
Abstract
Background and objective
AMG 986 is a first-in-class, novel apelin receptor small molecule agonist initially developed for the treatment of heart failure. The current phase I study was conducted to evaluate the pharmacokinetics and safety of a single-dose 200-mg capsule formulation of AMG 986 relative to the tablet formulation in 12 healthy subjects.
Methods
In a two-period, two-way crossover design, eligible subjects were randomized 1:1 to tablet/capsule or capsule/tablet treatment sequences; each treatment sequence lasted for approximately 6 days and comprised six subjects.
Results
Following a single oral dose of AMG 986, the geometric mean maximum observed concentration (Cmax) values were 9670 ng/mL and 6920 ng/mL and the geometric mean area under the curve from time zero to 120 h (AUC0–120h) values were 68,000 ng*h/mL and 59,900 ng*h/mL for the tablet and capsule, respectively. The geometric least squares means (90% confidence interval [90% CI]) for the ratios of capsule/tablet were 0.88 (90% CI 0.81–0.96) and 0.72 (90% CI 0.57–0.91) for AUC0–120h and Cmax, respectively. AMG 986 had an acceptable safety profile; all adverse events were grade 1 or 2 in severity.
Conclusion
There was a modest 12% decrease in AUC0–120h and a 28% decrease in Cmax with the AMG 986 capsule versus the tablet. These differences are not considered to be clinically relevant, suggesting the capsule formulation can be used in subsequent clinical studies of AMG 986.
Similar content being viewed by others


The current study compared the in vivo characteristics of 200-mg AMG 986 capsule and tablet formulations in healthy adult subjects. |
Following a single oral dose of AMG 986, no clinically significant differences were observed in the pharmacokinetic properties of the capsule and tablet formulations. |
The results suggest the capsule formulation can be administered in subsequent studies for AMG 986 development. |
1 Introduction
Chronic heart failure (HF) is a complex syndrome that results in inadequate systemic blood flow when neurohormonal mechanisms are no longer able to deliver an adequate physiological response [1]. The prognosis for patients with HF remains poor and there is an unmet need for novel therapies [2]. The apelin receptor is a G-protein-coupled receptor that counteracts the pressor effect of angiotensin II, attenuates ischemic injury, and contributes to neovascularization in HF [3, 4]. AMG 986 is a novel apelin receptor small molecule agonist that activates the apelin receptor to improve cardiac function by increasing cardiac contractility without affecting heart rate [5]. AMG 986 was initially developed as a treatment for patients with HF and its chemical structure is presented in Fig. 1.

Chemical structure of AMG 986
Recently, a phase Ib, first-in-human (FIH) study in healthy and HF subjects (ClinicalTrials.gov identifier: NCT03276728) reported that AMG 986 exposure increases non-linearly with increasing oral doses of 5 mg to 650 mg [3]. The oral bioavailability of AMG 986 ranges from 40 to 80% and the terminal half-life (t½) is approximately 20 h [3]. AMG 986 exposure is similar between subjects with severe renal impairment and normal renal function [6]. Additional studies indicate AMG 986 is highly protein bound (99.6% bound in human plasma) and does not preferentially distribute into blood cells. The metabolism of AMG 986 in vitro is principally catalyzed by human cytochrome P450 3A (CYP3A). AMG 986 is an in vitro inducer of CYP3A4, as determined by increases in CYP3A4 messenger RNA levels in primary human hepatocytes, and AMG 986 is not an in vitro inhibitor of any of the major drug-metabolizing human CYP enzymes. Additionally, AMG 986 is characterized in vitro as a substrate of P-glycoprotein and organic anion-transporting polypeptide 1B3 (Amgen, data on file).
The FIH clinical development of AMG 986 started with a tablet formulation manufactured by dry granulation via roller compaction. Although acceptable chemical stability is observed for the tablet formulation, including data from 6-month accelerated and 18-month long-term storage conditions, further solid-state characterization shows a 25–30% drug substance solid-state phase transformation (Amgen, data on file). This solid-state phase transformation is caused by shear force imparted during roller compaction and tablet compression. This could potentially be mitigated through a change of AMG 986 manufacturing process from roller compaction and tablet compression to wet granulation and encapsulation.
The aim of the current phase I study was to evaluate the pharmacokinetics (PK) and safety of a single-dose 200-mg capsule formulation of AMG 986 relative to the tablet in healthy subjects. The 200-mg dose was selected because it was well tolerated in the FIH study [3] and is anticipated to be near the upper range of doses that would be evaluated in phase II studies.
2 Materials and Methods
2.1 Study Design
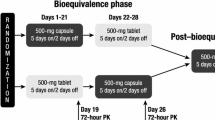
This was a phase I, open-label, single-dose, single-center, randomized, two-period, two-way crossover study conducted in healthy adult subjects in the United States. The study consisted of a 21-day screening period, after which eligible subjects were randomized 1:1 to AMG 986 capsule/tablet or tablet/capsule treatment sequences (Fig. 2). Each treatment sequence consisted of two periods, with a single dose of 8 × 25-mg AMG 986 tablets or 1 × 200-mg AMG 986 capsule administered in each period. The composition of the AMG 986 tablet and capsule formulations are listed in Tables 1 and 2, respectively.

Study design. N number of subjects, PK pharmacokinetic, R randomization
AMG 986 was administered orally on the morning of Days 1 and 6 under the supervision of clinic staff. Doses of AMG 986 were separated by a washout period of 5 days. Based on preliminary PK data available when the current study was planned, this period was calculated to be sufficient to eliminate 97% of the drug from systemic circulation. AMG 986 tablets and capsules were swallowed whole (not chewed, split, or crushed) and taken with water. Dosing was preceded by an overnight fast (except water) of at least 10 h. Subjects refrained from drinking water for at least 1 h before and after dosing and abstained from food for at least 4 hours after dosing.
2.2 Participants
Eligible subjects were healthy non-smoking men and women not of childbearing potential, between 18 and 55 years of age, and with a body mass index between 18 and 30 kg/m2 at the time of screening. Exclusion criteria included the concurrent or prior use of strong CYP3A4 inhibitors or strong CYP3A4 inducers within 14 days and 28 days, respectively, of Day 1.
2.3 PK Sampling and Assessments
On Days 1 and 6, blood samples for the PK analysis of AMG 986 were collected from each subject at pre-dose, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72, 96, and 120 h post-dose. The PK parameters assessed were maximum observed concentration (Cmax), median time to Cmax (tmax), area under the curve (AUC) from time zero to the last quantifiable time point post-dose (AUClast), AUC from time zero to infinity (AUCinf), AUC from time zero to 120 h (AUC0–120h), and t½. All PK parameters were estimated using noncompartmental analysis by Phoenix® WinNonlin® version 6.4 (Certara, Princeton, NJ, US).
In vitro dissolution of the tablet and capsule formulation was tested using a phase-appropriate, quality control method. Dissolution tests were performed using a United States Pharmacopeia Apparatus II (Varian VK 7025; Agilent Technologies Inc., CA, US) in 900 mL of 0.05 N hydrochloric acid at 37.0 ± 0.5 °C and an agitation speed of 75 revolutions per minute. For tablets, 8 × 25-mg tablets were dropped into each vessel for a total strength of 200 mg. Three vessels were used for a triplicate of measurement (N = 3). For capsules, 1 × 200-mg capsule was dropped into each vessel (N = 6) with a Japanese sinker. Sampling was performed at 10, 20, 30, 45, and 60 minutes with a sampling volume of 1 mL. Samples were filtered using 10-µm polyethylene filters and analyzed by high-performance liquid chromatography (HPLC) equipped with a Zorbax® SB-C18 column (4.6 × 50 mm, 3.5 µm) (Agilent Technologies Inc., CA, US) using an isocratic method.
Plasma concentrations of AMG 986 were determined using validated HPLC mass spectrometry. The analyte AMG 986 and internal standard (IS) 13C6-AMG 986 were extracted from 0.050 mL of human plasma by a protein precipitation extraction procedure. The extraction began by adding 50.0 μL of calibration standards, quality control samples, and study samples to appropriate wells of a 96-well plate, with 50.0 μL of blank human plasma added to each blank well. The samples were diluted with 50 μL of methanol/water (50/50, volume per volume [v/v]). Following the addition of 150 μL of IS to all appropriate wells (or 150 μL of acetonitrile to blank wells), the plate was covered, vortexed, and centrifuged. A Tomtec Quadra4™ (Tomtec Inc., CT, US) 96-well pipettor system was used to transfer 100 μL of the supernatant to a new 96-well plate. After 400 μL of acetonitrile/water (80/20, v/v) was added to all wells, the plate was covered and vortexed. The extracts were chromatographed under reverse-phase conditions on a Kinetex® C8 (Phenomenex Inc., CA, US) HPLC column using a gradient system with 0.1% formic acid in water and 0.1% formic acid in acetonitrile. The compounds were detected and quantified by tandem mass spectrometry in positive ion mode on an API 4000™ (AB Sciex, MA, US) equipped with a Turbo IonSpray® interface. The m/z transition values for AMG 986 and IS were 524.3–312.2 and 530.3–318.2, respectively. The assay had a lower limit of quantitation (LLOQ) of 1.0 ng/mL and an upper limit of quantitation of 1000 ng/mL. Calibration curves were obtained by performing a linear regression (weighted 1/x2) on the calibration standards.
2.4 Safety Outcomes
The safety and tolerability of AMG 986 capsule and tablet formulations was assessed via the subject incidence of treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), clinical laboratory tests, 12-lead electrocardiogram (ECG), and vital signs. All adverse events were graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
2.5 Statistical Analysis
Statistical analyses were performed using SAS version 9.3 (SAS Institute Inc., Cary, NC, US). No formal statistical hypothesis testing was performed. The study was designed to characterize the PK after a single administration of AMG 986 by descriptive summaries based on the derived PK parameters. The sample size of the study was based on practical considerations and was consistent with the number of subjects enrolled in similar studies. The PK analysis set included all randomized subjects who received at least one dose of AMG 986 and had at least one evaluable AMG 986 PK parameter. The safety analysis set included all study subjects who received at least one dose of AMG 986. Concentrations below the LLOQ (1.00 ng/mL) were set to zero before analysis. Geometric least squares means (GLSMs) and 90% confidence intervals (90% CIs) for the ratio of the GLSM (capsule/tablet) were estimated using a mixed-effects model. The model included the log-transformed PK parameter as the dependent variable and formulation (capsule or tablet), period, and treatment sequence as the independent variables. Subjects nested within sequence were included as the random effect. Safety results were summarized using descriptive statistics.
3 Results
3.1 In Vitro Dissolution Profiles
The two dosage forms had comparable dissolution profiles as shown in Fig. 3.

AMG 986 dissolution profiles for 8 × 25-mg tablets and 1 × 200-mg capsule. Method: USP Apparatus II, 900 mL, 0.05 N HCl, 75 rpm, 37 °C. HCl hydrochloric acid, rpm revolutions per minute, USP United States Pharmacopeia
3.2 Baseline Characteristics
Overall, 12 subjects were enrolled in the study and six subjects each were randomized to the two treatment sequences. All subjects received a single dose of 8 × 25-mg AMG 986 tablets and 1 × 200-mg AMG 986 capsule administered in each treatment period and all subjects completed the study. All 12 subjects were male, and the mean (SD) age was 38.8 (8.7) years (Table 3).
3.3 PK Evaluation of AMG 986 Capsule Formulation
Mean plasma concentration–time profiles for AMG 986 tablet and capsule formulations are presented in Fig. 4. After oral administration of 200-mg AMG 986, the median (range) tmax was 0.75 (0.48–2.0) h and 1.0 (0.75–4.0) h for the tablet and capsule formulations, respectively. The half-life was similar between the tablet and capsule formulations, with geometric mean t½ values of 33.7 h and 26.9 h, respectively. The geometric means for Cmax were 9670 and 6920 ng/mL for the tablet and capsule formulations, respectively. The AMG 986 concentrations prior to the second single dose on Day 6 were <1% of the mean Cmax concentrations. The geometric means for AUC0–120h, and AUCinf after administration of the AMG 986 tablet were 68,000 ng*h/mL and 69,400 ng*h/mL, respectively; and were 59,900 ng*h/mL and 60,800 ng*h/mL, respectively, after administration of the capsule. The GLSM ratios (90% CI) of capsule/tablet were 0.88 (0.81–0.96) and 0.72 (0.57–0.91) for AUC0–120h and Cmax, respectively (Table 4). Individual PK parameter estimates are reported in Supplementary Table 1 (see electronic supplementary material).

Linear scale (A) and log-linear scale (B) mean (standard deviation) plasma concentration–time profiles after oral administration of 200-mg AMG 986 tablet and capsule formulations (inset = time 0 to 24 h). LLOQ lower limit of quantitation, N number of subjects
3.4 Safety
A total of three subjects (25.0%) had TEAEs. By preferred term, TEAEs were grade 1 dry lip, contusion, and pain in extremity, and grade 2 procedural pain and headache. One subject had grade 2 headache that was considered related to AMG 986 by the investigator; all other TEAEs were considered by the investigator to be unrelated to AMG 986. There were no SAEs, fatal adverse events, or adverse events leading to the discontinuation of treatment. No clinically important changes from baseline in laboratory values, ECG parameters, or vital signs were observed in either group.
4 Discussion
AMG 986 is a first-in-class, novel apelin receptor small molecule initially developed as a treatment for HF [3, 5]. The current, phase I, two-way crossover study was conducted to confirm that the in vivo performance of a capsule formulation of AMG 986 was similar to the tablet formulation in healthy subjects. The results from this study have the potential to inform subsequent phase II studies and beyond in the AMG 986 clinical development program.
As observed in the oral dosing cohorts (tablet) of the FIH study [3], AMG 986 was rapidly absorbed following oral administration of 200 mg, and we observed similar tmax values of 0.75 and 1.0 h for the tablet and capsule formulations, respectively. The AUC0–120h of the AMG 986 capsule and tablet formulations was comparable, with a modest 12% decrease in AUC0–120h observed with the capsule formulation relative to the tablet and the 90% CI for the GLSM ratio (capsule/tablet) contained within the 0.80–1.25 range. Of note, there was a 28% decrease in Cmax with the AMG 986 capsule relative to the tablet, with the 90% CI for the GLSM ratio (capsule/tablet) outside the 0.80–1.25 range. However, the decrease in Cmax with the capsule formulation is not considered to have any clinically relevant ramifications for AMG 986 dose selection. The PK profile of the capsule formulation appears to have a longer distribution phase than the tablet formulation. Although both the formulations are immediate release with similar in vitro release mechanisms, the prolonged absorption in the capsule PK profile as seen in Fig. 4 may be contributed by variability in the in vivo release from the capsule formulation.
In a corroboration of findings from the FIH study [3], AMG 986 had an acceptable safety profile and all adverse events reported during this study were CTCAE grade 1 or 2 in severity. Moreover, no SAEs or fatal adverse events were reported and no trends indicative of clinically important laboratory or vital sign abnormalities were observed in healthy subjects.
There are limitations to the current study. Specifically, the AMG 986 formulations were only tested as a 200-mg dose; thus, future assessment of the capsule formulation at >200-mg dose levels may be warranted. Further, while consistent with the number of subjects enrolled in similar studies, the sample size of the current study was relatively small.
5 Conclusion
There was a modest 12% decrease in AUC0–120h and a 28% decrease in Cmax with the AMG 986 capsule versus the tablet. These differences are not considered to be clinically relevant. AMG 986 had an acceptable safety profile. The results of this study suggest the capsule formulation can be administered in subsequent studies for AMG 986 development.
References
Francis GS. Pathophysiology of chronic heart failure. Am J Med. 2001;110(Suppl 7A):37S–46S.
Jones NR, Roalfe AK, Adoki I, et al. Survival of patients with chronic heart failure in the community: a systematic review and meta-analysis. Eur J Heart Fail. 2019;21(11):1306–25.
Hellawell J, Abbasi S, Trivedi A, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of AMG 986, a novel small molecule apelin receptor agonist, in healthy subjects and heart failure patients. J Cardiac Fail. 2020;26(10):S68.
Ma Z, Song J-J, Martin S, et al. The Elabela-APJ axis: a promising therapeutic target for heart failure. Heart Fail Rev. 2020;2020:1–10.
Ason B, Chen Y, Guo Q, et al. Cardiovascular response to small-molecule APJ activation. JCI Insight. 2020;5(8):e132898.
Trivedi A, Mather O, Vega S, et al. Effect of severe renal impairment on the safety, tolerability, and pharmacokinetics of AMG 986. Drugs R D. 2022;22(1):89–94.
Acknowledgments
The authors would like to thank Robina Smith, Principal Investigator, WCCT Global. Medical writing support, which was in accordance with Good Publication Practice (GPP3) guidelines, was provided by Liam Gillies, PhD, CMPP, of Cactus Life Sciences (part of Cactus Communications), funded by Amgen Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study was funded by Amgen Inc.
Conflicts of interest
AT, Y-HK, RES, SV, JH, and EL are employees and stockholders of Amgen Inc. OM and GCC were employed by, and stockholders of, Amgen Inc. at the time of trial completion.
Availability of data and material
Qualified researchers may request data from Amgen clinical studies; complete details are available at http://www.amgen.com/datasharing.
Code availability
Not applicable.
Author contributions
All authors provided substantial contributions to the interpretation of data for the work, drafted and revised the manuscript critically for important intellectual content, and approved the final version to be published.
Ethics approval
The study was conducted at WCCT Global (Cypress, CA 90630, USA) following ethical guidelines of the Declaration of Helsinki and Council for International Organizations of Medical Sciences, applicable Good Clinical Practice guidelines of the International Council for Harmonization, and applicable local laws and regulations. The locally appointed ethics review board (IRBCo, Buena Park, CA 90621, USA) approved the research protocol.
Consent to participate
Written informed consent was obtained from all subjects.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Trivedi, A., Kiang, YH., Saw, R.E. et al. Evaluation of the Pharmacokinetics and Safety of AMG 986 Tablet and Capsule Formulations in Healthy Adult Subjects: A Phase I, Open-Label, Randomized Study. Drugs R D 22, 147–154 (2022). https://doi.org/10.1007/s40268-022-00388-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-022-00388-1