
Abstract
Background and Objective
Upacicalcet sodium hydrate is a novel small-molecule calcimimetic and has potential as a therapeutic agent for secondary hyperparathyroidism. We assessed the pharmacokinetics, pharmacodynamics, safety, and tolerability of a single intravenous dose of upacicalcet in Japanese healthy adults.
Method
This was a single-center, double-blinded, randomized, placebo-controlled, dose-escalation study. For each cohort, eight subjects were randomly assigned at a ratio of 3:1 to receive a single injection of placebo or upacicalcet 0.01, 0.1, 1.0, or 2.5 mg.
Result
The plasma concentration of upacicalcet increased in a dose-dependent manner. Upacicalcet rapidly disappeared from plasma after administration. The half-life of upacicalcet was approximately 1–2 h. The major excretion route of upacicalcet was via urine. Serum intact parathyroid hormone decreased in accordance with the upacicalcet dose, from the lowest dose of 0.01 mg. Gastrointestinal disorders occurred in one patient in the 1.0 mg group and in five patients in the 2.5 mg group. All adverse events were nonserious, and no symptomatic hypocalcemia occurred.
Conclusion
This study showed that upacicalcet acted as a calcimimetic and was excreted in the urine unchanged with little metabolism. Moreover, upacicalcet is a small molecule and has a small volume of distribution. In addition, less than 50% of upacicalcet binds to human plasma proteins. These findings suggest that upacicalcet administered to patients undergoing hemodialysis might be expected to have a long excretion period and sustained pharmacological effect.
Similar content being viewed by others

This study demonstrated upacicalcet was well-tolerated in healthy Japanese participants. |
The dose linearity of upacicalcet pharmacokinetics was confirmed in this study. |
Upacicalcet was almost unmetabolized and rapidly excreted in the urine when administered to healthy adults. |
1 Introduction
Secondary hyperparathyroidism (SHPT) is a major pathological condition in patients with chronic kidney disease (CKD)–mineral and bone disorder [1,2,3]. As CKD progresses, phosphorus excretion by the kidney is suppressed and calcium absorption from the intestinal tract is reduced because of reduced vitamin D activation. All these factors increase parathyroid hormone (PTH) secretion from the parathyroid glands [1, 4]. The abnormal increase in PTH secretion accelerates bone turnover, causing bone lesions such as osteitis fibrosa and promotes vascular calcification, resulting in cardiovascular disorders [5]. Thus, SHPT is one of the major factors in the reduced quality of life and increased mortality in patients with end-stage CKD [1, 6].
Vitamin D receptor activators (VDRAs) were the first medical treatment for SHPT. Active VDRAs decrease PTH levels but increase serum calcium and phosphorus levels, resulting in calcium and phosphorus overload [7, 8]. To resolve these adverse effects, calcimimetics, which act as allosteric modulators of calcium-sensing receptors (CaSRs) on the parathyroid gland, have been developed. Calcimimetics decrease PTH levels without increasing serum calcium and phosphorus levels [9,10,11,12]. Further, the combination of VDRAs and calcimimetics has been reported to improve the control of PTH, calcium, and phosphorus [12,13,14].
However, cinacalcet, the first-in-class calcimimetic agent, has been reported to exhibit several clinical issues (e.g., hypocalcemia and gastrointestinal disorders, including nausea and vomiting [15,16,17,18]), leading to low patient adherence [19], and drug–drug interactions because it inhibits cytochrome P450 (CYP)-2D6 and is a substrate for multiple CYP enzymes, primarily CYP3A4, CYP2D6, and CYP1A2 [20]. To resolve these issues, new calcimimetics have been developed: etelcalcetide is an injectable drug composed of d-amino acid peptides [21], and evocalcet is an oral drug with fewer gastrointestinal side effects than cinacalcet [15, 22].
Upacicalcet sodium hydrate (AJT-240/SK-1403) is a novel small-molecule calcimimetic agent containing an L-amino acid structure. It is designed as an injectable drug to be administered via a hemodialysis circuit to avoid gastrointestinal side effects and improve patient adherence. Results from nonclinical studies revealed the following properties of upacicalcet, an allosteric modulator of human CaSR: the ability to decrease serum intact PTH (iPTH) levels in animal models of SHPT in a dose-dependent manner; minimal metabolism by the liver; little inhibition or induction of major drug-metabolizing enzymes such as CYP; noncovalent binding to human plasma proteins to the extent of 44.2–45.6%; little inhibitory effects on or substrate properties for major drug transporters; and little binding activity for 60 receptors, ion channels, and transporters, excluding CaSR.
Here, we report the results of the first-in-human study of upacicalcet. In this study, we evaluated the pharmacokinetics, pharmacodynamics, and safety of upacicalcet in healthy Japanese adults.
2 Methods
2.1 Study Design
This study was a phase I, single-center, double-blinded, randomized, placebo-controlled, dose-escalation study. The primary objective was to assess the safety and tolerability of upacicalcet. In the first dose cohort, eight eligible participants were enrolled and randomly assigned to receive upacicalcet (n = 6) or placebo (n = 2). At the end of the cohort study, the investigator determined whether it was safe to start the next cohort study, after which the second dose cohort was initiated. Each subsequent cohort underwent the same procedure. Participants who participated in one cohort were not allowed to participate in another cohort.
To ensure safety before starting the next dose cohort, the blinded data were opened and assessed to determine whether any of the following had occurred: (1) moderate or severe adverse events (AEs) in which a causal relationship to the study drug in three or more participants could not be ruled out; (2) serious AEs in which a causal relationship to the study drug in any subject could not be ruled out; (3) corrected serum calcium level < 8.0 mg/dL in two or more participants; (4) corrected serum calcium level < 7.0 mg/dL and presence of symptoms associated with low serum calcium (tetany, arrhythmia, psychiatric symptoms) in any subject; and (5) Fridericia-corrected QT interval (QTcF) > 500 ms or QTcF increase > 60 ms compared with baseline (before treatment) in any subject. The enrollment and assignment of participants in this study are shown in Fig. 1.

Enrollment, assignment of subject of this study. Study outline for the dose escalation and expansion. After the previous cohort step was completed and the investigator judged, the next cohort step was started. Participants who participated in one cohort were not allowed to participate in another cohort. At dose cohort 2, adverse events of vomiting, nausea were observed. Although these adverse events did not violate the safety criteria, higher doses of upacicalcet were expected to increase the incidence of side effects due to overreaction. Therefore, the study protocol was amended to confirm the safety, pharmacokinetics, and pharmacodynamics of the lower dose of upacicalcet
2.2 Inclusion and Exclusion Criteria and Randomization
Participants who met all the inclusion criteria and did not meet any exclusion criteria at the screening test were enrolled in the study. The inclusion criteria were as follows: healthy male Japanese adults; aged 20–40 years; body weight 50–80 kg; body mass index 18.5–25.0 kg/m2; and able to provide written informed consent. The main exclusion criteria were as follows: serum albumin-corrected calcium (cCa) < 8.5 mg/dL; estimated glomerular filtration rate < 90 mL/min/1.73 m2; QTcF > 450 ms; serum iPTH < 10 pg/mL; dependence on alcohol or drugs; allergy or hypersensitivity to drugs or food; smoking habit of more than ten cigarettes/day; alcohol consumption of > 20 g/day; intake of any medication or supplements within 2 weeks prior to study drug administration; unable to consent to contraceptive use during the study duration; positive for HIV antigen/antibody, anti-hepatitis B antibody, hepatitis C virus antigen, or syphilis; or judged to be clinically inappropriate by the investigators or subinvestigators. The participants were randomly assigned according to an assignment form prepared by the external organization for case registration (EPS Corporation, Tokyo, Japan).
2.3 Interventions
For any dose cohort, participants who were eligible for the screening test were hospitalized 1 day before study drug administration; this day was termed as day −1. Upacicalcet or placebo was administered intravenously once on day 1. The starting dosage of upacicalcet for this first-in-human clinical trial was determined using the following procedure. The no-observed-adverse-effect level (NOAEL) of upacicalcet in preclinical 4-week repeated-dose studies was 10 mg/kg in rats and 3 mg/kg in dogs. The human equivalent dose (HED), calculated by converting the NOAEL to body surface area, was calculated as 1.61 mg/kg in rats and 1.67 mg/kg in dogs. Based on the HED calculated from the NOAEL in rats, the maximum recommended starting dose (MRSD) was 0.161 mg/kg with a safety factor of 10. Based on these calculations, the starting dosage of upacicalcet was set at 1.0 mg, assuming that a dose one-tenth of the MRSD was administered to a human weighing approximately 60 kg. The planned dosages of upacicalcet or placebo were 1.0, 2.5, 5, 10, 20, 30, and 40 mg for each cohort. Changes in dosing regimens during the study meant that two cohorts—the 0.01 and 0.1 mg cohorts—were added, whereas the cohorts receiving doses of ≥5 mg were discontinued (as described in Sect. 3 and in Fig. 1). The participants underwent medical examinations and tests for up to 48 h after administration and were discharged on day 3 when the investigator or subinvestigator judged that there were no clinical abnormalities. The participants returned to the hospital on day 7 for final observations, during which observation of the participants was completed if no abnormalities were found.
2.4 Pharmacokinetic Evaluations
To determine the plasma concentrations of upacicalcet and its predominant metabolites (M1, M2, and M3), blood samples were collected before administration and at 5, 10, 20, 30 min and 1, 2, 4, 6, 8, 12, 24, and 48 h after administration. To evaluate the urinary excretion rate of upacicalcet and its metabolites, pooled urine samples were collected from immediately after administration to 6, 6–12, 12–24, and 24–48 h after administration.
Blood samples were collected in a vacuum blood collection tube containing sodium heparin 3 mL (Terumo Co., Tokyo, Japan) at each time point. The collected blood was immediately centrifuged (4 °C, 3000 rpm, 10 min), and plasma was separated into two 0.5-mL bottles. The urine volume was measured, and two samples of 0.5 mL each were taken from the urine samples at each time point.
Both plasma and urine samples were frozen below − 20 °C for storage and transported to a drug concentration assay laboratory (Shin Nippon Biomedical Laboratories, Ltd., Tokyo, Japan). The concentrations of upacicalcet and its metabolites (M1, M2, and M3) in plasma and urine were measured using liquid chromatography–tandem mass spectrometry. Thereafter, the pharmacokinetic parameters (maximum plasma drug concentration [Cmax], time to Cmax, area under the plasma concentration–time curve [AUC]0-t, AUCinf, elimination half-life [t½], mean residence time [MRT]0-t, MRT0-∞, plasma clearance [CLp], volume of distribution at steady [Vdss] state, and urinary excretion rate) of upacicalcet were calculated.
2.5 Pharmacodynamic Evaluations
As upacicalcet is a calcimimetic developed for the treatment of SHPT, the pharmacodynamic parameters determined were serum iPTH, cCa, and phosphorus levels. Blood samples for determining plasma levels of iPTH, cCa, and phosphorus were collected before administration and 10 and 30 min and 1, 2, 4, 6, 8, 12, 24, and 48 h after administration. To determine the urinary excretion of calcium and phosphorus, urine samples were collected from immediately after administration to 6, 6–12, 12–24, and 24–48 h. Pharmacodynamic samples were stored below 10 °C or below −20 °C and assayed by an outsourced laboratory (LSI Medience Corporation, Tokyo, Japan).
2.6 Safety evaluations
AEs were recorded from the start of study drug administration to the last observation. AEs judged to be related to the investigational drug were defined as adverse drug reactions (ADRs). AEs and ADRs were classified according to the Japanese Medical Dictionary for Regulatory Activities system organ class and preferred term (version 17.1).
Laboratory tests were performed at the screening tests on days −1, 2, 3, and 7. Twelve-lead electrocardiograms (ECGs) were performed at the screening tests on day −1, day 1 (just before administration and 20 min and 2, 4, 8, and 12 h after administration), day 2, day 3, and day 7. Vital signs, such as systolic and diastolic blood pressure, heart rate, and body temperature, were measured during the screening tests on day −1, day 1 (just before administration and 1 h after administration), day 2, day 3, and day 7.
2.7 Statistical Analysis
Data from all randomized participants were included in the analysis. Data from the participants assigned to the placebo groups in each cohort were combined into the placebo dose group.
The pharmacokinetic parameters were calculated using noncompartmental methods with WinNonlin version 6.1 (Pharsight Corp, Mountain View, CA, USA). SAS version 9.3 (SAS Institute, Cary, NC, USA) was used to perform other statistical analyses and summarize the data.
3 Results
3.1 Study Population
In dose cohort 2, vomiting and nausea were observed with the administration of upacicalcet 2.5 mg. Although these AEs were mild and nonserious, further increases in the dosage of upacicalcet were considered likely to increase the incidence of adverse effects and have poor tolerability. Thus, the study protocol was amended to confirm the safety, pharmacokinetics, and pharmacodynamics of upacicalcet at doses lower than 1.0 mg (shown in Fig. 1).
Data from all randomized participants were included in the safety, pharmacokinetic, and pharmacodynamic analyses. The characteristics of participants in each group were similar (Table 1). All participants were appropriately assigned and completed the study.
3.2 Pharmacokinetic Findings
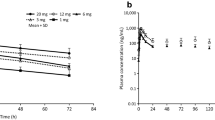
Figure 2 shows the mean plasma concentrations of upacicalcet and its metabolites over time. The mean t½ was 1.05–2.06 h. Further, 78.9–95.0% of upacicalcet was excreted in the urine within 48 h. The only metabolite present in the blood was M2, which was found in the 1.0 mg and 2.5 mg dose groups. M2 in all urine samples was less than the lower limit of quantification. Plasma concentrations of upacicalcet increased with increasing dose from a Cmax of 1.08 ng/mL in the 0.01 mg group to a Cmax of 280 ng/mL in the 2.5 mg dose group. The Cmax and AUCinf values of upacicalcet increased in a dose-dependent manner (Fig. 2A and Table 2). Table 3 shows the results of the dose-proportionality assessment for Cmax and AUCinf based on the power model.

Plasma concentration-time profile of upacicalcet (A) and M2 (B). Values are mean + standard deviation (N = 6 in each dose)
3.3 Pharmacodynamic Findings
3.3.1 Serum Intact Parathyroid Hormone
Serum iPTH levels in the upacicalcet group decreased within 10 min of drug administration (Fig. 3A). The lowest serum iPTH levels observed after administration in the placebo group and the 0.01, 0.1, 1.0, and 2.5 mg groups were 36.6 pg/mL at 2 h, 18.2 pg/mL at 10 min, 10.8 pg/mL at 30 min, 8.2 pg/mL at 1 h, and 9.3 pg/mL at 1 h, respectively. In the upacicalcet group, the duration of lowering of iPTH levels tended to increase with increasing dose but disappeared after 12 h of administration.

Mean serum intact PTH (A), corrected calcium (B), and phosphorus (C) time course after administration (n = 6)
3.3.2 Serum Albumin-Corrected Calcium
Serum cCa levels in the upacicalcet group decreased gradually 6–12 h after administration (Fig. 3B). The lowest serum cCa levels observed after administration in the placebo and 0.01, 0.1, 1.0, and 2.5 mg groups were 8.85 mg/dL at 12 h, 8.80 mg/dL at 12 h, 8.72 mg/dL at 6 and 8 h, 8.50 mg/dL at 8 h, and 8.32 mg/dL at 12 h, respectively. After 24 h of administration, this effect of lowering serum cCa levels disappeared.
3.3.3 Serum Phosphorus
There was no clear difference in serum phosphorus levels between the upacicalcet and placebo groups (Fig. 3C). The lowest serum phosphorus levels observed after administration in the placebo and 0.01, 0.1, 1.0, and 2.5 mg groups were 2.96 mg/dL at 6 h, 3.07 mg/dL at 6 h, 2.90 mg/dL at 6 h, 2.78 mg/dL at 6 h, and 2.73 mg/dL at 6 h, respectively.
3.3.4 Urinary Excretion of Calcium and Phosphorus
The urinary calcium excretion levels increased 0–6 h after administration in a dose-dependent manner (Fig. 4A). However, after 6–12 h, the degree of increase in urinary calcium excretion decreased in the upacicalcet group (Fig. 4B).

Urinary excretion of calcium (A: 0–6h after administration, B: 6–12 h after administration) and phosphorus (C: 0–6 h after administration, D: 6–12 h after administration). Values are mean + standard deviation (N = 6)
The urinary phosphorus excretion levels decreased 0–6 h after administration in the 1.0 mg and 2.5 mg upacicalcet groups (Fig. 4C). Moreover, these decreases in urinary phosphorus excretion persisted even after 6–12 h of administration (Fig. 4D).
3.3.5 Safety Analysis
Table 4 lists all reported AEs. No AEs occurred in participants in the placebo and upacicalcet 0.01 mg groups. Gastrointestinal disorders were observed in one patient and five participants in the 1.0 and 2.5 mg groups, respectively. All AEs were nonserious and mild or moderate in severity. No abnormalities were detected in laboratory tests, vital signs, or ECG findings that were reported as ADRs. The QTcF values obtained from the 12-lead ECG results are shown in Fig. 5. Although QTcF tended to be prolonged by upacicalcet administration, QTcF values correlated more with serum cCa than with plasma upacicalcet concentration (Fig. 6).

Mean QTcF time course after administration (n = 6)

Correlation between QTcF and plasma upacicalcet concentration (A) or serum calcium (B). A; Y = 411.3 + 0.003*X, X: plasma upacicalcet concentration (ng/mL), Y: QTcF (ms), correlation coefficient = 0.006. B in placebo group; Y=413.8 − 1.5*X, X: serum corrected calcium (mg/dL), Y: QTcF (ms), correlation coefficient = − 0.023. B in upacicalcet groups; Y=544.9 − 14.7*X, X: serum corrected calcium (mg/dL), Y: QTcF (ms), correlation coefficient = − 0.341
4 Discussion
This study demonstrates the pharmacokinetic, pharmacodynamic, and safety parameters of upacicalcet, a new calcimimetic agent. The dose linearity of upacicalcet pharmacokinetics was confirmed in this study. Further, upacicalcet was almost unmetabolized and rapidly excreted in the urine when administered to healthy adults. The small distribution volume of upacicalcet may have contributed to its rapid excretion when administered to healthy participants. The molecular weight of upacicalcet sodium is 373.75. Moreover, our previous studies have confirmed that upacicalcet is barely a substrate for liver metabolism and only has about a 45% binding rate to plasma proteins. Therefore, when upacicalcet was administered to patients with end-stage renal disease undergoing dialysis, plasma concentration of upacicalcet was expected to be maintained until the next dialysis and to be eliminated thereafter by dialysis. In addition, upacicalcet may be removed by dialysis and not accumulate in the body. This hypothesis should be confirmed by pharmacokinetic studies in patients undergoing dialysis.
Administration of upacicalcet decreased serum iPTH level rapidly, and the duration of this decrease was dose dependent. Changes in serum iPTH levels were followed by a decrease in serum calcium levels. Further, the highest decrease in calcium levels was observed approximately 6–12 h after upacicalcet administration. This implied that upacicalcet exerted its pharmacological effects as a calcimimetic in a drug concentration-dependent manner. In contrast, serum phosphorus was not significantly altered by upacicalcet administration. PTH regulates serum calcium in humans, and the bones and kidneys are its main targets of action. PTH triggers a positive flow of calcium and phosphorus from the bone to serum by promoting bone resorption [1, 4]. The renal action of PTH is to promote calcium reabsorption in the renal tubules and inhibit phosphorus reabsorption [23]. The decrease in serum PTH caused by upacicalcet may affect the bones and kidneys, resulting in contrasting blood dynamics of calcium and phosphorus. The study results related to urinary excretion of calcium and phosphorus support this notion. In patients undergoing dialysis who have almost no renal function, the renal effects of PTH are negligible. Therefore, the administration of upacicalcet to such patients undergoing dialysis is not expected to promote an increase in serum phosphorus via inhibition of urinary phosphorus excretion.
In this study, no major safety problems associated with upacicalcet were identified. However, upacicalcet dosing > 1.0 mg could cause side effects such as nausea and vomiting, as observed with other calcimimetics [15, 24], and this would make the drug less tolerable. On the other hand, pharmacodynamic results showed that upacicalcet tended to decrease serum iPTH levels, even at a dose of 0.01 mg. Therefore, the effective and well-tolerated dose range of upacicalcet might be wide.
Determination of the optimal dose range of upacicalcet is a topic of interest for the next study. Further, the results of this study could imply that a dose range of upacicalcet between 0.01 and 1.0 mg is effective, safe, and well tolerated. However, within this specified dose range, the study considered only upacicalcet 0.1 mg in addition to these two doses. Therefore, more detailed dose-finding studies are needed to clarify the optimal dose range of upacicalcet. This is one of the limitations of the present study.
Another limitation is that this study confirmed pharmacokinetics in only Japanese people. Upacicalcet is mainly excreted renally in an unchanged form. In addition, nonclinical studies have indicated that upacicalcet is not a substrate for drug-metabolizing enzymes or transporters in the liver. However, further studies are needed to determine whether racial differences affect the pharmacokinetics of upacicalcet.
5 Conclusion
This study showed that upacicalcet is well tolerated in healthy Japanese participants. Upacicalcet is a calcimimetic agent that is excreted renally; therefore, when used after a dialysis session in patients with SHPT, it is expected to maintain its effect until the next dialysis. Furthermore, upacicalcet may not accumulate and may be easily removed by dialysis should adverse effects occur. The efficacy and safety of upacicalcet, as a new calcimimetic agent, should be assessed in further clinical studies.
References
Cunningham J, Locatelli F, Rodriguez M. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011;6(4):913–21. https://doi.org/10.2215/CJN.06040710.
Moorthi RN, Moe SM. CKD–mineral and bone disorder: core curriculum 2011. Am J Kidney Dis. 2011;58(6):1022–36. https://doi.org/10.1053/j.ajkd.2011.08.009.
Komaba H, Kakuta T, Fukagawa M. Diseases of the parathyroid gland in chronic kidney disease. Clin Exp Nephrol. 2011;15(6):797–809. https://doi.org/10.1007/s10157-011-0502-5.
Fraser WD. Hyperparathyroidism. Lancet. 2009;374(9684):145–58. https://doi.org/10.1016/S0140-6736(09)60507-9.
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15(8):2208–18. https://doi.org/10.1097/01.ASN.0000133041.27682.A2.
Fernández M, Morales E, Gutierrez E, Polanco N, Hernández E, Mérida E, et al. Calciphylaxis: beyond CKD–MBD. Nefrologia. 2017;37(5):501–7. https://doi.org/10.1016/j.nefro.2017.02.006.
Kandula P, Dobre M, Schold JD, Schreiber MJ Jr, Mehrotra R, Navaneethan SD. Vitamin D supplementation in chronic kidney disease: a systematic review and meta-analysis of observational studies and randomized controlled trials. Clin J Am Soc Nephrol. 2011;6(1):50–62. https://doi.org/10.2215/CJN.03940510.
Goodman WG. Recent developments in the management of secondary hyperparathyroidism. Kidney Int. 2001;59(3):1187–201. https://doi.org/10.1046/j.1523-1755.2001.0590031187.x.
Chertow GM, Block GA, Correa-Rotter R, Drüeke TB, Floege J, EVOLVE Trial Investigators, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367(26):2482–94. https://doi.org/10.1056/NEJMoa1205624.
Fukagawa M, Fukuma S, Onishi Y, Yamaguchi T, Hasegawa T, Akizawa T, et al. Prescription patterns and mineral metabolism abnormalities in the cinacalcet era: results from the MBD-5D study. Clin J Am Soc Nephrol. 2012;7(9):1473–80. https://doi.org/10.2215/CJN.13081211.
Wetmore JB, Gurevich K, Sprague S, Da Roza G, Buerkert J, Reiner M, et al. Randomized Trial of Cinacalcet versus Vitamin D Analogs as Monotherapy in Secondary Hyperparathyroidism (PARADIGM). Clin J Am Soc Nephrol. 2015;10(6):1031–40. https://doi.org/10.2215/CJN.07050714.
Komaba H, Fukagawa M, KRN1493 Study Group. Impact of cinacalcet hydrochloride on the achievement of the Japanese Society for Dialysis Therapy (JSDT) guideline targets: a post-hoc analysis of the KRN1493 study. Ther Apher Dial. 2008;12(1):44–9. https://doi.org/10.1111/j.1744-9987.2008.00631.x.
Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K, et al. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transpl. 2011;26(4):1327–39. https://doi.org/10.1093/ndt/gfq725.
Fishbane S, Shapiro WB, Corry DB, Vicks SL, Roppolo M, Rappaport K, et al. Cinacalcet HCl and concurrent low-dose vitamin D improves treatment of secondary hyperparathyroidism in dialysis patients compared with vitamin D alone: the ACHIEVE study results. Clin J Am Soc Nephrol. 2008;3(6):1718–25. https://doi.org/10.2215/CJN.01040308.
Fukagawa M, Shimazaki R, Akizawa T, Evocalcet study group. Head-to-head comparison of the new calcimimetic agent evocalcet with cinacalcet in Japanese hemodialysis patients with secondary hyperparathyroidism. Kidney Int. 2018;94(4):818–25. https://doi.org/10.1016/j.kint.2018.05.013.
Block GA, Martin KJ, de Francisco AL, Turner SA, Avram MM, Suranyi MG, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350(15):1516–25. https://doi.org/10.1056/NEJMoa031633.
Fukagawa M, Yumita S, Akizawa T, Uchida E, Tsukamoto Y, Iwasaki M, et al. Cinacalcet (KRN1493) effectively decreases the serum intact PTH level with favorable control of the serum phosphorus and calcium levels in Japanese dialysis patients. Nephrol Dial Transpl. 2008;23(1):328–35. https://doi.org/10.1093/ndt/gfm534.
Palmer SC, Nistor I, Craig JC, Pellegrini F, Messa P, Tonelli M, et al. Cinacalcet in patients with chronic kidney disease: a cumulative meta-analysis of randomized controlled trials. PloS Med. 2013;10(4):e1001436. https://doi.org/10.1371/journal.pmed.1001436.
Y. Gincherman, K. Moloney, C. McKee, Coyne DW. Assessment of adherence to cinacalcet by prescription refill rates in hemodialysis patients. Hemodial Int. 2010; 14(1); 68–72. https://doi.org/10.1111/j.1542-4758.2009.00397.x
Padhi D, Harris R. Clinical pharmacokinetic and pharmacodynamic profile of cinacalcet hydrochloride. Clin Pharmacokinet. 2009;48(5):303–11. https://doi.org/10.2165/00003088-200948050-00002.
Eidman KE, Wetmore JB. Treatment of secondary hyperparathyroidism: how do cinacalcet and etelcalcetide differ? Semin Dial. 2018;31(5):440–4. https://doi.org/10.1111/sdi.12734.
Akizawa T, Ikejiri K, Kondo Y, Endo Y, Fukagawa M. Evocalcet: a new oral calcimimetic for dialysis patients with secondary hyperparathyroidism. Ther Apher Dial. 2019;24(3):248–57. https://doi.org/10.1111/1744-9987.13434.
Segawa H, Yamanaka S, Onitsuka A, Tomoe Y, Kuwahata M, Ito M, et al. Parathyroid hormone-dependent endocytosis of renal type Iic Na-Pi cotransporter. Am J Physiol Renal Physiol. 2007;292(1):F395-403. https://doi.org/10.1152/ajprenal.00100.2006.
Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA. 2017;317(2):156–64. https://doi.org/10.1001/jama.2016.19468.
Acknowledgements
Sanwa Kagaku Kenkyusho Co., Ltd. (SKK) was involved in the collection, management, analysis, and interpretation of the study data. The authors thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
SKK sponsored this study. SKK paid the Open Access fee for this article.
Conflict of interest
Fumihiko Koiwa and Masafumi Fukagawa received honoraria from SKK for consulting and lectures. Daisuke Honda is an employee of SKK. Tadao Akizawa received consulting fees from SKK.
Ethics approval
The study was conducted at SOUSEIKAI Sumida Hospital, Tokyo, Japan. All the study protocols and informed consent documents were reviewed and approved by the Japanese Pharmaceuticals and Medical Devices Agency and the investigation review board of the Hakata Clinic. This study was conducted in compliance with the Helsinki Declaration and comparable ethical standards, Good Clinical Practice guidelines, and other regulatory requirements.
Availability of data material
The dataset from this study is not available in any open data repository.
Code availability
Not applicable.
Consent to participate
All participants provided written informed consent before participating in the study.
Consent for publication
Not applicable.
Author contributions
Fumihiko Koiwa, Rie Yazawa, and Tadao Akizawa substantially contributed to the study design and the acquisition and interpretation of the data. Masafumi Fukagawa and Daisuke Honda substantially contributed to the analysis and interpretation of data. All authors critically revised the report, commented on drafts of the manuscript, and approved the final report.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Koiwa, F., Yazawa, R., Fukagawa, M. et al. First-in-Human Phase I Study of the Novel Injectable Calcimimetic Agent Upacicalcet in Healthy Adult Japanese Participants. Drugs R D 22, 131–140 (2022). https://doi.org/10.1007/s40268-022-00385-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-022-00385-4