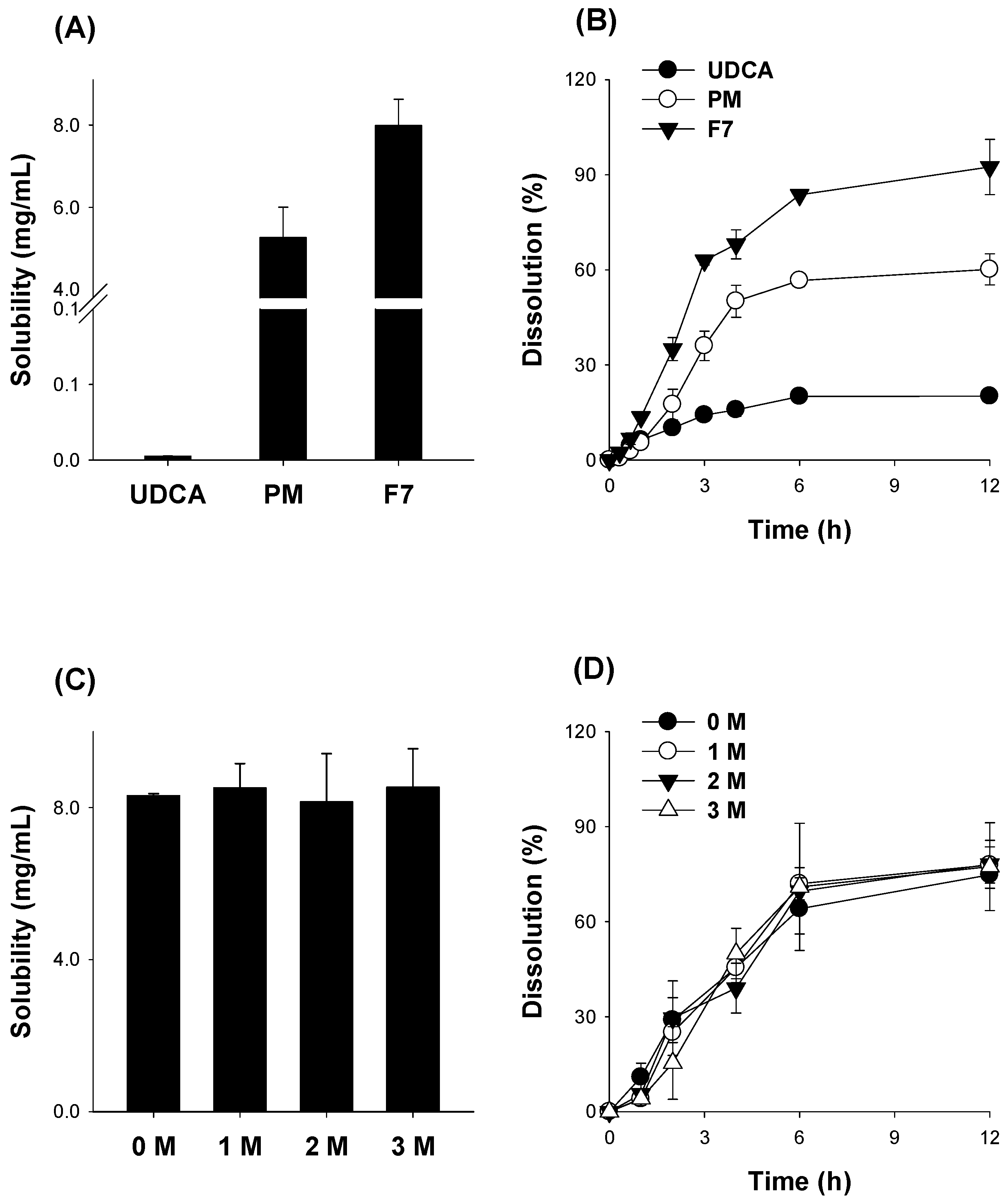
3.3. Optimization of pH-Modified Controlled Release Formulation of UDCA
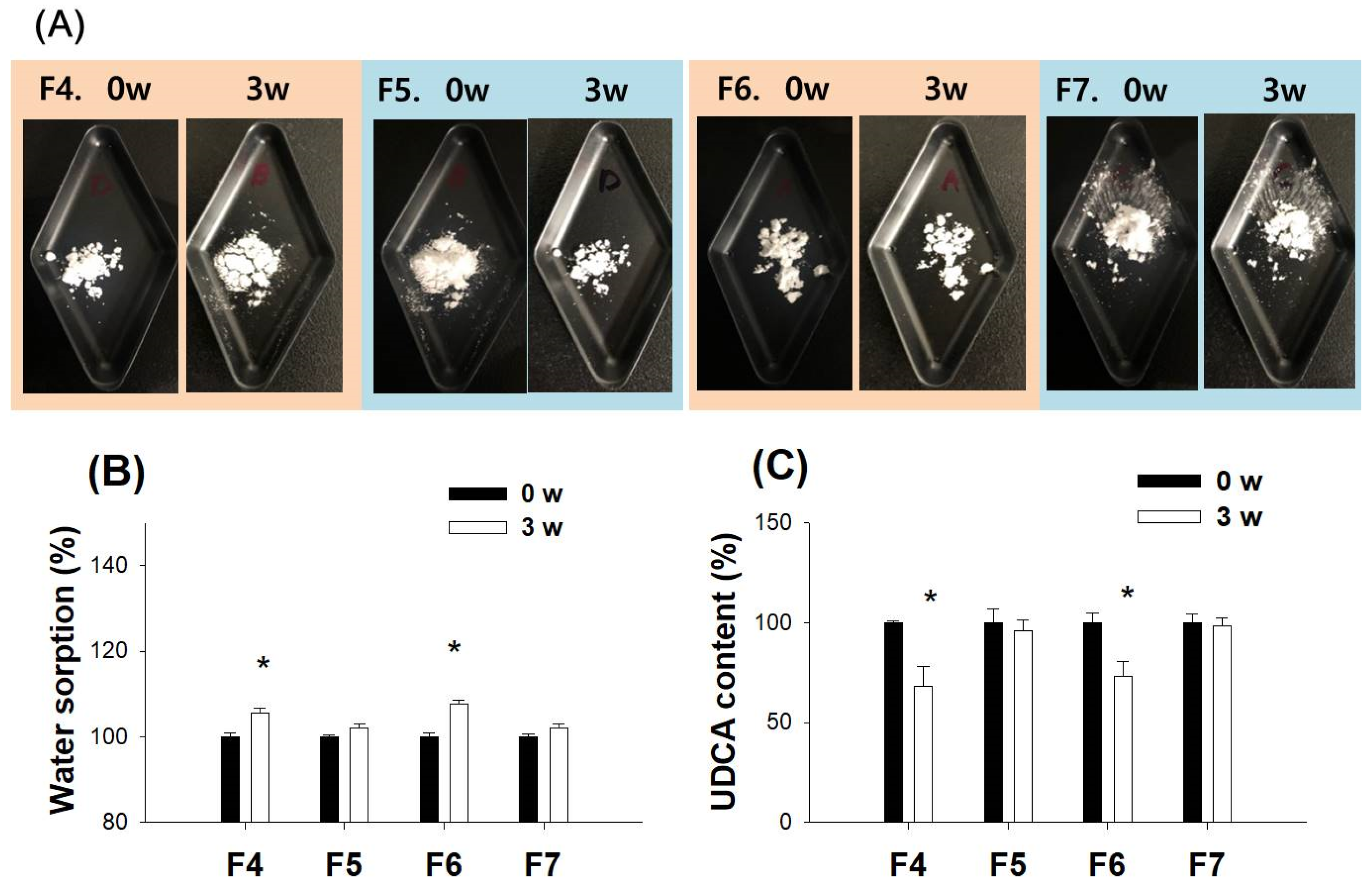
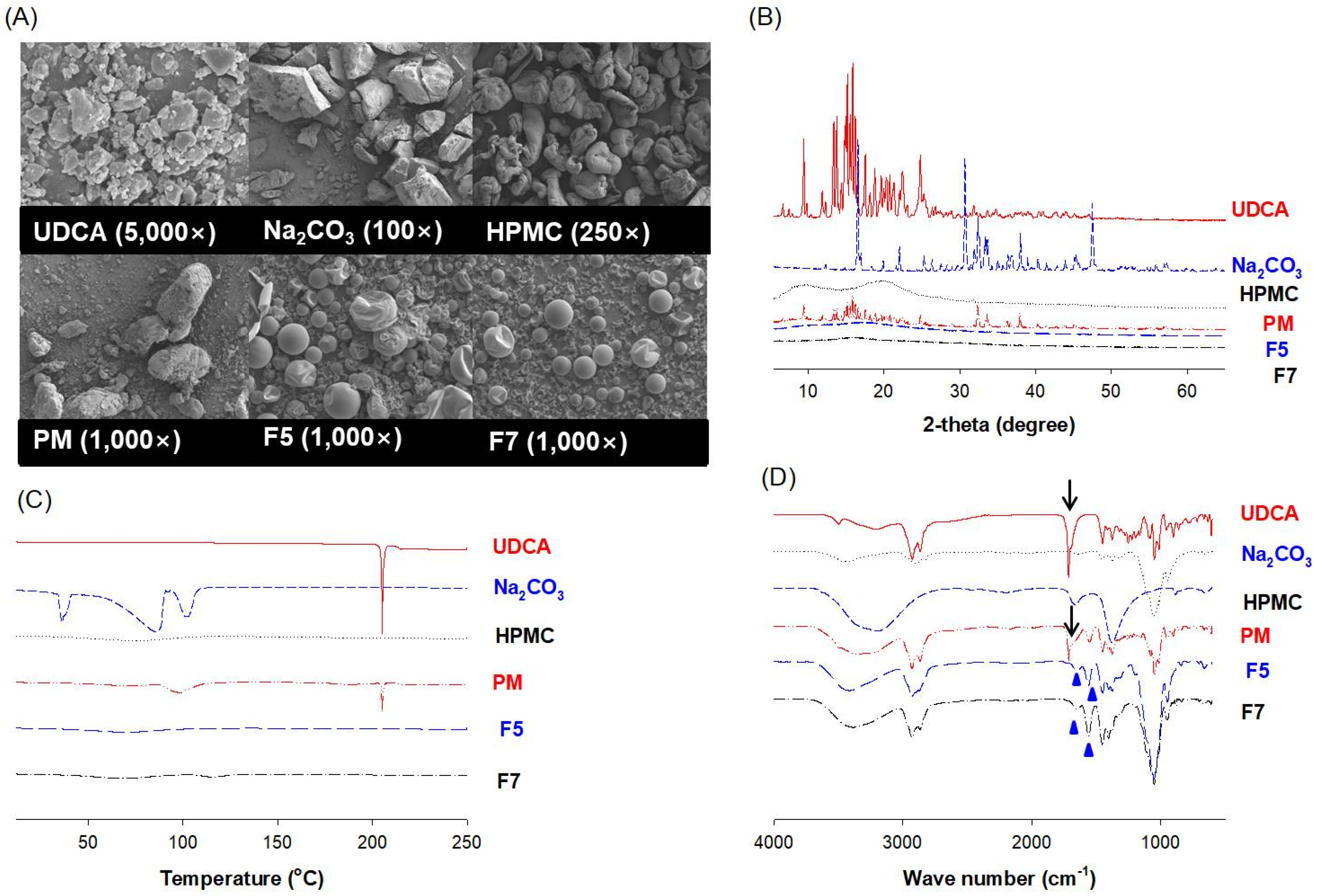
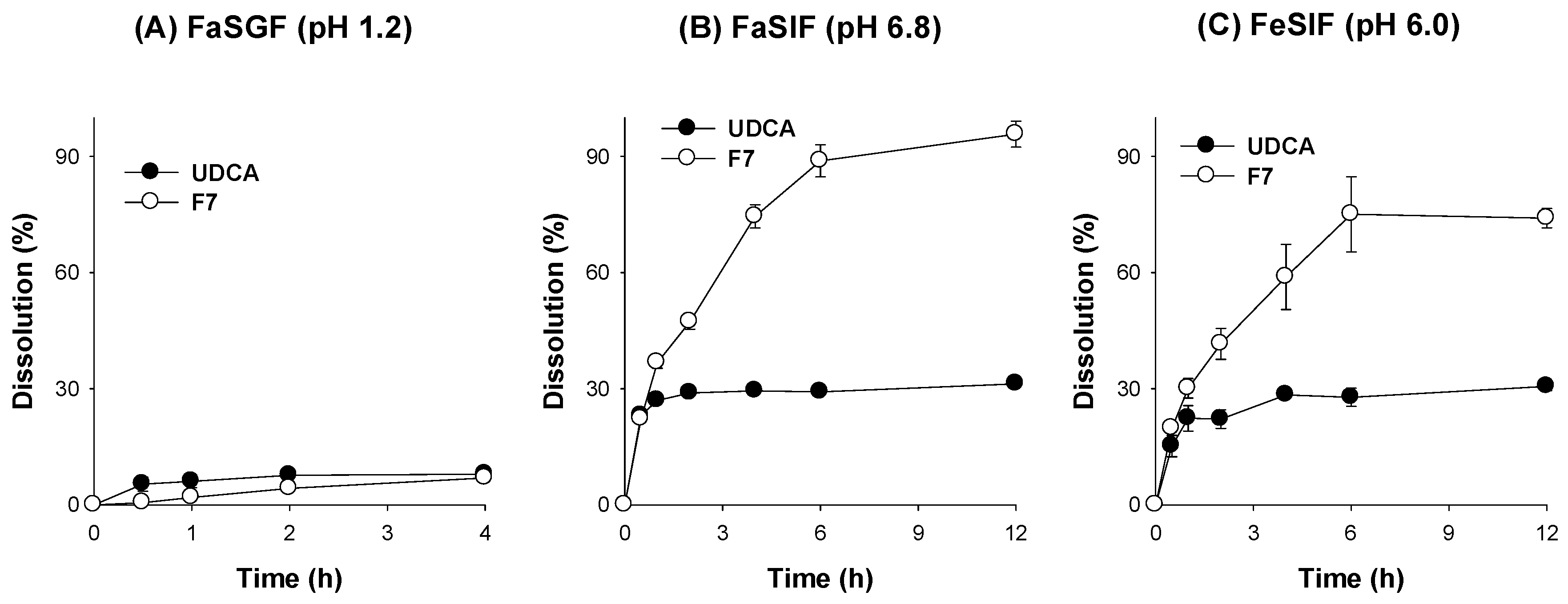
3.4. Characterization of pH-Modified Controlled Release Formulation of UDCA
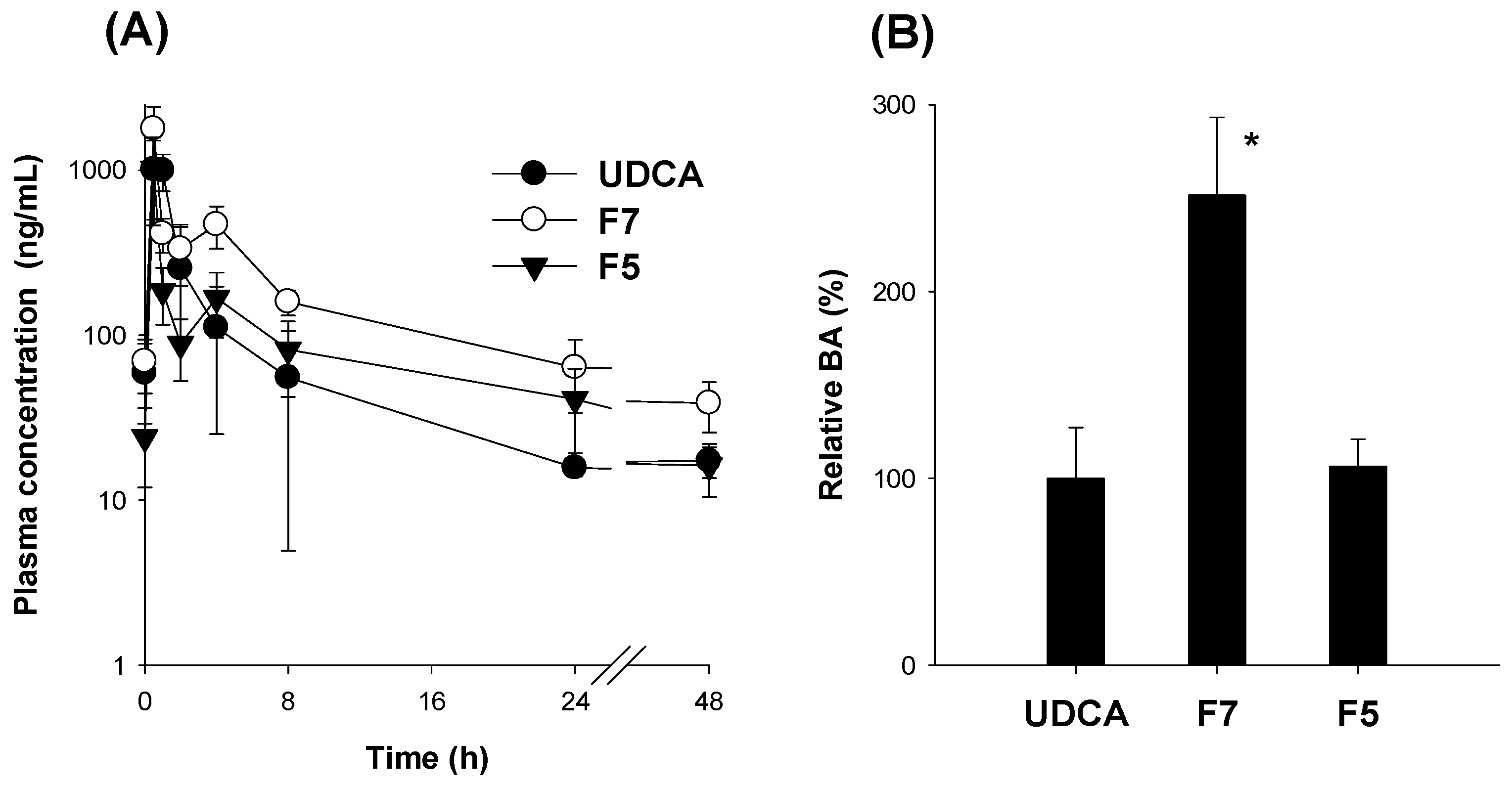
3.5. Pharmacokinetics of UDCA from UDCA Formulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, M.J.; Kang, Y.J.; Kwon, M.; Choi, Y.A.; Choi, M.-K.; Chi, H.-Y.; Yoo, H.H.; Shim, C.-K.; Song, I.-S. Ursodeoxycholate Restores Biliary Excretion of Methotrexate in Rats with Ethinyl Estradiol Induced-Cholestasis by Restoring Canalicular Mrp2 Expression. Int. J. Mol. Sci. 2018, 19, 1120. [Google Scholar] [CrossRef] [Green Version]
- Petr, T.; Smíd, V.; Kučerová, V.; Váňová, K.; Leníček, M.; Vítek, L.; Smíd, F.; Muchová, L. The effect of heme oxygenase on ganglioside redistribution within hepatocytes in experimental estrogen-induced cholestasis. Physiol. Res. 2014, 63, 359–367. [Google Scholar] [CrossRef]
- Zhou, F.; Gao, B.; Deng, C.; Huang, G.; Xu, T.; Wang, X. Dynamic expression of corticotropin-releasing hormone and urocortin in estrogen induced-cholestasis pregnant rat. Reprod. Toxicol. 2016, 65, 179–186. [Google Scholar] [CrossRef]
- Heuman, D.M.; Pandak, W.M.; Hylemon, P.B.; Vlahcevic, Z.R. Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts: In vitro studies in rat hepatocytes and human erythrocytes. Hepatology 1991, 14, 920–926. [Google Scholar] [CrossRef]
- Trauner, M.; Graziadei, I.W. Review article: Mechanisms of action and therapeutic applications of ursodeoxycholic acid in chronic liver diseases. Aliment. Pharmacol. Ther. 1999, 13, 979–996. [Google Scholar] [CrossRef]
- Okada, K.; Shoda, J.; Taguchi, K.; Maher, J.M.; Ishizaki, K.; Inoue, Y.; Ohtsuki, M.; Goto, N.; Takeda, K.; Utsunomiya, H.; et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G735–G747. [Google Scholar] [CrossRef]
- Lapenna, D.; Ciofani, G.; Festi, D.; Neri, M.; Pierdomenico, S.D.; Giamberardino, M.A.; Cuccurullo, F. Antioxidant properties of ursodeoxycholic acid. Biochem. Pharmacol. 2002, 64, 1661–1667. [Google Scholar] [CrossRef]
- Paumgartner, G.; Beuers, U. Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease. Clin. Liver Dis. 2004, 8, 67–81. [Google Scholar] [CrossRef]
- Paumgartner, G.; Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology 2002, 36, 525–531. [Google Scholar] [CrossRef]
- Beuers, U.; Trauner, M.; Jansen, P.; Poupon, R. New paradigms in the treatment of hepatic cholestasis: From UDCA to FXR, PXR and beyond. J. Hepatol. 2015, 62, S25–S37. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, D.H.; Rhee, H.; Choi, S.-H.; Sung, M.J.; Choi, W.S. Improvement of bioavailability of water insoluble drugs: Estimation of intrinsic bioavailability (Short Communication). Korean J. Chem. Eng. 2008, 25, 171–175. [Google Scholar] [CrossRef]
- Igimi, H.; Carey, M.C. pH-Solubility relations of chenodeoxycholic and ursodeoxycholic acids: Physical-chemical basis for dissimilar solution and membrane phenomena. J. Lipid Res. 1980, 21, 72–90. [Google Scholar] [CrossRef]
- Walker, S.; Rudolph, G.; Raedsch, R.; Stiehl, A. Intestinal absorption of ursodeoxycholic acid in patients with extrahepatic biliary obstruction and bile drainage. Gastroenterology 1992, 102, 810–815. [Google Scholar] [CrossRef]
- Parquet, M.; Metman, E.H.; Raizman, A.; Rambaud, J.C.; Berthaux, N.; Infante, R. Bioavailability, gastrointestinal transit, solubilization and faecal excretion of ursodeoxycholic acid in man. Eur. J. Clin. Investig. 1985, 15, 171–178. [Google Scholar] [CrossRef]
- Panini, R.; Vandelli, M.A.; Forni, F.; Pradelli, J.M.; Salvioli, G. Improvement of ursodeoxycholic acid bioavailability by 2-hydroxypropyl-β-cyclodextrin complexation in healthy volunteers. Pharmacol. Res. 1995, 31, 205–209. [Google Scholar] [CrossRef]
- Ma, Y.-Q.; Li, G.; Xu, J.-H.; Zhang, J.; Zhang, Z.-Z.; Xiao, H.-Y.; Li, X.-F. Combination of submicroemulsion and phospholipid complex for novel delivery of ursodeoxycholic acid. Pharm. Dev. Technol. 2014, 19, 363–372. [Google Scholar] [CrossRef]
- Yue, P.-F.; Yuan, H.-L.; Xie, H.; Xiao, X.-H.; Yang, M.; Liao, M.-X.; Zhu, W.-F.; Cai, P.-L. Preparation, Characterization, and Bioavailability of Ursodeoxycholic Acid–Phospholipid Complex In Vivo. Drug Dev. Ind. Pharm. 2008, 34, 708–718. [Google Scholar] [CrossRef]
- Scalia, S.; Giunchedi, P.; Pazzi, P.; Conte, U. Enhancement of Ursodeoxycholic Acid Bioavailability by Cross-linked Sodium Carboxymethyl Cellulose. J. Pharm. Pharmacol. 2000, 52, 383–388. [Google Scholar] [CrossRef]
- Scalia, S.; Scagliarini, R.; Pazzi, P. Evaluation of ursodeoxycholic acid bioavailability from immediate- and sustained-release preparations using gas chromatography-mass spectrometry and high-performance liquid chromatography. Arzneimittelforschung 2000, 50, 129–134. [Google Scholar] [CrossRef]
- Simoni, P.; Sabatini, L.; Baraldini, M.; Mirasoli, M.; Roda, A.; Roda, E. Pharmacokinetics and bioavailability of four modified-release ursodeoxycholic acid preparations for once-a-day administration. Int. J. Clin. Pharmacol. Res. 2002, 22, 37–45. [Google Scholar]
- Zhou, D.; Law, D.; Reynolds, J.; Davis, L.; Smith, C.; Torres, J.L.; Dave, V.; Gopinathan, N.; Hernandez, D.T.; Springman, M.K.; et al. Understanding and Managing the Impact of HPMC Variability on Drug Release from Controlled Release Formulations. J. Pharm. Sci. 2014, 103, 1664–1672. [Google Scholar] [CrossRef]
- Ma, D.; Djemai, A.; Gendron, C.M.; Xi, H.; Smith, M.; Kogan, J.; Li, L. Development of a HPMC-based controlled release formulation with hot melt extrusion (HME). Drug Dev. Ind. Pharm. 2013, 39, 1070–1083. [Google Scholar] [CrossRef]
- Song, I.-S.; Jeong, H.-U.; Choi, M.-K.; Kwon, M.; Shin, Y.; Kim, J.H.; Lee, H.-S. Interactions between cyazofamid and human drug transporters. J. Biochem. Mol. Toxicol. 2020, 34, e22459. [Google Scholar] [CrossRef]
- Jeon, J.-H.; Lee, J.; Park, J.-H.; Lee, C.-H.; Choi, M.-K.; Song, I.-S. Effect of Lactic Acid Bacteria on the Pharmacokinetics and Metabolism of Ginsenosides in Mice. Pharmaceutics 2021, 13, 1496. [Google Scholar] [CrossRef]
- Kwon, M.; Lim, D.Y.; Lee, C.H.; Jeon, J.-H.; Choi, M.-K.; Song, I.-S. Enhanced Intestinal Absorption and Pharmacokinetic Modulation of Berberine and Its Metabolites through the Inhibition of P-Glycoprotein and Intestinal Metabolism in Rats Using a Berberine Mixed Micelle Formulation. Pharmaceutics 2020, 12, 882. [Google Scholar] [CrossRef]
- Nam, S.J.; Lee, T.; Choi, M.K.; Song, I.S. Characterization of preclinical in vitro and in vivo pharmacokinetic properties of KPLA-012, a benzopyranyl 1,2,3-triazole compound, with anti-angiogenetic and anti-tumor progressive effects. Mass Spectrom. Lett. 2018, 9, 61–65. [Google Scholar]
- Leigh, M.; Kloefer, B.; Schaich, M. Comparison of the Solubility and Dissolution of Drugs in FastedState Biorelevant Media (FaSSIF and FaSSIF-V2). Dissolution Technol. 2013, 20, 44–50. [Google Scholar] [CrossRef]
- Salihović, S.; Dickens, A.M.; Schoultz, I.; Fart, F.; Sinisalu, L.; Lindeman, T.; Halfvarson, J.; Orešič, M.; Hyötyläinen, T. Simultaneous determination of perfluoroalkyl substances and bile acids in human serum using ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2020, 412, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Kaneko, R.; Nomura, M.; Naito, H.; Kitamori, K.; Nakajima, T.; Ogawa, T.; Hattori, H.; Seno, H.; Ishii, A. Simple and rapid quantitation of 21 bile acids in rat serum and liver by UPLC-MS-MS: Effect of high fat diet on glycine conjugates of rat bile acids. Nagoya J. Med. Sci. 2013, 75, 57–71. [Google Scholar]
- Luo, L.; Schomaker, S.; Houle, C.; Aubrecht, J.; Colangelo, J.L. Evaluation of Serum Bile Acid Profiles as Biomarkers of Liver Injury in Rodents. Toxicol. Sci. 2014, 137, 12–25. [Google Scholar] [CrossRef] [Green Version]
- Bathena, S.P.R.; Mukherjee, S.; Olivera, M.; Alnouti, Y. The profile of bile acids and their sulfate metabolites in human urine and serum. J. Chromatogr. B 2013, 942–943, 53–62. [Google Scholar] [CrossRef]
- Roda, A.; Fini, A. Effect of Nuclear Hydroxy Substituents on Aqueous Solubility and Acidic Strength of Bile Acids. Hepatology 1984, 4, 72S–76S. [Google Scholar] [CrossRef]
- Yonemochi, E.; Ueno, Y.; Ohmae, T.; Oguchi, T.; Nakajima, S.; Yamamoto, K. Evaluation of amorphous ursodeoxycholic acid by thermal methods. Pharm. Res. 1997, 14, 798–803. [Google Scholar] [CrossRef]
- Yue, P.-F.; Zhang, W.-J.; Yuan, H.-L.; Yang, M.; Zhu, W.-F.; Cai, P.-L.; Xiao, X.-H. Process Optimization, Characterization and Pharmacokinetic Evaluation in Rats of Ursodeoxycholic Acid–Phospholipid Complex. AAPS PharmSciTech 2008, 9, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.; Lee, J.-H.; Oh, Y.-J.; Bin Choy, Y.; Park, M.C.; Chang, H.C.; Choy, J.-H. Inorganic-polymer nanohybrid carrier for delivery of a poorly-soluble drug, ursodeoxycholic acid. Int. J. Pharm. 2010, 402, 117–122. [Google Scholar] [CrossRef]
- Jiao, F.; Chen, X.; Fu, Z.; Hu, Y.; Wang, Y. Intercalation of Mg–Al layered double hydroxides by (+)-dibenzoyl-d-tartaric acid: Preparation and characterization. J. Mol. Struct. 2009, 921, 328–332. [Google Scholar] [CrossRef]
- Kutbi, H.I.; Asfour, H.Z.; Kammoun, A.K.; Sirwi, A.; Cavalu, S.; Gad, H.A. Optimization of Hyaluronate-Based Liposomes to Augment the Oral Delivery and the Bioavailability of Berberine. Materials 2021, 14, 5759. [Google Scholar] [CrossRef]
- Pund, S.; Borade, G.; Rasve, G. Improvement of anti-inflammatory and anti-angiogenic activity of berberine by novel rapid dissolving nanoemulsifying technique. Phytomedicine 2014, 21, 307–314. [Google Scholar] [CrossRef]
- Stiehl, A. Intestinal absorption of bile acids: Effect of ursodeoxycholic acid treatment. Ital. J. Gastroenterol. 1995, 27, 193–195. [Google Scholar]
- Gao, P.; Fagerness, P.E. Diffusion in HPMC Gels. I. Determination of Drug and Water Diffusivity by Pulsed-Field-Gradient Spin-Echo NMR. Pharm. Res. 1995, 12, 955–964. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research. Dissolution Testing of Immediate Release Solid Oral Dosage Forms. FDA Guidance Documents 1997. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/dissolution-testing-immediate-release-solid-oral-dosage-forms (accessed on 1 February 2022).
- Jin, S.; Lee, C.; Lim, D.; Lee, J.; Park, S.-J.; Song, I.-S.; Choi, M.-K. Improved Hygroscopicity and Bioavailability of Solid Dispersion of Red Ginseng Extract with Silicon Dioxide. Pharmaceutics 2021, 13, 1022. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.; Ensom, M. Post Hoc Power Analysis: An Idea Whose Time Has Passed? Pharmacotherapy 2001, 21, 405–409. [Google Scholar] [CrossRef]
- Deng, J.W.; Song, I.-S.; Shin, H.; Yeo, C.-W.; Cho, D.-Y.; Shon, J.-H.; Shin, J.-G. The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: The contribution of transporting activity changes by SLCO1B1*15. Pharm. Genom. 2008, 18, 424–433. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | IV (UDCA 5 mg/kg) | PO (UDCA 30 mg/kg) | |||
|---|---|---|---|---|---|
| Parameters | |||||
| Co (μg/mL) | 4.17 ± 2.31 | ||||
| Cmax (μg/mL) | 1.76 ± 0.84 | ||||
| Tmax (h) | 0.44 ± 0.13 | ||||
| AUC24h (μg∙h/mL) | 5.91 ± 3.19 | 5.42 ± 1.84 | |||
| AUC∞ (μg∙h/mL) | 7.35 ± 5.29 | 5.72 ± 2.10 | |||
| t1/2 (h) | 7.31 ± 2.11 | 5.45 ± 2.96 | |||
| MRT (h) | 4.86 ± 2.80 | 5.68 ± 2.19 | |||
| Absolute BA (%) | 15.2 | ||||
| Treatment | PO (UDCA 30 mg/kg) | ||||
| Parameters | Control (pH 6.3) | +Na citrate 50 mM (pH 2.7) | +NaOH 30 mM (pH 11.4) | +NaOH 100 mM (pH 12.4) | |
| Cmax (μg/mL) | 1.81 ± 0.73 | 2.86 ± 1.11 | 1.33 ± 0.13 | 1.90 ± 0.69 | |
| Tmax (h) | 0.45 ± 0.11 | 0.42 ± 0.14 | 0.75 ± 0.43 | 0.69 ± 0.88 | |
| AUC24h (μg∙h/mL) | 5.77 ± 1.67 | 4.39 ± 1.65 | 6.78 ± 3.05 | 6.07 ± 2.50 | |
| AUC∞ (μg∙h/mL) | 6.06 ± 2.01 | 4.63 ± 2.05 | 7.63 ± 2.84 | 6.66 ± 2.89 | |
| t1/2 (h) | 4.26 ± 1.18 | 4.23 ± 2.04 | 4.61 ± 2.31 | 5.56 ± 2.99 | |
| MRT (h) | 5.26 ± 2.50 | 3.99 ± 3.63 | 6.21 ± 2.91 | 6.89 ± 3.69 | |
| Relative BA (%) | 100 | 76.4 ± 33.8 | 125 ± 46.8 | 109 ± 47.6 | |
| Step | Formulations | UDCA (mg) | HPMC (mg) | NaOH (mg) | Na2CO3 (mg) |
|---|---|---|---|---|---|
| Step I | F1 | 200 | 100 | 10 | |
| F2 | 200 | 200 | 10 | ||
| F3 | 200 | 600 | 10 | ||
| Step II | F4 | 200 | 200 | 50 | |
| F5 | 200 | 200 | 150 | ||
| F6 | 200 | 600 | 50 | ||
| F7 | 200 | 600 | 150 | ||
| Step III | F8 | 200 | 600 | 30 | |
| F9 | 200 | 600 | 90 | ||
| F7 | 200 | 600 | 150 | ||
| F10 | 200 | 600 | 200 | ||
| PM | 200 | 600 | 150 |
| Parameters | UDCA | F7 | F5 |
|---|---|---|---|
| Cmax (μg/mL) | 1.19 ± 0.39 | 1.79 ± 0.65 | 1.02 ± 0.56 |
| Tmax (h) | 0.87 ± 0.25 | 0.5 ± 0.0 * | 0.5 ± 0.0 * |
| AUC48h (μg∙h/mL) | 3.03 ± 0.99 | 6.53 ± 0.95 * | 3.03 ± 0.75 |
| AUC∞ (μg∙h/mL) | 3.34 ± 0.90 | 8.39 ± 1.53 * | 3.54 ± 0.49 |
| t1/2 (h) | 13.7 ± 8.93 | 14.2 ± 3.55 | 15.6 ± 5.12 |
| MRT (h) | 14.9 ± 6.3 | 18.2 ± 5.12 | 20.3 ± 6.97 |
| Relative BA (%) | 100 ± 27 | 251 ± 45.9 | 106 ± 14.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Lee, C.H.; Lee, J.-G.; Jeon, S.Y.; Choi, M.-K.; Song, I.-S. Enhancing Dissolution and Oral Bioavailability of Ursodeoxycholic Acid with a Spray-Dried pH-Modified Extended Release Formulation. Pharmaceutics 2022, 14, 1037. https://doi.org/10.3390/pharmaceutics14051037
Lee J, Lee CH, Lee J-G, Jeon SY, Choi M-K, Song I-S. Enhancing Dissolution and Oral Bioavailability of Ursodeoxycholic Acid with a Spray-Dried pH-Modified Extended Release Formulation. Pharmaceutics. 2022; 14(5):1037. https://doi.org/10.3390/pharmaceutics14051037
Chicago/Turabian StyleLee, Jaehyeok, Chul Haeng Lee, Jong-Geon Lee, So Yeon Jeon, Min-Koo Choi, and Im-Sook Song. 2022. "Enhancing Dissolution and Oral Bioavailability of Ursodeoxycholic Acid with a Spray-Dried pH-Modified Extended Release Formulation" Pharmaceutics 14, no. 5: 1037. https://doi.org/10.3390/pharmaceutics14051037