
Abstract
Inflammation plays an important role in the response to danger signals arising from damage to our body and in restoring homeostasis. Dysregulated inflammatory responses occur in many diseases, including cancer, sepsis and autoimmunity. The efficacy of anti-inflammatory drugs, developed for the treatment of dysregulated inflammation, can be potentiated using biomaterials, by improving the bioavailability of drugs and by reducing side effects. In this Review, we first outline key elements and stages of the inflammatory environment and then discuss the design of biomaterials for different anti-inflammatory therapeutic strategies. Biomaterials can be engineered to scavenge danger signals, such as reactive oxygen and nitrogen species and cell-free DNA, in the early stages of inflammation. Materials can also be designed to prevent adhesive interactions of leukocytes and endothelial cells that initiate inflammatory responses. Furthermore, nanoscale platforms can deliver anti-inflammatory agents to inflammation sites. We conclude by discussing the challenges and opportunities for biomaterial innovations in addressing inflammation.
Similar content being viewed by others
Introduction
Inflammation is the body’s natural and essential response to signals arising from tissue damage or pathogenic infection1. Following trauma or infection, inflammation drives the restoration of homeostasis by protecting the host from exogenous pathogens and by repairing damaged tissue2. Inflammation usually occurs as a sequence of events, starting with a rapid induction phase, which leads to a pro-inflammatory response, gradually followed by a resolution phase3. Therefore, inflammation is crucial to wound healing. However, if this well-orchestrated response is dysregulated, inflammation may become uncontrolled or chronic, which can ultimately lead to the development and progression of various inflammatory diseases, such as cancer, obesity, sepsis, cardiovascular, neuronal and autoimmune diseases4.
Dysregulated chronic or local inflammation occurs in several stages. In the early stage, danger signals, such as damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs), which are released from damaged cells or pathogens, activate resident immune cells to generate cytokines, chemokines, proteases, growth factors and oxygen-free radicals5. Circulating immune cells then home to the site of the inflamed tissue through endothelial cell adhesion to boost pro-inflammatory responses6. This coordinated immune response ultimately restores homeostasis. However, if the response is insufficient or excessive, upsetting the balance between the innate and adaptive arms of the immune system, it can lead to late-stage inflammation. Such a dysregulated immune response can cause a catastrophic cascade, characterized by local or systemic tissue damage and excessive danger signal production. Shifts in the inflammatory response from acute to chronic can also cause a breakdown of immune tolerance, leading to the progression of inflammatory disease, which may even result in death7.
The connection between inflammation and homeostasis is increasingly understood, and a holistic consideration of the inflammatory process offers opportunities for the design of specific and efficacious therapeutic strategies to target and regulate inflammation8. Drug therapy is commonly used to treat inflammation, and biomaterials can be applied as drug carrier to enable controlled drug delivery and release with high efficacy and minimal side effects. In addition to drug delivery, biomaterials can also scavenge pro-inflammatory factors or block undesired leukocyte–endothelial cell interactions to inhibit inflammation8. Biomaterials can, thus, complement conventional drug therapies, offering design versatility, targeting of different pathways and high spatiotemporal control of anti-inflammatory activities.
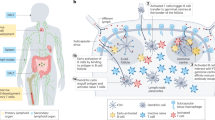
In this Review, we discuss three major biomaterials-based strategies for modulating the different stages of dysregulated inflammation (Fig. 1); biomaterials as scavengers of danger signals in early inflammation; biomaterials as inhibitors of leukocyte–endothelial cell adhesion in the middle stages of inflammation; and biomaterials as delivery systems for anti-inflammatory agents in the middle and late stages of inflammation. Finally, we highlight future directions and challenges for the clinical translation of anti-inflammatory biomaterials.

Biomaterials can be applied to control sterile inflammation in the early stage (scavenging strategy), middle stage (blockage strategy) and late stage (delivery strategy). cfNA, cell-free nucleic acid; DAMP, damage-associated molecular pattern; EPR, enhanced permeability and retention; RONS, reactive oxygen and nitrogen species.
The inflammatory environment
Danger signals
PAMPs released by bacteria and viruses trigger inflammation in infection; DAMPs are the endogenous counterpart of PAMPs, inducing sterile inflammation (that is, inflammation owing to trauma rather than infection)9. DAMPs and PAMPs are recognized by different innate immune pattern recognition receptors, such as Toll-like receptors (TLRs), which are expressed on immune cells9,10. Upon binding extracellular DAMPs and PAMPs, TLRs activate cytoplasmic adapter molecules that initiate a cascade of activation pathways, including nuclear factor-κB (NF-κB), interferon regulatory factor and a link to MAPK pathways, leading to the production of pro-inflammatory cytokines (for example, tumour necrosis factor (TNF), interleukin-1 (IL-1) and IL-6) and chemokines through transcriptional and post-transcriptional mechanisms9,10. Furthermore, reactive oxygen and nitrogen species (RONS) can activate IκB kinases and/or inhibit phosphotyrosine and phosphoserine/threonine phosphatases to upregulate redox-sensitive NF-κB, further exacerbating inflammation11. In addition to TLRs, immune cells possess NOD-like receptors (NLRs) to specifically identify pathogenic patterns in the cytoplasm, resulting in an inflammasome-mediated activation of pro-inflammatory cytokine release2. An auxiliary mechanism in dealing with inflammation involves cyclic GMP–AMP synthase and its downstream effector, stimulator of interferon genes, which recognizes intracellular DNAs and promotes the release of type I interferons and other inflammatory cytokines12. Therefore, immune cells can recognize and respond to various danger signals in the initial stage of inflammation and transmit signals to the nucleus for the production of cytokines.
Inflammatory cells
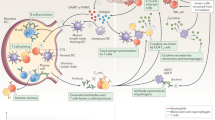
Innate immune cells, that is, monocytes, macrophages and neutrophils, orchestrate early inflammation13,14 (Fig. 2). Within minutes of an injury, tissue-resident macrophages and circulating neutrophils are activated by DAMPs and release inflammatory mediators that recruit circulating innate immune cells to the injury site15. Additional mechanosignalling is triggered by direct interaction between surface molecules in adjacent cells16,17. In response to macrophage-produced cytokines and PAMPs or DAMPs, released by injured or invading cells, the postcapillary venular endothelium upregulates adhesion molecules involved in the leukocyte recruitment cascade18,19. This cascade involves mechanosignalling between adhesion molecules on the endothelium and various integrins on leukocytes, which can direct the recruitment of circulating leukocytes, eventually leading to their extravasation. Neutrophils are the first immune cells to arrive at the affected site16,17,18,19, and intravascular neutrophils adhered to the endothelium can modify the endothelial barrier20. In response to chemoattractants, neutrophils release TNF in close proximity to endothelial junctions to locally increase microvascular permeability21,22. Circulating neutrophils migrate to the site as early as 20 min after injury and accumulate for the next 2 h (ref.23). Circulating monocytes subsequently infiltrate the site, 24 h after injury, and increase in number for up to 72 h post-injury24. These infiltrating monocytes produce inflammatory mediators, clear dead cells, stimulate extracellular matrix production and angiogenesis, and regenerate parenchymal cells15. In early inflammation, CD4+ T helper 1 (TH1) cells infiltrate the injury site and release pro-inflammatory cytokines24,25. In late inflammation, regulatory T (Treg) cells and TH2 cells, along with regulatory M2-like macrophages, increase in number and resolve inflammation by producing anti-inflammatory cytokines, such as transforming growth factor-β (TGFβ) and IL-10 (refs26,27).

The inflammatory microenvironment comprises invasive pathogens, damaged cells and vasculature, infiltrating immune cells, danger signals, such as pathogen-associated molecular patterns and endogenous tissue damage-associated molecular patterns, and a plethora of pro-inflammatory molecules, such as cytokines, chemokines, enzymes, leukotrienes and eicosanoids. In particular, reactive oxygen and nitrogen species (RONS) can activate Toll-like-receptor-mediated nuclear factor-κB (NF-κB) and interferon regulatory factor pathways during inflammation. Localized inflammation induces the activation of microvascular endothelial cells, causing changes in vascular permeability to promote leukocyte homing, such as neutrophil adhesion and transmigration, as well as activation of platelets and monocytes. Nanoparticles can accumulate in the inflammatory microenvironment owing to the enhanced permeability and retention (EPR) effect, transcytosis or nanomaterials-induced endothelial leakiness (NanoEL). COX2, cyclooxygenase 2; HMGB1, high-mobility group box 1; IL-1β, interleukin-1β; IL-6, interleukin-6; PGE2, prostaglandin E2; TNF, tumour necrosis factor.
Inflammatory mediators
Activated inflammatory cells release acute mediators of inflammation28, such as the pro-inflammatory cytokines TNF, IL-1β and IL-6, which have profound effects on tissue regeneration, response to infection or pain, and neuronal activity28,29 (Fig. 2). When dysregulated, these pro-inflammatory cytokines can contribute to the pathogenesis of diseases, such as systemic inflammatory response syndrome, atherosclerosis, rheumatoid arthritis, multiple sclerosis and septic shock30.
RONS, including oxygen and nitrogen free radicals, such as superoxide radical (O2•−), hydroxyl radical (•OH) and nitric oxide radical (•NO), are pro-inflammatory molecules that cause lipid peroxidation and oxidative stress31,32. RONS can have deleterious effects on tissues and are, thus, regulated by endogenous antioxidant mechanisms that involve enzymes (superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx)) and antioxidants (ascorbic acid, α-tocopherol and glutathione)31,32. Overproduction of RONS results in oxidative stress and tissue damage.
Enzymes play essential roles in inflammation and wound healing33,34. Cyclooxygenase 1 and cyclooxygenase 2 are upregulated in leukocytes and metabolize arachidonic acid into prostaglandins (PGs) (for example, PGE2, PGD2, PGF2), which regulate vascular permeability, neuron activity, bronchial reactivity and cardiovascular smooth muscles33. Activated inflammatory cells produce proteolytic enzymes that activate the complement, kallikrein–kinin and coagulation cascades, as well as proteinase-activated receptors, integrins and other adhesion receptors and ion channels, triggering the five cardinal signs of inflammation: increased blood flow and redness, fluid leakage and swelling, pain, heat and loss of tissue function34.
Neutrophil extracellular traps (NETs) are large molecular complexes produced by activated neutrophils35. NETs fight infection by immobilizing and killing bacteria, viruses and fungi, ultimately activating the innate immune response and coagulation. Abnormal NETs formation is associated with chronic autoimmunity and sterile and infectious inflammation36.
Biomaterials for inflammation targeting
Biomaterials can be used to target anti-inflammatory therapeutics to desired cells, tissues and organs, to improve drug delivery and anti-inflammatory efficacy, and to reduce toxicity37,38. Targeting strategies can be divided into passive and active targeting.
Passive targeting
Passive targeting is the transport of drug formulations through leaky fenestrated capillaries by passive diffusion or convection39. Enhanced permeability is often observed in vasculatures of inflamed tissue and at tumour sites39,40. Therapeutic nanoparticles can, thus, accumulate in the interstitial space, a phenomenon termed the enhanced permeability and retention (EPR) effect, which was first established in the development of anticancer nanomedicines39. The EPR effect can also be exploited to passively deliver biomaterial–drug formulations to the inflamed site through the leaky vasculature surrounding inflamed tissue39,40. However, the clinical significance of the EPR effect in cancer is under debate41,42 and, thus, the clinical impact of nanoparticle-mediated anti-inflammatory therapy must be viewed with caution. Nevertheless, the EPR effect is well established in animal models. Size-dependent accumulation of nanoparticles has been observed in a variety of inflamed tissues, including intestine, heart and tumour tissue40,43,44. In general, medium-sized (20–200-nm-diameter) nanoparticles are optimal for passively targeting drugs to inflamed tissue44. Furthermore, nanoparticle shape can have an effect on endocytosis by immune cells45, influencing the binding and phagocytosis by macrophages, as well as binding by targeted dendritic cells through electrostatic interactions46. Biomaterial surface charge may modulate or overshadow the effects of nanoparticle size or shape in passive targeting47,48 and can affect therapeutic uptake and the toxicity profiles of immune cells48.
Active targeting
Active targeting involves the conjugation of moieties that specifically bind inflammatory molecules and cells to the surface of biomaterials-based drug formulations. Inflammatory targets include inflamed vasculature, immune cells, pathogens, enzymes and mediators or products of inflammation.
Vascular inflammation is common in the pathogenesis of atherosclerosis49. Vascular inflammation is a result of the interaction between circulating leukocytes and endothelial cells, which is mediated by endothelial cell adhesion molecules (CAMs), including selectins and immunoglobulins, that are expressed on endothelial cells following activation by cytokines50. Therapeutic targeting of endothelial CAMs is a promising approach to managing vascular inflammation. P-selectin and E-selectin are usually expressed at low levels on the endothelial surface, but are highly expressed during the early and middle stages of inflammation51. Vascular cell adhesion molecule 1 (VCAM1) and intercellular adhesion molecule 1 (ICAM1) are endothelial transmembrane proteins that are also upregulated during vascular inflammation52. Magnetic particles decorated with anti-VCAM1 antibodies can localize specifically to the ischaemically injured brain by targeting inflamed endothelial tissue53. Alternatively, nanoparticles can be coated with leukocyte plasma membranes38. Proteins in the leukocyte plasma membrane specifically bind endothelial cell receptors, allowing targeted delivery to inflamed tissues54.
Active delivery may also be achieved by cellular transcytosis, enabling nanoparticle delivery and tumour penetration55. Here, macromolecules and nanoparticles can permeate the endothelium through endocytosis and exocytosis instead of going through the gap junctions between endothelial cells. Transcytosis is divided into adsorptive transcytosis, which can be exploited for cationic nanoparticles and albumin, and receptor-mediated transcytosis56,57, which involves ligands, such as chlorotoxin, transferrin and TAT peptide57. There is ample evidence that inflammation opens up tight junctions and enhances transcytosis at the blood–brain barrier58. The central nervous system endothelium undergoes alterations during neuroinflammation, including disruption of tight junctions, leading to increased permeability, which allows uptake of iron oxide nanoparticles59. Inflammation also plays a major role in endothelial dysfunction, contributing to the accelerated endocytosis of iron oxide nanoparticles into the plaque endothelium60.
The integrity of vasculature is intrinsically sensitive to biophysical cues spanning the microscale to nanoscale61,62. Some inorganic nanomaterials can randomly enter the nanometre-wide gaps of the adherens junctions between endothelial cells to subsequently produce micrometre-sized gaps between them63,64, leading to a phenomenon called nanomaterials-induced endothelial leakiness (NanoEL). NanoEL is dependent on the physical properties of inorganic nanoparticles (size, density and charge) and facilitates active targeting of nanomedicines65,66. For example, gold nanoparticles with a negative charge and a size of 10–30 nm, as well as silica nanoparticles with a density between 1.57 and 1.72 g cm−3 can induce endothelial leakiness63. NanoEL might also promote cancer cell intravasation and extravasation66, which raises the question about the risk of cancer nanomedicines. However, given that inflammation can also cause vascular leakage, NanoEL in an inflammatory but non-cancerous scenario might be beneficial for the delivery of anti-inflammation nanomedicines, such as nanoparticulate scavengers67.
Receptors or ligands expressed on leukocytes at inflamed sites provide targets for anti-inflammatory biomaterial–drug formulations68,69. A variety of ligands have been targeted to simulate leukocyte–endothelial cell receptor–ligand interactions, track the fate of neutrophils in the inflammatory microenvironment and facilitate targeted drug delivery51,70. Likewise, a variety of membrane proteins, including integrins, folic acid receptors, lipid scavenger receptors, chemokine receptors and CAMs71, have been used to target and image neutrophils and macrophages. Targeted immunotherapy of atherosclerosis can be achieved using nanoparticles coated with high-density lipoproteins (HDL) with a high affinity for monocytes and macrophages to modulate the CD40–TRAF6 interaction72. The impairment of the migratory capacity of monocytes inhibits their recruitment to the inflammatory site, consequently reducing plaque macrophage content, which is helpful to ameliorate inflammation and atherosclerosis. Exosomes or cell membranes containing targeting proteins can also be applied as nanoparticle coatings to achieve selective homing to an inflamed site38. Moreover, engineered nanoparticles can target platelets, T cells, B cells and dendritic cells for the diagnosis and treatment of inflammatory diseases38.
Inflammatory molecules can also be used as stimuli for biomaterials degradation to achieve degradation-mediated targeting. High levels of reactive oxygen species (ROS) are associated with chronic inflammation32. Biomaterials designed with ROS-responsive frameworks can, thus, be degraded in inflamed tissue for the site-specific release of anti-inflammatory drugs73. Inflammatory enzymes can also be exploited to trigger biomaterial activity74. For example, myeloperoxidase-responsive biomaterials exhibiting luminescent signals can be applied to estimate neutrophil count and allow specific imaging of neutrophils in inflammation-associated diseases75,76. Similarly, biomaterials with a structure sensitive to proteolytic matrix metalloproteinases, which are abundant in inflammatory sites, can deliver drugs to inflamed tissues77,78. In addition, low pH is a feature of the inflammatory microenvironment79,80. Thus, biomaterials can be designed to be pH-sensitive to respond specifically to inflammatory molecules and achieve precise diagnosis and treatment80.
Scavenging strategies
RONS scavengers
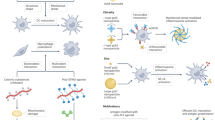
RONS are produced as metabolites or signal molecules in living organisms during cellular metabolism or in response to environmental stimulation81. Low levels of RONS can act as cellular signalling messengers by reversibly oxidizing thiol groups in proteins, thereby, modifying the protein structure and function. However, high levels of RONS disrupt cellular processes by non-specifically attacking proteins, lipids and DNA81. Excess RONS are produced by cells involved in the host defence response, for example, by polymorphonuclear neutrophils, and promote endothelial dysfunction by oxidizing crucial cellular signalling proteins, such as tyrosine phosphatases82. Under inflammatory conditions, oxidative stress produced by polymorphonuclear neutrophils leads to the opening of inter-endothelial junctions, promoting the migration of inflammatory cells across the endothelial barrier. These inflammatory cells not only participate in the clearance of pathogens and foreign particles but also lead to tissue injury. For example, M1 macrophages contribute to tissue injury by releasing large quantities of highly reactive cytotoxic oxidants to destroy pathogens27. Therefore, regulation of RONS levels by antioxidant therapy can maintain intracellular redox homeostasis and prevent oxidative-stress-related diseases82,83. Scavengers with antioxidative effects serve as enzyme mimetics to remove overexpressed RONS for the alleviation of inflammation82. For example, antioxidant enzymes and natural small molecules (for example, vitamin E and vitamin C) can prevent oxidative damage and inflammatory disease82. However, these naturally occurring antioxidants are limited in their ability to balance RONS levels, because they are not stable in harsh conditions (for example, acidic pH or redox environment), show poor pharmacokinetics, non-specific tissue accumulation and potentially harmful effects at high doses82,84. Alternatively, biomaterials with enzyme-like catalytic antioxidant activities can protect cells from oxidative damage and reduce inflammation82,83,85. Antioxidative biomaterials fall into two main categories: artificial-selenoenzyme-based scavengers and catalytic-nanomaterial-based scavengers (Fig. 3a).

In the early stage of inflammation, pathogen-associated molecular patterns (PAMPs) and endogenous tissue damage-associated molecular patterns (DAMPs) are released from injured cells. Reactive oxygen and nitrogen species (RONS), cell-free nucleic acids (cfNAs) and PAMPs or DAMPs can induce an inflammatory response and the recruitment of immune cells. a | RONS scavengers, such as artificial selenoenzymes and catalytic nanomaterials. b | cfNA scavengers, such as nucleic-acid-binding polymers, fibres and particles, can reduce the level of RONS and cfNAs in PAMPs or DAMPs and attenuate inflammation development.
Artificial-selenoenzyme-based scavengers
Organoselenium compounds are natural molecules (for example, glutathione oxidase and selenium) widely used in organic synthesis. Artificial selenoenzymes that exhibit GPx activity have been used therapeutically to catalyse the reduction of hydroperoxides and maintain a metabolic balance of ROS86,87. The GPx-mimicking drug Ebselen (2-phenyl-1,2-benzoisoselenazol-3(2H)-one)86,87 is a promising therapeutic with anti-inflammatory, antioxidant and cytoprotective activity, and is currently being tested in clinical trials as an anti-inflammatory drug (Phase II, NCT04484025 and NCT04483973). Inspired by the success of Ebselen, other organoselenium compounds with GPx activity have been developed, falling into two categories based on their structure: compounds with a direct Se-N–Se-O bond and compounds with intramolecular non-covalent Se•••N–Se•••O interactions87,88,89.
Catalytic-nanomaterial-based scavengers
Metal-based, carbon-based and polymer-based nanomaterials with antioxidant-enzyme-like catalytic activity can also serve as RONS scavengers82. The antioxidative activities of nanomaterials are affected by many factors, including size, morphology, composition, surface modification groups, substrate selectivity, pH and temperature, as well as the ions or molecules in the reaction system83. Different nanomaterials might have different antioxidative mechanisms82,83. Nanomaterials composed of noble metals (Pt, Au and Pd) are useful as ROS scavengers owing to their excellent catalytic activity and low cytotoxicity90,91,92,93. For example, Pt nanoparticles modified with citrate possess a similar catalytic activity as SOD, CAT and GPx. They have been used to scavenge ROS induced by KRIT1 loss of function in a cellular model of the human cerebral cavernous malformation disease90. Au nanoclusters modified with amine-terminated polyamidoamine (PAMAM) dendrimers have CAT-like activity and can, thus, be used to reduce intracellular H2O2 levels and protect neuronal cells from H2O2-mediated cytotoxicity91. Pd nanomaterials with CAT-like and SOD-like catalytic activity can moderate mitochondrial injury and protect cells from oxidative damage92.
Cerium oxide (CeO) nanoparticles have antioxidative activity owing to the variable oxidation state of cerium (Ce3+/Ce4+) and have been explored as RONS scavengers in treating spinal cord injury, inflammation, sepsis, Alzheimer disease, ischaemic stroke and Parkinson disease (in mouse and rat models)94,95,96,97,98,99. For example, small, positively charged CeO nanoparticles conjugated with triphenylphosphonium, which targets mitochondria, mitigated neuronal death through ROS scavenging and attenuated reactive gliosis and mitochondrial damage in a mouse model of Alzheimer disease96. Nanomaterials containing other metal oxides, such as Fe3O4, V2O5, Mn3O4, MnO2, CuxO and mixed metal oxides, also exhibit enzymatic activity and can, therefore, be used as RONS scavengers100,101,102,103,104,105,106. For example, Fe3O4 nanoparticles can protect neurons from H2O2-induced oxidative damage and ameliorate ROS-induced neurodegeneration in a Drosophila model of Alzheimer disease101. Manganese ferrite and ceria-anchored mesoporous silica nanoparticles can act synergistically to scavenge ROS and generate oxygen, inducing M1 to M2 polarization of macrophages and alleviating inflammation in knee joints in a rat model of rheumatoid arthritis105.
2D transition-metal dichalcogenides, which are graphene analogues with a flexible sheet-like structure, high surface-to-volume ratio and low toxicity in vivo, have also been explored as ROS scavengers107. For example, MoS2 nanosheets can scavenge ROS and have been investigated for treating oxidative-stress-related diseases108,109. Similarly, Mo-based polyoxometalate nanoclusters can eliminate detrimental ROS in the treatment of acute kidney injury in mice110,111. In addition, single-atom nanozymes112,113, metal-organic frameworks114 and black phosphorus115 have been explored in antioxidative stress applications. Moreover, carbon-based nanomaterials and naturally occurring biopolymers have been studied as antioxidants in the treatment of inflammation in mouse and rat models116,117,118,119.
cfNA scavengers
cfNA-binding polymers
There is growing evidence that cell-free nucleic acids (cfNAs) released by dying cells activate immune cells via TLRs (TLR3, TLR7, TLR8 and TLR9)10. Overactivation of these TLRs can lead to inflammatory and autoimmune diseases10. Nucleic-acid-binding polymers can be applied as cfNA scavengers in anti-inflammatory treatments (Fig. 3b). For example, polycationic polymers can attenuate the activation of all nucleic-acid-sensing TLRs120. Upon their systemic administration, these polycationic polymers prevented fatal liver injury in an acute toxic shock mouse model.
cfNAs also activate blood coagulation and, thus, cfNA-binding polymers show anticoagulant and antithrombotic activity in vitro and in vivo121. For example, the cationic third-generation PAMAM dendrimer (PAMAM-G3) inhibited thrombosis-induced pulmonary thromboembolism and carotid artery injury in mice121. Pathologic cutaneous scarring, which affects millions of people worldwide, is strongly correlated with excess cfNAs122. In a mouse model, PAMAM-G3 could reduce wound contraction and angiogenesis, while inducing more random collagen deposition and, hence, less scarring compared with untreated mice122. cfNAs also play a role in autoimmune disorders, such as lupus erythematosus, by promoting antibody generation. Cationic polymers have been applied as scavengers to limit cfNA-associated autoimmunity123. Furthermore, TLR signalling can induce tumour progression and metastasis by upregulating the secretion of pro-inflammatory cytokines. PAMAM-G3 can reduce liver tumour metastasis in a mouse model of pancreatic cancer by scavenging cfNA and inhibiting the activation of TLRs124. The nucleic-acid-binding and TLR-inhibitory activities of PAMAM can be potentiated by grafting PAMAM onto other polymer chains. For example, PAMAM-grafted polycaprolactone platforms were designed with different backbone lengths and charge densities125; grafted polymers with long backbones and high charge densities exhibited greater accumulation in inflamed joints of arthritic rats and enhanced therapeutic effects as compared with polymers with short backbones and low charge densities.
Other cationic polymers, such as heparin reversal agents and hexadimethrine bromide, can also act as cfNA scavengers. Heparin reversal agents are composed of a dendritic core with cationic-heparin-binding groups and short poly(ethylene glycol) (PEG) chains on the periphery126. The interaction of heparin reversal agents with DNA has been optimized by improving the interaction of DNA with these polymers126. Hexadimethrine bromide has a high affinity for mitochondrial DNA, which is similar to bacterial DNA and can provoke inflammation via the TLR9 pathway127. Hexadimethrine bromide could, therefore, mitigate the severity of multiple organ injuries in a rat model by scavenging mitochondrial DNA.
Thiazole orange is a cyanine dye that specifically binds nucleic acids and can, thus, be used as cfDNA scavenger. For example, thiazole-orange-modified dextran can be applied as cfNA scavenger to attenuate macrophage infiltration in the infarct area in a myocardial ischaemia reperfusion mouse model128.
cfNA-binding nanoparticles and nanofibres
Although nucleic-acid-binding polymers exhibit anti-inflammatory activity, their unfavourable biodistribution and rapid excretion from the body limit their application. Alternatively, nucleic-acid-binding nanoparticles and nanofibres show higher scavenging capacity, favourable biodistribution and clearance129,130,131,132,133,134 (Fig. 3b). For example, a cationic micelle containing poly(lactic-co-glycolic acid) (PLGA) and poly(2-(diethylamino)ethyl methacrylate) can be applied as anti-inflammatory treatment in animal models with CpG-induced inflammation and collagen-induced arthritis129. Intravenous injection of these micelles reduced bone and cartilage damage, as well as ankle and tissue swelling, with significantly better therapeutic results compared with the corresponding soluble polymer. Similarly, polyethyleneimine (PEI)-functionalized, biodegradable mesoporous silica nanoparticles can be designed as cfNA scavengers with different charge densities131. These silica nanoparticles showed superior performance compared with nucleic-acid-binding polymers in inhibiting cfNA-induced inflammation and subsequent multiple organ injury caused by sepsis in mice. Of note, nucleic-acid-binding nanoparticles exhibited higher accumulation and retention in the inflamed caecum, and a more desirable in vivo safety profile, compared with their soluble polymer counterparts. Cationic PEI can also be immobilized onto nanofibrous meshes and electrospun microfibre meshes to produce nucleic-acid-binding fibres132,133. These meshes attenuated an inflammatory response triggered by negatively charged agonists or DAMPs in the serum of post-trauma patients132. The nucleic-acid-binding meshes suppressed the NF-κB response of cells stimulated with DAMPs released from doxorubicin-treated tumour cells in vitro133.
Scavengers of other danger signals
In addition to RONS and cfNAs, other danger signals are associated with inflammatory diseases, including lipopolysaccharide (LPS), chemokines, autoantibodies and PAMPs135,136,137,138. LPS is a component of Gram-negative bacterial cell walls and is a TLR4 agonist. Inspired by the observation that HDL can bind LPS, a series of HDL-like nanoparticles were synthesized and shown to be effective in scavenging LPS and inhibiting activation of TLR4 receptors135. Inflammatory chemokines, including monocyte chemoattractant protein 1 (MCP1) and IL-8, play important roles in chronic inflammation and wound healing136,137. A modular hydrogel based on glycosaminoglycan derivatives and star-shaped PEG was designed for chemokine scavenging and chronic wound treatment136. In animal models, this hydrogel outperformed Promogran, a commercial anti-inflammatory product, in reducing inflammation, increasing angiogenesis and advancing wound closure. Nanofibres composed of pullulan, chondroitin sulfate and tannic acid have been used as wound dressings for recessive dystrophic epidermolysis bullosa137. The scavenging fibres removed over 99% of the inflammatory MCP1 from the solution in vitro within 2 h.
Challenges and opportunities of scavenging strategy
Several challenges remain to be overcome for scavenging biomaterials to optimally control inflammation. The mechanisms by which nanomaterial properties, such as size, shape and surface chemistry, affect its ability to scavenge danger signals are unclear. Size determines in vivo biodistribution, particularly accumulation in inflamed sites139, in addition to intracellular trafficking, particularly interaction with endosomal TLR receptors139. Shape determines the extracellular and intracellular fate of a scavenger140. However, shape may be more important than size in terms of scavenging performance; for example, a 2D nanomaterial might be more effective than a nanoparticle in binding a danger signal associated with a protein or embedded in a secreted vesicle. Surface chemistry governs the type of danger signals that can be removed. A structure–property relationship between the composition of scavengers and types of danger signals to be removed would be invaluable for addressing inflammatory diseases that demand diverse scavenging characteristics.
Studies in animal models showed that nucleic-acid-binding polymers and nanoparticles can achieve therapeutic effects in treating inflammatory diseases, ranging from acute liver injury to lupus and sepsis; however, little information is available on the types of danger signals that are neutralized, which is one of the most interesting and pressing questions in biomaterials-mediated inflammation modulation. The design of retrievable scavenging biomaterials coupled with advanced analytical chemistry would be needed to address this question. If successful, such mechanistic studies might identify new biomarkers that would pave the way for the development of potent and precision therapeutics for inflammation therapy. Of note, cationic materials are often toxic, as documented in many non-viral gene delivery studies129,130. Therefore, the danger-signal-binding affinity needs to be balanced in terms of toxicity, which might require systematic screening and optimization.
Blockage strategies
Blocking leukocyte–endothelial cell interactions
The adhesive interaction of leukocytes and endothelial cells enables leukocyte migration to inflamed sites, an important element in immune surveillance18,141. Calcium-dependent transmembrane glycoproteins, called selectins, are expressed on both endothelial cells and leukocytes, and are involved in their interaction. P-selectin is expressed by platelets and endothelial cells, and L-selectin by leukocytes51,142. Compounds that block these selectins can prevent leukocyte migration and reduce inflammation (Fig. 4a).

In the middle stage of inflammation, immune cells are recruited to the inflammatory site, generating excessive pro-inflammatory cytokines and inducing severe inflammation. a | Anionic biomaterials, such as linear polymers, dendritic polymers and nanomaterials, can block the interaction between L-selectins on leukocytes and L-selectin ligands on the endothelium, or P-selectin on the endothelium and P-selectin ligands on leukocytes, to inhibit the migration of immune cells to the inflammation sites. b | Toll-like receptor (TLR) antagonists can be applied to block the activation of TLRs on immune cells to prevent the activation of immune cells.
Heparin is an anticoagulant that also possesses anti-inflammatory activity by blocking P-selectin and L-selectin143. The use of heparin as an anti-inflammatory agent is limited, owing to the risks of heparin-induced thrombocytopenia and uncontrolled bleeding. However, non-anticoagulant heparin-like molecules that block selectin interactions can also reduce inflammation144.
Polysulfates
Dendritic polyglycerol sulfate (dPGS), which is widely used in biomaterials research, was initially considered as a heparin mimetic144,145,146,147,148,149. The dendritic structure and peripheral sulfate groups of dPGS mimic naturally occurring ligands in heparin through a multivalent binding mechanism. dPGS caused a tenfold shorter clotting time than heparin at concentrations above those needed to inhibit inflammation144. Moreover, dPGS exhibits high multivalent binding affinities to positively charged proteins, such as L-selectin and P-selectin145. Administration of dPGS could reduce leukocyte extravasation and oedema formation in a contact dermatitis mouse model. Daily treatment of arthritis rats with dPGS is chondroprotective and can inhibit arthritis symptoms and the presence of inflammatory cells in synovial fluid146.
To increase the biodegradability of dPGS for clearance, a biodegradable form of sulfated poly(glycidol-co-caprolactone) was developed147, which also has a competitive inhibitory effect on the leukocytic cell adhesion receptor, L-selectin. Similarly, shell-degradable polysulfates can be prepared by incorporating hydrolytically or enzymatically cleavable linkers148. These shell-degradable dPGS derivatives activate the complement pathway149; here, rapidly degrading carbamate-functionalized dPGS showed the highest binding affinity to L-selectin.
Anisotropic gold colloids, such as gold nanorods, are ideal contrast agents owing to their non-radiation de-excitation150. dPGS can be conjugated onto gold nanorods to design compounds with low toxicity and high inflammation-targeting performance, allowing imaging of inflammation in a mouse model of rheumatoid arthritis with high contrast151.
Sulfate interacts with selectins through electrostatic interactions. In addition to dPGS, other polysulfates have been studied for their anti-inflammatory activity, including sulfated PEG dendritic macromolecules152 and sulfated polysaccharides153. These polysulfates inhibit L-selectin binding and sulfated polysaccharides could prevent lymphocyte homing from peripheral blood to the spleen and lymph nodes in mice.
Polyphosphonates
Like sulfates, phosphonates and bisphosphonates are negatively charged and can be used to block leukocyte migration and modulate inflammation154,155,156,157. For example, an azabisphosphonate (ABP)-capped dendrimer can specifically target monocytes and induce their anti-inflammatory conversion in mice with rheumatoid arthritis154. ABP dendrimers can also inhibit the development of autoimmune encephalomyelitis in a mouse model155. Here, an analogue dendrimer capped with 12 azabiscarboxylate end groups was used as control to confirm the interaction between ABP dendrimers and monocytes156. The ABP binds to the membrane of monocytes through both non-specific and specific binding, whereas the azabiscarboxylate-functionalized dendrimer showed little binding activity, indicating the importance of the phosphonate in blocking immune cell migration.
Polyanionic macromolecules can also inhibit L-selectin157, with the inhibition efficiency increasing in the order of carboxylate < phosphate < phosphonate ≈ sulfonate < bisphosphonate < sulfate. Although polysulfates, such as dPGS, exhibit excellent L-selectin binding, further research is needed to elucidate the correlation between the microstructure (surface charge and particle size) of the polyanions and their ability to block leukocyte migration.
Polycarboxylates
Little is known about the anti-inflammatory effects of polycarboxylates. A degradable conjugate of dendritic polycarboxylate and morphine has been shown to specifically target peripheral nerve receptors and dampen pain locally without central adverse effects in a rat model158. In contrast to conventional morphine, the intravenously administered conjugate exclusively activated peripheral opioid receptors to produce analgesia in inflamed rat paws without major side effects, such as sedation or constipation.
Glycoconjugates
Polysaccharides and glycoproteins, such as polyanions, can also selectively bind selectins159,160. The sialyl LewisX tetrasaccharide (sLeX) is a common carbohydrate structure used in selectin-binding studies. For example, sLeX-modified poly(2-hydroxypropyl) methacrylamide can act as cell adhesion inhibitor, with excellent binding affinity for E-selectin, L-selectin and P-selectin159. Poly(phosphorhydrazone) dendrimers can be grafted with mannose units160 to achieve selectin binding, with a G3 dendrimer with 48 trimannosides and a G4 dendrimer with 96 dimannosides showing the highest binding avidity for C-type lectins on dendritic cells.
Blockage of TLRs
Blockage of immune cell TLRs is another promising strategy to reduce inflammation161 (Fig. 4b). For example, peptide–gold nanoparticle hybrids can suppress the activity of several TLRs162,163; here, the hydrophobicity and structure of the amino acids in the peptides are crucial for modulating TLR4 responses. In a murine intestinal inflammation model, peptide–gold nanoparticles improved the disease activity index, reduced weight loss and ameliorated colonic inflammation. The anti-inflammatory activity of the peptide–gold nanoparticles can be further enhanced by modulating the physiochemical parameters of the nanoparticles, in particular, their size164. Nanoparticles with a 20-nm nanoparticle core exhibit the strongest inhibition of TLR4 activation, which can be attributed to high cell uptake and strong endosomal pH buffering capacity. It was accidentally discovered that adding cigarette smoke extract improved TLR4 inhibition of peptide–gold nanoparticles165. Cigarette smoke extract components are adsorbed on the peptide–gold nanoparticles, which increases their cellular uptake, thus, avoiding endosomal acidification usually required for TLR4 activation, thereby, enhancing the anti-inflammatory activity of the peptide–gold nanoparticles.
Challenges and opportunities of blockage strategy
Blockage strategies have proven useful in reducing inflammation; however, the mechanisms by which biomaterials interact with selectins and other CAMs have not yet been fully elucidated. Understanding these interactions is important for developing biomaterials that regulate the homing of circulating immune cells to inflammatory sites. Electrostatic forces play an important role in these binding events; however, binding affinity is not simply proportional to the charge density on the biomaterial157. The types of anions also matter. For example, polymers with a sulfate group are more effective than those with a phosphate group in binding L-selectin and blocking the recruitment of immune cells to inflammatory sites. Other anionic structures may potentially be even more potent. As in scavenging strategies, the size and structure of the binding group influence the interactions with CAMs; for example, multivalency endowed by a dendritic morphology or surface functionalization of a particle. Interestingly, the softness of the particle may also matter, because the flexibility or shape adaption could facilitate multivalent interactions. However, the exact relation between these interactions and particle size and softness remains elusive. Additionally, most selectin-binding studies are based on electrostatic interactions, which are not specific, similar to nucleic acid scavenging strategies. Non-specific binding, however, cannot claim the benefit of blocking an unspecified upstream signal, as in the case of scavenging, where inhibition of the TLR‒ligand interaction attenuates the activation of the MYD88–NF-κB pathway166. Using selectin-binding antibodies (against P-selectin, L-selectin and E-selectin) would be a more specific blockage strategy167. These antibodies would need to be combined with biomaterials to maintain their bioactivity and achieve high blockage efficacy.
Delivery strategies
Unlike scavenging and blockage strategies, which are based on the intrinsic properties of the biomaterials, delivery of anti-inflammatory agents by biomaterials also depends on the drug properties. A variety of materials, including polymers, lipids and inorganic materials, have been explored as drug carriers168,169 that deliver anti-inflammatory agents, such as small-molecule drugs, biomacromolecules (nucleic acids and proteins) and therapeutic gas (Fig. 5).

In the final stage of uncontrolled inflammation, an excess of immune cells produces an inflammatory response, which can result in a cytokine storm. Biomaterials can be applied as drug delivery platforms, in particular, as nanocarriers, to extracellularly or intracellularly deliver therapeutic agents to the inflammatory sites through the enhanced permeability and retention effect, transcytosis or nanomaterials-induced endothelial leakiness.
Small-molecule drug delivery
Small-molecule drugs (for example, resveratrol, dexamethasone, irbesartan and celecoxib) are not only suited for intracellular targets but also applicable to cell-surface or extracellular targets for anti-inflammatory therapy170. However, their bioavailability and bioactivity are limited by poor water solubility and rapid metabolism. More importantly, their off-target toxicity (for example, immunosuppression and gastrointestinal toxicity) can cause serious side effects and is a major cause of failure in clinical trials171. Drug delivery platforms based on biomaterials as drug carriers can help to overcome these limitations172,173,174,175.
PLGA is biocompatible and biodegradable, and can be used for the design of drug delivery carriers for treating inflammatory diseases, including osteoarthritis, nasal polyp, inflammatory bowel disease and myocardial ischaemia–reperfusion injury176,177,178. Dexamethasone sodium phosphate a synthetic corticosteroid, has been widely applied to alleviate inflammation and reduce the loss of cartilage extracellular matrix. ROS-responsive hollow microspheres, composed of dexamethasone sodium phosphate and PLGA, can be locally injected into inflamed osteoarthritic tissue to treat arthritis and joint destruction in mice177. Liposome-based nanoplatforms allow targeted delivery of anti-inflammatory small-molecular drugs (for example, morin, dexamethasone, quercetin and resveratrol) for the treatment of rheumatoid arthritis, sepsis, vascular inflammation and inflammatory liver disease in animal models179,180,181. Other synthetic polymer-based carriers, including poly(ε-caprolactone) nanoparticles, poly(ester amide) microspheres, parylene-C nanostructured films, PAMAM dendrimers and polydimethylsiloxane-based 3D scaffolds have also been explored for the delivery of anti-inflammatory drugs182,183,184,185.
Compared with synthetic polymers, natural biopolymers tend to have higher biocompatibility and their biodegradation is less likely to cause tissue irritation compared with the acidic breakdown products of PLGA186,187,188,189,190. For example, alginate hydrogels can be used to deliver mesenchymal stem cells for implantation, preserving the viability of the cells and delivering anti-inflammatory agents to improve the interaction of mesenchymal stem cells with T lymphocytes and, thus, tissue regeneration188. A pH-responsive, dexamethasone-loaded DNA platform can deliver therapeutic agents to macrophages, resulting in inhibition of sterile inflammation after ischaemia–reperfusion injury in mice189,190.
Compared with polymer-based carriers, inorganic drug carriers such as mesoporous silica nanoparticles have a higher surface-to-volume ratio, higher drug loading capacities and different controlled release profiles because of their porous structure191. For example, mesoporous silica nanoparticles can serve as excellent stealth nanocarriers for the delivery of hydrophobic anti-inflammatory drugs192. Similarly, resveratrol can be encapsulated in silica nanoparticles to improve the transport of the drug across the tight junctions of the Caco-2 (human epithelial colorectal adenocarcinoma cell) monolayer193.
Delivery of therapeutic biomacromolecules
Nucleic acid delivery
Inhibition of pro-inflammatory cytokine generation by siRNA delivery is a promising therapeutic approach to control inflammation. PEI-based cationic nanomaterials are widely used for nucleic acid delivery194,195,196,197,198,199. Multi-shell nanoparticles containing a calcium phosphate core and PLGA/PEI layer allow intrarectal delivery of TNF, keratinocide-derived cytokine and interferon gamma-induced protein 10 siRNA to inhibit their gene expression and reduce intestinal inflammation in a mouse model194. Signal transducer and activator of transcription 1 siRNA, cyclooxygenase 2 siRNA and NF-κB siRNA can also be delivered for inflammation treatment195,196,197. Co-delivery of drugs and siRNA has shown better anti-inflammatory performance than drug or siRNA delivery alone198,199. The cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) can be mixed with TNF siRNA and then covered with poly-(1,4-phenyleneacetone dimethylene thioketal), a polymer consisting of ROS-sensitive thioketal linkages200. Owing to the high ROS levels at sites of inflammation, the DOTAP nanoparticles release encapsulated siRNA in inflamed tissues following oral administration in mice with ulcerative colitis. Similarly, acid-responsive DOTAP-containing nanoparticles enable delivery of siRNA to inflammatory sites, avoiding unwanted burst release201. A DOTAP-containing PEG-b-PLGA cationic-lipid-assisted nanoparticle allows delivery of Bruton’s tyrosine kinase siRNA for the treatment of rheumatoid arthritis202. To reduce the toxicity of positively charged gene delivery vectors, pH-sensitive nanoparticles203 can be designed with multi-armed PEG and acid-responsive linkers to alleviate inflammation-induced liver damage in mice.
Plasmids and the CRISPR gene editing system can also be delivered to modulate inflammation. For example, endothelial nitric oxide synthase, a pro-angiogenic enzyme, promotes angiogenesis and reduces IL-6 expression204,205, and endothelial nitric oxide synthase plasmid can be delivered together with IL-10 in an elastin-like polypeptide-based injectable system204. Cholesterol-modified PAMAM allows co-delivery of resveratrol and haem oxygenase 1 plasmid205. Inhalation of the cholesterol-modified PAMAM nanoplatform achieved a therapeutic effect in animals with acute lung injury. Cationic-lipid-assisted nanoparticles can deliver CRISPR–Cas9 mRNA and gRNA to suppress NOD-, LRR- and pyrin domain-containing 3 (NLRP3) expression206, demonstrating promising therapeutic efficacy for NLRP3-dependent inflammatory diseases in vitro and in vivo. Despite promising results in preclinical studies, several issues should be taken into careful consideration for clinical translation, including the large size of plasmids, the potentially high off-target effects owing to the persistent expression of the Cas9 enzyme and delivery inefficiency of non-viral vectors207.
Protein delivery
Anti-inflammatory proteins can also be delivered to control inflammation. Several interleukins, including IL-4 (refs208,209), IL-10 (refs210,211) and IL-27 (ref.212), induce the transition of macrophages to the anti-inflammatory M2 phenotype. For example, IL-4 can be delivered by mesoporous silica nanoparticles with an ultra-large pore size (30 nm) to extend the relatively short bioactivity of IL-4 and improve its anti-inflammatory therapeutic efficacy208. Alternatively, using microfluidics, a microchannel can be engineered in a hydrogel, surrounded by a porous membrane, which can then be dried; subsequent reswelling of the hydrogel triggers the release of IL-4 and dexamethasone, and the release can last for up to 96 h (ref.209). A variety of nanocarriers have been used for IL-10 delivery for inflammation modulation, including pluronic-based nanocarriers, mineral-coated microparticles and hyaluronan and heparin-based hydrogel systems210,211.
Neurotrophin 3 (NT3), stromal-derived factor 1α, TGFβ1 and other proteins213,214,215,216 have also demonstrated anti-inflammatory activity. For example, an NT3/fibroin-coated gelatin sponge scaffold was engineered for spinal cord injury therapy in rat and canine models213. An N-desulfated heparin-containing PEG-diacrylate hydrogel was developed to deliver stromal-derived factor 1α214. Biomaterial scaffolds that enable local delivery of proteins such as TGFβ1 can improve the transplantation of islets in a mouse model by reducing local cytokine production and leukocyte infiltration215. Furthermore, temporal control of the immunomodulatory microenvironment is important. In an in vitro study, staged delivery of pro-inflammatory cytokines, such as MCP1 and interferon gamma, followed by the release of the anti-inflammatory cytokines IL-4 and IL-10, could polarize macrophages from an M1 to an M2 phenotype to direct an initially pro-inflammatory response, which is beneficial for wound healing by clearing debris and preventing infection at the site, to a subsequent pro-healing response217. This was accomplished with a two-compartment biomaterial system containing a magnetically responsive biphasic ferrogel, which can be externally activated to trigger the release of the cytokines on demand. Although these findings have to be validated in vivo, this study highlights the potential advantages of biomaterials in the temporal-spatial control of inflammation, which cannot be easily achieved by conventional drug therapy.
Delivery of anti-inflammatory gas
Gas therapy using carbon monoxide (CO)218,219,220,221,222,223, hydrogen (H2)224,225,226,227 or hydrogen sulfide (H2S)228 can be applied for the treatment of inflammatory diseases. CO selectively inhibits the generation of pro-inflammatory cytokines (for example, TNF, IL-1β and macrophage inflammatory protein 1β) and increases the expression of anti-inflammatory cytokines (for example, IL-10). Despite its promising therapeutic activity, the use of CO in clinical applications is limited, owing to a lack of targeting and potential toxicity. Photoactive CO-releasing nanoplatforms can overcome this limitation. For example, ferritin-containing protein cages and 3-hydroxybenzo[g]quinolone frameworks218,219 release CO under light irradiation, and the amount of released gas can be regulated by the duration of irradiation. Alternatively, esterase and pH-responsive CO prodrugs can be administered for the treatment of LPS-induced systemic inflammation in a mouse model220. Prolonged CO release can be achieved by a syringe-injectable peptide hydrogel, enabling a CO-releasing half-lifetime of 20.2 ± 0.6 min, which is longer than the commonly used CO-releasing molecule Ru(CO)3Cl(glycinate) (t1/2 ≈ 3.6 min)221.
H2 gas can downregulate pro-inflammatory cytokines and transcription factors, thereby, alleviating inflammatory-response-induced tissue injury. Semiconducting polymer dots and liposomes enveloping chlorophyll a, 1-ascorbic acid and gold nanoparticles can deliver H2 gas224,225. Both nanoplatforms reduced ROS and the concentration of pro-inflammatory cytokines in the paws of mice. High H2 concentration can also be generated in inflammatory tissue by cyclic passivation and activation of Mg in Mg-PLGA microparticles226.
Challenges and opportunities of delivery
These lipidic, polymeric and inorganic nanocarriers all have distinct advantages and disadvantages in drug delivery applications168,169. Lipidic nanoparticles are easy to formulate, and many different lipids are available with well-defined composition and properties, which can be screened to identify the ideal lipid mixture for optimal delivery performance229. The efficacy of the COVID-19 mRNA vaccine is partly related to such an optimized lipid formulation230. However, encapsulating small hydrophilic drugs and functionalization of lipid nanoparticles remain challenging, but could be addressed through modification of lipids. Polymeric nanoparticles show high compositional and structural diversity, in terms of molecular weight and architecture, enabling the encapsulation of almost any drug. In addition, biodegradability, stimulus responsiveness and targeting can be incorporated into polymeric nanoparticles. However, batch-to-batch variations in composition and molecular weight of the polymeric carrier may be problematic for clinical translation. For example, a wide molecular weight distribution leads to polydispersity in nanoparticle size and surface potential, which, in turn, promotes aggregation and affects performance. Inorganic nanoparticles are usually more uniform in size, composition and structure, and can possess electrical, magnetic or optical properties for external control release or imaging. Inorganic nanoparticles with a porous structure, such as mesoporous silica nanoparticles, allow loading of drug molecules into the internal pores with high efficiency231,232. If drug loading is confined to surface immobilization, however, the loading level is lower, and surface-exposed drugs are more prone to degradation.
Several strategies can be applied to improve the delivery performance of nanocarriers168,169. For example, more specific inflammation targeting can be achieved by designing carriers that respond to inflammatory molecules. Furthermore, the delivery of multiple cargoes with one vehicle (all-in-one drug delivery platform) at different stages of inflammation would be valuable to treat inflammation. For example, by adapting to the characteristics of the inflammatory microenvironment at different stages, the delivery system could initially release scavengers to remove cfDNA or mitigate RONS, followed by the release of drugs that can block leukocyte–endothelial cell interactions, supported by anti-inflammation agents to strengthen the therapeutic efficacy at the late stage of inflammation. To reduce the complexity associated with multiple drugs, a proactive carrier could be designed that can perform scavenging and blockage functions in addition to delivery.
In addition to inflammatory molecules, other targets could be explored; for example, cells and cellular communication of the microvascular compartment, which collaborate to recruit leukocytes to inflamed tissue. Local signals regulating inflammation are transmitted between cells by paracrine signalling, mechanosignalling, direct signal transduction via gap junctions (connexins) and tunnelling nanotubes, and through release and uptake of cell-derived vesicles, such as exosomes. The recruitment of circulating leukocytes is directed by mechanosignalling between integrins on leukocytes and adhesion molecules on the endothelium. Distinct leukocyte populations and vascular beds express different combinations of adhesion molecules, which could inspire the design of drug delivery platforms to realize tissue-specific targeting233. Connexins are membrane proteins that form gap junctions and enable direct signal transduction between adjacent cells by allowing rapid transfer of ions, second messengers and metabolites. Connexin 43 channels in gap junctions are involved in the communication between leukocytes and epithelial cells in the lung and intestine234. The synthetic peptide ACT1, which mimics the carboxyl terminus of connexin 43, stabilizes gap junctions and decreases infiltration of inflammatory neutrophils into wounds235. These connexins can be targeted to modulate the communication between epithelial or endothelial cells and leukocytes236. Cell–cell signalling can also involve exosomes that shuttle proteins, lipids, microRNA and mRNAs between cells237. Exosomes are released from most cell types, including leukocytes and endothelial cells, in response to various stimuli, including inflammation. As drug delivery vehicles, exosomes could be designed to express surface molecules to deliver them to specific vascular beds in a targeted manner238.
Perspectives
Infectious diseases and sterile inflammatory diseases exhibit common pathophysiological features, such as monocyte recruitment, macrophage polarization and enhanced vascular permeability1,2,6. cfNAs released by dead or damaged cells can recruit immune cells and trigger inflammatory responses35. Furthermore, many pathogens, including viruses and bacteria, can activate immune cells by themselves5,9. Pathogen-induced inflammation can be alleviated through eradication of pathogens; however, elimination of sterile inflammation that can develop into chronic inflammation, which is a cause of tumour occurrence, is more difficult2,7. Inflammation can be classified into local and systemic inflammation, depending on the sites of the inflammatory response. However, local and systemic inflammatory responses are connected239,240, and many local inflammatory diseases, including hepatitis, pneumonia and periodontitis, can induce serious systemic inflammation if they are not controlled at an early stage239,240. Subsequent systemic inflammatory responses, such as a cytokine storm, can lead to multi-organ failure and even death.
The complexity, variability and heterogeneity of the inflammatory environment and corresponding immune response can lead to heterogeneous treatment responses among patients1,7. Inflammation is a highly dynamic process7, and, thus, an anti-inflammatory treatment would be desirable that can adapt to the progression of inflammation. For example, sepsis is a complex condition induced by infection and underpinned by an intricate interplay of pro-inflammatory and anti-inflammatory responses131 (Fig. 6a). Patients with sepsis first undergo a phase of excessive systemic inflammation mediated by various pro-inflammatory mediators of the innate immune system, followed by either recovery or re-entering of the inflammatory phase. Therefore, immunostimulation and immunosuppression need to be balanced for sepsis therapy.

a | Inflammation is a highly dynamic process, exemplified here by the development of sepsis. Sepsis is characterized by an intricate interplay of pro-inflammatory and anti-inflammatory responses. An optimal anti-inflammatory therapy should, therefore, adapt to the progression of inflammation. b | Various parameters of biomaterials can be designed for scavenging, blockage and delivery strategies for inflammation modulation. Composition, size, shape, charge and mechanical compliance affect the behaviour of the biomaterial in vitro and in vivo, including tissue distribution, trafficking and cellular interactions. The mechanical compliance of biomaterials, whether as scaffold to interact with leukocytes or as a nanoparticulate injectable to interact with cells and tissues, is also relevant. Finally, biomaterials can play a passive or active role in inflammation-modulation strategies. DAMP, damage-associated molecular pattern; GSH, glutathione; IL-1β, interleukin-1β; IL-6, interleukin-6; mtDNA, mitochondrial DNA; NET, neutrophil extracellular trap; ΝF-κB, nuclear factor-κB; PAMP, pathogen-associated molecular pattern; RONS, reactive oxygen and nitrogen species; ROS, reactive oxygen species; TNF, tumour necrosis factor.
Homeostasis and dysregulation of inflammation are the result of multiple interactions of various cell types with their microenvironment. Real-time monitoring of these interaction dynamics would allow the design of therapeutic strategies tailored to the different stages of the inflammation. Biomaterials offer a vast parametric space to design inflammation-modulation strategies (Fig. 6b), including chemical properties, such as hydrophobicity and charge characteristics, physical properties, such as size, shape and softness, as well as environmental responsiveness to achieve active targeting.
The chemistry of biomaterials is related to their immune-modulatory activities and can, thus, be tailored to different anti-inflammatory strategies. Biomaterials with antioxidant-enzyme-like catalytic activity can be used for RONS scavenging, the potency of which is affected by many factors, including size, morphology, composition, surface modification, pH, temperature and the ions and molecules in the reaction system82,83. Most cfNA-scavenging materials are positively charged to bind negative cfNA through electrostatic interactions120. Negatively charged materials can also interact with nucleic acids through metal coordination. Negatively charged biomaterials can be applied to block P-selectin and L-selectin on leukocytes144,145 to inhibit their homing; here, the blockage efficiency is not directly proportional to the charge density or zeta potential of the material157. Nanomaterials used for anti-inflammatory drug delivery are often amphiphilic to allow the formation of nanoparticles through self-assembly to encapsulate drugs or biomacromolecular agents168,169. These nanoparticles can then release the loaded drugs in response to the intracellular microenvironment in the targeted disease area. Materials can also be designed to generate therapeutic gas under external stimuli or to react with intracellular components220,224. Furthermore, different anti-inflammatory strategies may be incorporated into the biomaterial design to achieve a synergistic effect.
Biomaterials can be designed to passively or actively target specific features of the inflammatory environment for therapeutic and diagnostic applications8. Biomaterials with nucleic-acid-binding, ROS-neutralization and mediator-absorption activities can be used to scavenge danger signals and block early inflammation. Biomaterials that block CAMs can prevent leukocyte–endothelial cell adhesion to reduce immune cell recruitment during the middle stage of inflammation. Biomaterials capable of delivering anti-inflammatory drugs in a stimuli-responsive manner can best regulate the inflammatory environment during the middle and late stages of inflammation (Fig. 6b).
However, the vast parametric space of nanomaterial properties makes the establishment of structure–property relationships challenging. To limit the number of animal experiments and to increase the relevance to human responses, microphysiological systems or human tissue chips can be applied for nanomaterial testing. Often constructed with human-induced pluripotent stem cells, engineered 3D tissues can be connected with physiologically relevant flows to form tissues on a chip241,242, which can be applied for disease modelling and drug discovery. Inflammation models can then be generated by adding the appropriate stimulus; for example, addition of TNF to the culture medium perfusing a tissue-engineered blood vessel activates the endothelium and significantly increases transendothelial events of immune cells241. Similarly, an ex vivo multi-organ model of ulcerative colitis can be designed by connecting microphysiological systems of the human gut, liver and circulating Treg cells and TH17 cells242. Such physiologically relevant in vitro and ex vivo systems allow the investigation of the relationship between metabolism, immunity and tissue homeostasis. However, there is ample room to improve such tissue chips; for example, including blood vessel endothelium in the chip design would enable the study of transendothelial immune cell migration241,243,244.
Biomaterials, however, face different challenges to conventional drugs in terms of clinical translation. The majority of approved drugs have a low molecular weight and are uniform in size and composition. By contrast, biomaterials are macromolecular and can range from the nanoscale to the macroscale, with a wide distribution in molecular weight and size8. Therefore, considerations of biocompatibility and toxicity of biomaterials are more complicated. For example, biodegradability is beneficial for biomaterials intended as scavengers and drug carriers to avoid long-term complications8. However, the optimal biodegradation rate may vary depending on the application; if the biomaterial degrades too fast, the scavenging or delivery function may be compromised, and storage and shipping may be more costly than for non-degradable biomaterials; for example, if an anhydrous packaging is required. Finally, the complexity and heterogeneity of patient-specific inflammation pose a challenge for the clinical translation of biomaterials8.
Several biomaterials are currently in clinical trials for inflammation-related diseases (Table 1), with only a few approved by the US Food and Drug Administration. In some cases, in particular, for scavenging biomaterials, only ex vivo trials have been conducted thus far. In addition to using microphysiological systems based on induced pluripotent stem cells, relevant animal models would be required to enable clinical translation245. Many diseases, including cancer (origin, progression and metastasis), Alzheimer disease and acute organ injuries (for example, COVID-19-induced lung injury), are tightly linked to local or systemic inflammation7,239,246. Anti-inflammatory therapies for these diseases could be preclinically investigated in sophisticated animal models.
As the concept of biomaterials-assisted inflammation control develops, a future research focus should be on applications, for which such strategies provide a unique advantage, for example, in cancer therapy. A drug carrier could deliver a chemodrug in a controlled manner and, at the same time, also scavenge pro-metastatic factors. The spatiotemporal control of inflammation, which can be achieved by biomaterials-assisted strategies, is also particularly important for cancer treatment. Similarly, the dual functionality of a biomaterial, that is, delivery and scavenging, could be advantageous for the treatment of infectious diseases.
Biomaterials are not passive entities but active participants in the inflammation process. Therefore, a holistic consideration of interactions between biomaterials and the inflammatory microenvironment can inform the design of multifunctional biomaterials to tackle inflammation. Biomaterials could be engineered to induce a therapeutic or immunologic response by themselves, including biomaterials that have been optimized for drug delivery and tissue engineering. Initial translational barriers, such as toxicity, lack of biocompatibility, robustness and manufacturing, have been addressed for many implants and drug delivery devices. This wealth of knowledge can provide the basis for the clinical translation of biomaterials for inflammation treatment. Building on preclinical successes, bridging the gap between preclinical research and clinical translation will pave the way for biomaterials-based anti-inflammatory therapies for various diseases.
References
Kotas, M. E. & Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827 (2015).
Rock, K. L., Latz, E., Ontiveros, F. & Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 28, 321–342 (2009).
Schett, G. & Neurath, M. F. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat. Commun. 9, 3261 (2018).
Furman, D. et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 25, 1822–1832 (2019).
Zindel, J. & Kubes, P. DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu. Rev. Pathol. Mech. Dis. 15, 493–518 (2020).
Luster, A. D., Alon, R. & von Andrian, U. H. Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 6, 1182–1190 (2005).
Rajendran, P. et al. The multifaceted link between inflammation and human diseases. J. Cell. Physiol. 233, 6458–6471 (2018).
Darnell, M. & Mooney, D. J. Leveraging advances in biology to design biomaterials. Nat. Mater. 16, 1178–1185 (2017).
Tang, D., Kang, R., Coyne, C. B., Zeh, H. J. & Lotze, M. T. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158–175 (2012).
Cen, X., Liu, S. & Cheng, K. The role of toll-like receptor in inflammation and tumor immunity. Front. Pharmacol. 9, 878 (2018).
El-Kenawi, A. & Ruffell, B. Inflammation, ROS, and mutagenesis. Cancer Cell 32, 727–729 (2017).
Barber, G. N. STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770 (2015).
Kurts, C. & Meyer-Schwesinger, C. Protecting the kidney against autoimmunity and inflammation. Nat. Rev. Nephrol. 15, 66–68 (2019).
Neurath, M. F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 20, 970–979 (2019).
McDonald, B. & Kubes, P. Innate immune cell trafficking and function during sterile inflammation of the liver. Gastroenterology 151, 1087–1095 (2016).
Gargalionis, A. N., Basdra, E. K. & Papavassiliou, A. G. Mechanosignalling in tumour progression. J. Cell. Mol. Med. 22, 704 (2018).
Knapik, D. M. et al. Mechanosignaling in bone health, trauma and inflammation. Antioxid. Redox Signal. 20, 970–985 (2014).
Kreuger, J. & Phillipson, M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat. Rev. Drug Discov. 15, 125–142 (2016).
Kolaczkowska, E. & Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 (2013).
Németh, T., Sperandio, M. & Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 19, 253–257 (2020).
Finsterbusch, M., Voisin, M.-B., Beyrau, M., Williams, T. J. & Nourshargh, S. Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. J. Exp. Med. 211, 1307–1314 (2014).
Morikis, V. A. & Simon, S. I. Neutrophil mechanosignaling promotes integrin engagement with endothelial cells and motility within inflamed vessels. Front. Immunol. 9, 2774 (2018).
Ng, L. G. et al. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. J. Invest. Dermatol. 131, 2058–2068 (2011).
Baggiolini, M. Chemokines and leukocyte traffic. Nature 392, 565–568 (1998).
Sallusto, F. & Baggiolini, M. Chemokines and leukocyte traffic. Nat. Immunol. 9, 949–952 (2008).
Larouche, J., Sheoran, S., Maruyama, K. & Martino, M. M. Immune regulation of skin wound healing: mechanisms and novel therapeutic targets. Adv. Wound Care 7, 209–231 (2018).
Atri, C., Guerfali, F. Z. & Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 19, 1801 (2018).
Bäck, M., Yurdagul, A., Tabas, I., Öörni, K. & Kovanen, P. T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Mantovani, A., Dinarello, C. A., Molgora, M. & Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 50, 778–795 (2019).
McInnes, I. B., Buckley, C. D. & Isaacs, J. D. Cytokines in rheumatoid arthritis — shaping the immunological landscape. Nat. Rev. Rheumatol. 12, 63–68 (2016).
Yang, B., Chen, Y. & Shi, J. Reactive oxygen species (ROS)-based nanomedicine. Chem. Rev. 119, 4881–4985 (2019).
Tejero, J., Shiva, S. & Gladwin, M. T. Sources of vascular nitric oxide and reactive oxygen species and their regulation. Physiol. Rev. 99, 311–379 (2019).
Duffin, R. et al. Prostaglandin E2 constrains systemic inflammation through an innate lymphoid cell–IL-22 axis. Science 351, 1333–1338 (2016).
Ramachandran, R., Altier, C., Oikonomopoulou, K. & Hollenberg, M. D. Proteinases, their extracellular targets, and inflammatory signaling. Pharmacol. Rev. 68, 1110–1142 (2016).
Daniel, C. et al. Extracellular DNA traps in inflammation, injury and healing. Nat. Rev. Nephrol. 15, 559–579 (2019).
Boeltz, S. et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 26, 395–408 (2019).
Alaarg, A. et al. Applying nanomedicine in maladaptive inflammation and angiogenesis. Adv. Drug Deliv. Rev. 119, 143–158 (2017).
Yan, H. et al. Engineering cell membrane-based nanotherapeutics to target inflammation. Adv. Sci. 6, 1900605 (2019).
Park, J. et al. Alliance with EPR effect: combined strategies to improve the EPR effect in the tumor microenvironment. Theranostics 9, 8073–8090 (2019).
Youshia, J. & Lamprecht, A. Size-dependent nanoparticulate drug delivery in inflammatory bowel diseases. Expert Opin. Drug Deliv. 13, 281–294 (2016).
Wilhelm, S. et al. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 1, 16014 (2016).
Sindhwani, S. et al. The entry of nanoparticles into solid tumours. Nat. Mater. 19, 566–575 (2020).
Chen, K.-H. et al. Nanoparticle distribution during systemic inflammation is size-dependent and organ-specific. Nanoscale 7, 15863–15872 (2015).
Lundy, D. J., Chen, K.-H., Toh, E. K.-W. & Hsieh, P. C.-H. Distribution of systemically administered nanoparticles reveals a size-dependent effect immediately following cardiac ischaemia-reperfusion injury. Sci. Rep. 6, 25613 (2016).
Kinnear, C., Moore, T. L., Rodriguez-Lorenzo, L., Rothen-Rutishauser, B. & Petri-Fink, A. Form follows function: nanoparticle shape and its implications for nanomedicine. Chem. Rev. 117, 11476–11521 (2017).
Shen, Z., Ye, H., Yi, X. & Li, Y. Membrane wrapping efficiency of elastic nanoparticles during endocytosis: size and shape matter. ACS Nano 13, 215–228 (2018).
Tu, Z. et al. Combination of surface charge and size controls the cellular uptake of functionalized graphene sheets. Adv. Funct. Mater. 27, 1701837 (2017).
Dobrovolskaia, M. A., Aggarwal, P., Hall, J. B. & McNeil, S. E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 5, 487–495 (2008).
Siti, H. N., Kamisah, Y. & Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 71, 40–56 (2015).
Weber, C., Fraemohs, L. & Dejana, E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 7, 467–477 (2007).
Zahr, A. et al. Endomucin prevents leukocyte–endothelial cell adhesion and has a critical role under resting and inflammatory conditions. Nat. Commun. 7, 10363 (2016).
Sager, H. B. et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci. Transl Med. 8, 342ra80 (2016).
McAteer, M. A. et al. In vivo magnetic resonance imaging of acute brain inflammation using microparticles of iron oxide. Nat. Med. 13, 1253–1258 (2007).
Molinaro, R. et al. Biomimetic proteolipid vesicles for targeting inflamed tissues. Nat. Mater. 15, 1037–1046 (2016).
Tuma, P. L. & Hubbard, A. L. Transcytosis: crossing cellular barriers. Physiol. Rev. 83, 871–932 (2003).
Tee, J. K. et al. Nanoparticles’ interactions with vasculature in diseases. Chem. Soc. Rev. 48, 5381–5407 (2019).
Pandit, S., Dutta, D. & Nie, S. Active transcytosis and new opportunities for cancer nanomedicine. Nat. Mater. 19, 478–480 (2020).
Villaseñor, R., Lampe, J., Schwaninger, M. & Collin, L. Intracellular transport and regulation of transcytosis across the blood–brain barrier. Cell. Mol. Life Sci. 76, 1081–1092 (2019).
Berndt, D. et al. Inflammation-induced brain endothelial activation leads to uptake of electrostatically stabilized iron oxide nanoparticles via sulfated glycosaminoglycans. Nanomed. Nanotechnol. Biol. Med. 13, 1411–1421 (2017).
Poller, W. C. et al. Uptake of citrate-coated iron oxide nanoparticles into atherosclerotic lesions in mice occurs via accelerated transcytosis through plaque endothelial cells. Nano Res. 9, 3437–3452 (2016).
Dejana, E. Endothelial cell–cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5, 261–270 (2004).
Setyawati, M. I. et al. Titanium dioxide nanomaterials cause endothelial cell leakiness by disrupting the homophilic interaction of VE–cadherin. Nat. Commun. 4, 1673 (2013).
Setyawati, M. I., Tay, C. Y., Bay, B. H. & Leong, D. T. Gold nanoparticles induced endothelial leakiness depends on particle size and endothelial cell origin. ACS Nano 11, 5020–5030 (2017).
Wang, J., Zhang, L., Peng, F., Shi, X. & Leong, D. T. Targeting endothelial cell junctions with negatively charged gold nanoparticles. Chem. Mater. 30, 3759–3767 (2018).
Han, X. et al. Zwitterionic micelles efficiently deliver oral insulin without opening tight junctions. Nat. Nanotechnol. 15, 605–614 (2020).
Peng, F. et al. Nanoparticles promote in vivo breast cancer cell intravasation and extravasation by inducing endothelial leakiness. Nat. Nanotechnol. 14, 279–286 (2019).
Lim, J. et al. Inflammation drives retraction, stiffening, and nodule formation via cytoskeletal machinery in a three-dimensional culture model of aortic stenosis. Am. J. Pathol. 186, 2378–2389 (2016).
Myerson, J. W. et al. Non-affinity factors modulating vascular targeting of nano- and microcarriers. Adv. Drug Deliv. Rev. 99, 97–112 (2016).
Nicolás-Ávila, J. Á., Adrover, J. M. & Hidalgo, A. Neutrophils in homeostasis, immunity, and cancer. Immunity 46, 15–28 (2017).
Wang, Z., Li, J., Cho, J. & Malik, A. B. Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol. 9, 204–210 (2014).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011).
Lameijer, M. et al. Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nat. Biomed. Eng. 2, 279–292 (2018).
Ye, H. et al. Recent advances on reactive oxygen species-responsive delivery and diagnosis system. Biomacromolecules 20, 2441–2463 (2019).
Aratani, Y. Myeloperoxidase: its role for host defense, inflammation, and neutrophil function. Arch. Biochem. Biophys. 640, 47–52 (2018).
Guo, J. et al. A myeloperoxidase-responsive and biodegradable luminescent material for real-time imaging of inflammatory diseases. Mater. Today 20, 493–500 (2017).
Xu, X. et al. A self-illuminating nanoparticle for inflammation imaging and cancer therapy. Sci. Adv. 5, eaat2953 (2019).
Gallo, J. et al. CXCR4-targeted and MMP-responsive iron oxide nanoparticles for enhanced magnetic resonance imaging. Angew. Chem. Int. Ed. 53, 9550–9554 (2014).
Cai, C. et al. MMP-2 responsive unidirectional hydrogel-electrospun patch loading TGF-β1 siRNA polyplexes for peritendinous anti-adhesion. Adv. Funct. Mater. 31, 2008364 (2021).
Wong, C., Pratiwi, F. W., Chen, P., Mou, C. & Hsu, S. Revealing the phagosomal pH regulation and inflammation of macrophages after endocytosing polyurethane nanoparticles by a ratiometric pH nanosensor. Adv. Biol. 5, 2000200 (2021).
Zhang, C. Y. et al. pH-responsive nanoparticles targeted to lungs for improved therapy of acute lung inflammation/injury. ACS Appl. Mater. Interfaces 11, 16380–16390 (2019).
Zhao, C. et al. Oxidative-species-selective materials for diagnostic and therapeutic applications. Angew. Chem. Int. Ed. 60, 9804–9827 (2021).
Liu, Y. & Shi, J. Antioxidative nanomaterials and biomedical applications. Nano Today 27, 146–177 (2019).
Huang, Y., Ren, J. & Qu, X. Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem. Rev. 119, 4357–4412 (2019).
Suzuki, K. Anti-oxidants for therapeutic use: why are only a few drugs in clinical use? Adv. Drug Deliv. Rev. 61, 287–289 (2009).
Cao, F. et al. An enzyme-mimicking single-atom catalyst as an efficient multiple reactive oxygen and nitrogen species scavenger for sepsis management. Angew. Chem. Int. Ed. 132, 5146–5153 (2020).
Mugesh, G. & Singh, H. B. Synthetic organoselenium compounds as antioxidants: glutathione peroxidase activity. Chem. Soc. Rev. 29, 347–357 (2000).
Huang, X., Liu, X., Luo, Q., Liu, J. & Shen, J. Artificial selenoenzymes: designed and redesigned. Chem. Soc. Rev. 40, 1171–1184 (2011).
Sun, H. et al. Self-assembly of cricoid proteins induced by “soft nanoparticles”: an approach to design multienzyme-cooperative antioxidative systems. ACS Nano 9, 5461–5469 (2015).
Huang, X., Yin, Y. & Liu, J. Design of artificial selenoenzymes based on macromolecular scaffolds. Macromol. Biosci. 10, 1385–1396 (2010).
Moglianetti, M. et al. Platinum nanozymes recover cellular ROS homeostasis in an oxidative stress-mediated disease model. Nanoscale 8, 3739–3752 (2016).
Liu, C. et al. Tailoring enzyme-like activities of gold nanoclusters by polymeric tertiary amines for protecting neurons against oxidative stress. Small 12, 4127–4135 (2016).
Ge, C. et al. Facet energy versus enzyme-like activities: the unexpected protection of palladium nanocrystals against oxidative damage. ACS Nano 10, 10436–10445 (2016).
Miao, Z. et al. Ultrasmall rhodium nanozyme with RONS scavenging and photothermal activities for anti-inflammation and antitumor theranostics of colon diseases. Nano Lett. 20, 3079–3089 (2020).
Selvaraj, V. et al. Inhibition of MAP kinase/NF-kB mediated signaling and attenuation of lipopolysaccharide induced severe sepsis by cerium oxide nanoparticles. Biomaterials 59, 160–171 (2015).
Kim, J. et al. Functional recovery of contused spinal cord in rat with the injection of optimal-dosed cerium oxide nanoparticles. Adv. Sci. 4, 1700034 (2017).
Kwon, H. J. et al. Mitochondria-targeting ceria nanoparticles as antioxidants for Alzheimer’s disease. ACS Nano 10, 2860–2870 (2016).
Kwon, H. J. et al. Ceria nanoparticle systems for selective scavenging of mitochondrial, intracellular, and extracellular reactive oxygen species in Parkinson’s disease. Angew. Chem. Int. Ed. 57, 9408–9412 (2018).
Soh, M. et al. Ceria–zirconia nanoparticles as an enhanced multi-antioxidant for sepsis treatment. Angew. Chem. Int. Ed. 129, 11557–11561 (2017).
Choi, S. W., Cha, B. G. & Kim, J. Therapeutic contact lens for scavenging excessive reactive oxygen species on the ocular surface. ACS Nano 14, 2483–2496 (2020).
Lord, M. S. et al. Cellular uptake and reactive oxygen species modulation of cerium oxide nanoparticles in human monocyte cell line U937. Biomaterials 33, 7915–7924 (2012).
Zhang, Y. et al. Dietary iron oxide nanoparticles delay aging and ameliorate neurodegeneration in Drosophila. Adv. Mater. 28, 1387–1393 (2016).
Vernekar, A. A. et al. An antioxidant nanozyme that uncovers the cytoprotective potential of vanadia nanowires. Nat. Commun. 5, 5301 (2014).
Pereira, D. R. et al. Scavenging nanoreactors that modulate inflammation. Adv. Biosyst. 2, 1800086 (2018).
Yao, J. et al. ROS scavenging Mn3O4 nanozymes for in vivo anti-inflammation. Chem. Sci. 9, 2927–2933 (2018).
Kim, J. et al. Synergistic oxygen generation and reactive oxygen species scavenging by manganese ferrite/ceria co-decorated nanoparticles for rheumatoid arthritis treatment. ACS Nano 13, 3206–3217 (2019).
Liu, T. et al. Ultrasmall copper-based nanoparticles for reactive oxygen species scavenging and alleviation of inflammation related diseases. Nat. Commun. 11, 2788 (2020).
Tu, Z., Guday, G., Adeli, M. & Haag, R. Multivalent interactions between 2D nanomaterials and biointerfaces. Adv. Mater. 30, 1706709 (2018).
Yim, D. et al. Adjustable intermolecular interactions allowing 2D transition metal dichalcogenides with prolonged scavenging activity for reactive oxygen species. Small 14, 1800026 (2018).
Ji, D. et al. Targeted intracellular production of reactive oxygen species by a 2D molybdenum disulfide glycosheet. Adv. Mater. 28, 9356–9363 (2016).
Zhang, X.-D. et al. Highly catalytic nanodots with renal clearance for radiation protection. ACS Nano 10, 4511–4519 (2016).
Ni, D. et al. Molybdenum-based nanoclusters act as antioxidants and ameliorate acute kidney injury in mice. Nat. Commun. 9, 5421 (2018).
Jiao, L. et al. When nanozymes meet single-atom catalysis. Angew. Chem. Int. Ed. 132, 2585–2596 (2020).
Zhang, X. et al. Single-atom nanozymes: a rising star for biosensing and biomedicine. Coord. Chem. Rev. 418, 213376 (2020).
Ji, Z., Zhang, H., Liu, H., Yaghi, O. M. & Yang, P. Cytoprotective metal-organic frameworks for anaerobic bacteria. Proc. Natl Acad. Sci. USA 115, 10582–10587 (2018).
Chen, W. et al. Black phosphorus nanosheets as a neuroprotective nanomedicine for neurodegenerative disorder therapy. Adv. Mater. 30, 1703458 (2018).
Sun, H., Zhou, Y., Ren, J. & Qu, X. Carbon nanozymes: enzymatic properties, catalytic mechanism, and applications. Angew. Chem. Int. Ed. 57, 9224–9237 (2018).
Lee, H. J. et al. Amine-modified single-walled carbon nanotubes protect neurons from injury in a rat stroke model. Nat. Nanotechnol. 6, 120–124 (2011).
Jiang, D. et al. DNA origami nanostructures can exhibit preferential renal uptake and alleviate acute kidney injury. Nat. Biomed. Eng. 2, 865–877 (2018).
Bao, X., Zhao, J., Sun, J., Hu, M. & Yang, X. Polydopamine nanoparticles as efficient scavengers for reactive oxygen species in periodontal disease. ACS Nano 12, 8882–8892 (2018).
Lee, J. et al. Nucleic acid-binding polymers as anti-inflammatory agents. Proc. Natl Acad. Sci. USA 108, 14055–14060 (2011).
Jain, S. et al. Nucleic acid scavengers inhibit thrombosis without increasing bleeding. Proc. Natl Acad. Sci. USA 109, 12938–12943 (2012).
Holl, E. K. et al. The nucleic acid scavenger dendrimer polyamidoamine third-generation dendrimer inhibits fibroblast activation and inhibits granulation tissue contraction. Plast. Reconstr. Surg. 134, 420e–433e (2014).
Holl, E. K. et al. Scavenging nucleic acid debris to combat autoimmunity and infectious disease. Proc. Natl Acad. Sci. USA 113, 9728–9733 (2016).
Naqvi, I. et al. Polymer-mediated inhibition of pro-invasive nucleic acid DAMPs and microvesicles limits pancreatic cancer metastasis. Mol. Ther. 26, 1020–1031 (2018).
Peng, B. et al. Tuned cationic dendronized polymer: molecular scavenger for rheumatoid arthritis treatment. Angew. Chem. Int. Ed. 58, 4254–4258 (2019).
Meneksedag-Erol, D., Kizhakkedathu, J. N., Tang, T. & Uludağ, H. Molecular dynamics simulations on nucleic acid binding polymers designed to arrest thrombosis. ACS Appl. Mater. Interfaces 10, 28399–28411 (2018).
Aswani, A. et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front. Immunol. 9, 891 (2018).
Chen, H. H. et al. Theranostic nucleic acid binding nanoprobe exerts anti-inflammatory and cytoprotective effects in ischemic injury. Theranostics 7, 814–825 (2017).
Liang, H. et al. Cationic nanoparticle as an inhibitor of cell-free DNA-induced inflammation. Nat. Commun. 9, 4291 (2018).
Wu, J. et al. Cationic block copolymer nanoparticles with tunable DNA affinity for treating rheumatoid arthritis. Adv. Funct. Mater. 30, 2000391 (2020).
Dawulieti, J. et al. Treatment of severe sepsis with nanoparticulate cell-free DNA scavengers. Sci. Adv. 6, eaay7148 (2020).
Jackman, J. G. et al. Polycationic nanofibers for nucleic acid scavenging. Biomacromolecules 17, 3706–3713 (2016).
Lee, J. et al. Nucleic acid scavenging microfiber mesh inhibits trauma-induced inflammation and thrombosis. Biomaterials 120, 94–102 (2017).
Liu, F. et al. A cationic metal–organic framework to scavenge cell-free DNA for severe sepsis management. Nano Lett. 21, 2461–2469 (2021).
Foit, L. & Thaxton, C. S. Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding Toll-like receptor 4. Biomaterials 100, 67–75 (2016).
Lohmann, N. et al. Glycosaminoglycan-based hydrogels capture inflammatory chemokines and rescue defective wound healing in mice. Sci. Transl Med. 9, eaai9044 (2017).
Boyle, W. S. et al. Ternary composite nanofibers containing chondroitin sulfate scavenge inflammatory chemokines from solution and prohibit squamous cell carcinoma migration. ACS Appl. Bio Mater. 2, 619–624 (2019).
Puthia, M. et al. A dual-action peptide-containing hydrogel targets wound infection and inflammation. Sci. Transl Med. 12, eaax6601 (2020).
Ren, H. et al. Role of liposome size, surface charge, and PEGylation on rheumatoid arthritis targeting therapy. ACS Appl. Mater. Interfaces 11, 20304–20315 (2019).
Da Silva-Candal, A. et al. Shape effect in active targeting of nanoparticles to inflamed cerebral endothelium under static and flow conditions. J. Control. Release 309, 94–105 (2019).
Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 15, 692–704 (2015).
Liu, Z. et al. L-selectin mechanochemistry restricts neutrophil priming in vivo. Nat. Commun. 8, 15196 (2017).
Wang, L., Fuster, M., Sriramarao, P. & Esko, J. D. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 (2005).
Türk, H., Haag, R. & Alban, S. Dendritic polyglycerol sulfates as new heparin analogues and potent inhibitors of the complement system. Bioconjug. Chem. 15, 162–167 (2004).
Dernedde, J. et al. Dendritic polyglycerol sulfates as multivalent inhibitors of inflammation. Proc. Natl Acad. Sci. USA 107, 19679–19684 (2010).
Schneider, T. et al. Effects of dendritic polyglycerol sulfate on articular chondrocytes. Inflamm. Res. 64, 917–928 (2015).
Ferraro, M. et al. Biodegradable polyglycerol sulfates exhibit promising features for anti-inflammatory applications. Biomacromolecules 19, 4524–4533 (2018).
Reimann, S. et al. Shell cleavable dendritic polyglycerol sulfates show high anti-inflammatory properties by inhibiting L-selectin binding and complement activation. Adv. Healthc. Mater. 4, 2154–2162 (2015).
Silberreis, K., Niesler, N., Rades, N., Haag, R. & Dernedde, J. Sulfated dendritic polyglycerol is a potent complement inhibitor. Biomacromolecules 20, 3809–3818 (2019).
Chen, Y.-S., Zhao, Y., Yoon, S. J., Gambhir, S. S. & Emelianov, S. Miniature gold nanorods for photoacoustic molecular imaging in the second near-infrared optical window. Nat. Nanotechnol. 14, 465–472 (2019).
Vonnemann, J. et al. Polyglycerolsulfate functionalized gold nanorods as optoacoustic signal nanoamplifiers for in vivo bioimaging of rheumatoid arthritis. Theranostics 4, 629–641 (2014).
Rele, S. M. et al. Dendrimer-like PEO glycopolymers exhibit anti-inflammatory properties. J. Am. Chem. Soc. 127, 10132–10133 (2005).
Zhang, K. et al. Anti-inflammatory properties of GLPss58, a sulfated polysaccharide from Ganoderma lucidum. Int. J. Biol. Macromol. 107, 486–493 (2018).
Hayder, M. et al. A phosphorus-based dendrimer targets inflammation and osteoclastogenesis in experimental arthritis. Sci. Transl Med. 3, 81ra35 (2011).
Hayder, M. et al. Phosphorus-based dendrimer ABP treats neuroinflammation by promoting IL-10-producing CD4+ T cells. Biomacromolecules 16, 3425–3433 (2015).
Ledall, J. et al. Interaction studies reveal specific recognition of an anti-inflammatory polyphosphorhydrazone dendrimer by human monocytes. Nanoscale 7, 17672–17684 (2015).
Weinhart, M., Gröger, D., Enders, S., Dernedde, J. & Haag, R. Synthesis of dendritic polyglycerol anions and their efficiency toward L-selectin inhibition. Biomacromolecules 12, 2502–2511 (2011).
Gonzalez-Rodriguez, S. et al. Polyglycerol-opioid conjugate produces analgesia devoid of side effects. eLife 6, e27081 (2017).
Moog, K. E. et al. Polymeric selectin ligands mimicking complex carbohydrates: from selectin binders to modifiers of macrophage migration. Angew. Chem. Int. Ed. 56, 1416–1421 (2017).
Blattes, E. et al. Mannodendrimers prevent acute lung inflammation by inhibiting neutrophil recruitment. Proc. Natl Acad. Sci. USA 110, 8795–8800 (2013).
Gao, W., Xiong, Y., Li, Q. & Yang, H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front. Physiol. 8, 508 (2017).
Yang, H. et al. Amino acid-dependent attenuation of toll-like receptor signaling by peptide-gold nanoparticle hybrids. ACS Nano 9, 6774–6784 (2015).
Yang, H. et al. Endosomal pH modulation by peptide-gold nanoparticle hybrids enables potent anti-inflammatory activity in phagocytic immune cells. Biomaterials 111, 90–102 (2016).
Gao, W. et al. Size-dependent anti-inflammatory activity of a peptide-gold nanoparticle hybrid in vitro and in a mouse model of acute lung injury. Acta Biomater. 85, 203–217 (2019).
Gao, W. et al. Enhanced anti-inflammatory activity of peptide–gold nanoparticle hybrids upon cigarette smoke extract modification through TLR inhibition and autophagy induction. ACS Appl. Mater. Interfaces 11, 32706–32719 (2019).
Hu, Y.-H. et al. SPOP negatively regulates Toll-like receptor-induced inflammation by disrupting MyD88 self-association. Cell. Mol. Immunol. 18, 1708–1717 (2021).
Impellizzeri, D. & Cuzzocrea, S. Targeting selectins for the treatment of inflammatory diseases. Expert Opin. Ther. Targets 18, 55–67 (2014).
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20, 101–124 (2021).
Manzari, M. T. et al. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 6, 351–370 (2021).
Adams, J. L., Smothers, J., Srinivasan, R. & Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 14, 603–622 (2015).
Lin, A. et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl Med. 11, eaaw8412 (2019).
Zou, Y., Huang, B., Cao, L., Deng, Y. & Su, J. Tailored mesoporous inorganic biomaterials: assembly, functionalization, and drug delivery engineering. Adv. Mater. 33, 2005215 (2021).
Schudel, A., Francis, D. M. & Thomas, S. N. Material design for lymph node drug delivery. Nat. Rev. Mater. 4, 415–428 (2019).
Tu, Z. et al. Directed graphene-based nanoplatforms for hyperthermia: overcoming multiple drug resistance. Angew. Chem. Int. Ed. 57, 11198–11202 (2018).
Calin, M. & Manduteanu, I. Emerging nanocarriers-based approaches to diagnose and reduce vascular inflammation in atherosclerosis. Curr. Med. Chem. 24, 550–567 (2017).
Makadia, H. K. & Siegel, S. J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3, 1377–1397 (2011).
Chung, M.-F., Chia, W.-T., Wan, W.-L., Lin, Y.-J. & Sung, H.-W. Controlled release of an anti-inflammatory drug using an ultrasensitive ROS-responsive gas-generating carrier for localized inflammation inhibition. J. Am. Chem. Soc. 137, 12462–12465 (2015).
Di Francesco, M. et al. Engineering shape-defined PLGA microPlates for the sustained release of anti-inflammatory molecules. J. Control. Release 319, 201–212 (2020).
Bartneck, M. et al. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 37, 367–382 (2015).
Jia, M. et al. A novel dexamethasone-loaded liposome alleviates rheumatoid arthritis in rats. Int. J. Pharm. 540, 57–64 (2018).
Gao, J., Wang, S. & Wang, Z. High yield, scalable and remotely drug-loaded neutrophil-derived extracellular vesicles (EVs) for anti-inflammation therapy. Biomaterials 135, 62–73 (2017).
Badri, W. et al. Poly (ε-caprolactone) nanoparticles loaded with indomethacin and Nigella sativa L. essential oil for the topical treatment of inflammation. J. Drug Deliv. Sci. Technol. 46, 234–242 (2018).
Soiberman, U. et al. Subconjunctival injectable dendrimer-dexamethasone gel for the treatment of corneal inflammation. Biomaterials 125, 38–53 (2017).
Jiang, K. et al. Local release of dexamethasone from macroporous scaffolds accelerates islet transplant engraftment by promotion of anti-inflammatory M2 macrophages. Biomaterials 114, 71–81 (2017).
Zhang, C. Y., Gao, J. & Wang, Z. Bioresponsive nanoparticles targeted to infectious microenvironments for sepsis management. Adv. Mater. 30, 1803618 (2018).
Jacob, J., Haponiuk, J. T., Thomas, S. & Gopi, S. Biopolymer based nanomaterials in drug delivery systems: a review. Mater. Today Chem. 9, 43–55 (2018).
Formica, F. A., Barreto, G. & Zenobi-Wong, M. Cartilage-targeting dexamethasone prodrugs increase the efficacy of dexamethasone. J. Control. Release 295, 118–129 (2019).
Moshaverinia, A. et al. Regulation of the stem cell–host immune system interplay using hydrogel coencapsulation system with an anti-inflammatory drug. Adv. Funct. Mater. 25, 2296–2307 (2015).
Tang, W., Yang, J., Zhao, Z., Lian, Z. & Liang, G. Intracellular coassembly boosts the anti-inflammation capacity of dexamethasone. Nanoscale 9, 17717–17721 (2017).
Sellner, S. et al. Dexamethasone-conjugated DNA nanotubes as anti-inflammatory agents in vivo. Biomaterials 134, 78–90 (2017).
Brown, T. D., Whitehead, K. A. & Mitragotri, S. Materials for oral delivery of proteins and peptides. Nat. Rev. Mater. 5, 127–148 (2020).
Yan, Y. et al. Euryale ferox seed-inspired superlubricated nanoparticles for treatment of osteoarthritis. Adv. Funct. Mater. 29, 1807559 (2019).
Juère, E. et al. In vitro dissolution, cellular membrane permeability, and anti-inflammatory response of resveratrol-encapsulated mesoporous silica nanoparticles. Mol. Pharm. 14, 4431–4441 (2017).
Frede, A. et al. Colonic gene silencing using siRNA-loaded calcium phosphate/PLGA nanoparticles ameliorates intestinal inflammation in vivo. J. Control. Release 222, 86–96 (2016).
Scheinman, R. I., Trivedi, R., Vermillion, S. & Kompella, U. B. Functionalized STAT1 siRNA nanoparticles regress rheumatoid arthritis in a mouse model. Nanomedicine 6, 1669–1682 (2011).
Chen, Z., Krishnamachary, B., Penet, M.-F. & Bhujwalla, Z. M. Acid-degradable dextran as an image guided siRNA carrier for COX-2 downregulation. Theranostics 8, 1–12 (2018).
Stansel, T., Wickline, S. A. & Pan, H. NF-κB inhibition suppresses experimental melanoma lung metastasis. J. Cancer Sci. Clin. Ther. 4, 256–265 (2020).
Wang, Q. et al. Targeting NF-kB signaling with polymeric hybrid micelles that co-deliver siRNA and dexamethasone for arthritis therapy. Biomaterials 122, 10–22 (2017).
Xu, X. et al. Efficient and targeted drug/siRNA co-delivery mediated by reversibly crosslinked polymersomes toward anti-inflammatory treatment of ulcerative colitis (UC). Nano Res. 12, 659–667 (2019).
Wilson, D. S. et al. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 9, 923–928 (2010).
Aldayel, A. M. et al. Lipid nanoparticles with minimum burst release of TNF-α siRNA show strong activity against rheumatoid arthritis unresponsive to methotrexate. J. Control. Release 283, 280–289 (2018).
Zhao, G. et al. Nanoparticle-delivered siRNA targeting Bruton’s tyrosine kinase for rheumatoid arthritis therapy. Biomater. Sci. 7, 4698–4707 (2019).
Tang, Y. et al. SiRNA crosslinked nanoparticles for the treatment of inflammation-induced liver injury. Adv. Sci. 4, 1600228 (2017).
Dash, B. C. et al. An injectable elastin-based gene delivery platform for dose-dependent modulation of angiogenesis and inflammation for critical limb ischemia. Biomaterials 65, 126–139 (2015).
Kim, G., Piao, C., Oh, J. & Lee, M. Self-assembled polymeric micelles for combined delivery of anti-inflammatory gene and drug to the lungs by inhalation. Nanoscale 10, 8503–8514 (2018).
Xu, C. et al. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat. Commun. 9, 4092 (2018).
Wang, H.-X. et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem. Rev. 117, 9874–9906 (2017).
Kwon, D. et al. Extra-large pore mesoporous silica nanoparticles for directing in vivo M2 macrophage polarization by delivering IL-4. Nano Lett. 17, 2747–2756 (2017).
Frey, L. et al. A dual-layered microfluidic system for long-term controlled in situ delivery of multiple anti-inflammatory factors for chronic neural applications. Adv. Funct. Mater. 28, 1702009 (2018).
Kim, M. et al. Targeted delivery of anti-inflammatory cytokine by nanocarrier reduces atherosclerosis in Apo E−/-mice. Biomaterials 226, 119550 (2020).
Shamskhou, E. A. et al. Hydrogel-based delivery of IL-10 improves treatment of bleomycin-induced lung fibrosis in mice. Biomaterials 203, 52–62 (2019).
Meka, R. R., Venkatesha, S. H. & Moudgil, K. D. Peptide-directed liposomal delivery improves the therapeutic index of an immunomodulatory cytokine in controlling autoimmune arthritis. J. Control. Release 286, 279–288 (2018).
Li, G. et al. Graft of the NT-3 persistent delivery gelatin sponge scaffold promotes axon regeneration, attenuates inflammation, and induces cell migration in rat and canine with spinal cord injury. Biomaterials 83, 233–248 (2016).
Krieger, J. R. et al. Spatially localized recruitment of anti-inflammatory monocytes by SDF-1α-releasing hydrogels enhances microvascular network remodeling. Biomaterials 77, 280–290 (2016).
Liu, J. M. H. et al. Transforming growth factor-beta 1 delivery from microporous scaffolds decreases inflammation post-implant and enhances function of transplanted islets. Biomaterials 80, 11–19 (2016).
Wu, J. et al. A tumor microenvironment-responsive biodegradable mesoporous nanosystem for anti-inflammation and cancer theranostics. Adv. Healthc. Mater. 9, 1901307 (2020).
Tolouei, A. E., Dülger, N., Ghatee, R. & Kennedy, S. A magnetically responsive biomaterial system for flexibly regulating the duration between pro-and anti-inflammatory cytokine deliveries. Adv. Healthc. Mater. 7, 1800227 (2018).
Fujita, K., Tanaka, Y., Abe, S. & Ueno, T. A photoactive carbon-monoxide-releasing protein cage for dose-regulated delivery in living cells. Angew. Chem. Int. Ed. 55, 1056–1060 (2016).
Popova, M., Soboleva, T., Ayad, S., Benninghoff, A. D. & Berreau, L. M. Visible-light-activated quinolone carbon-monoxide-releasing molecule: prodrug and albumin-assisted delivery enables anticancer and potent anti-inflammatory effects. J. Am. Chem. Soc. 140, 9721–9729 (2018).
Ji, X. et al. Esterase-sensitive and pH-controlled carbon monoxide prodrugs for treating systemic inflammation. J. Med. Chem. 62, 3163–3168 (2019).
Kim, I. et al. Supramolecular carbon monoxide-releasing peptide hydrogel patch. Adv. Funct. Mater. 28, 1803051 (2018).
Yang, G. et al. A multifunctional anti-inflammatory drug that can specifically target activated macrophages, massively deplete intracellular H2O2, and produce large amounts CO for a highly efficient treatment of osteoarthritis. Biomaterials 255, 120155 (2020).
Wang, S.-B. et al. Near-infrared light responsive nanoreactor for simultaneous tumor photothermal therapy and carbon monoxide-mediated anti-inflammation. ACS Cent. Sci. 6, 555–565 (2020).
Wan, W.-L. et al. In situ nanoreactor for photosynthesizing H2 gas to mitigate oxidative stress in tissue inflammation. J. Am. Chem. Soc. 139, 12923–12926 (2017).
Zhang, B. et al. Polymer dots compartmentalized in liposomes as a photocatalyst for in situ hydrogen therapy. Angew. Chem. Int. Ed. 58, 2744–2748 (2019).
Wan, W. et al. An in situ depot for continuous evolution of gaseous H2 mediated by a magnesium passivation/activation cycle for treating osteoarthritis. Angew. Chem. Int. Ed. 130, 10023–10027 (2018).
Wan, W.-L. et al. Photosynthesis-inspired H2 generation using a chlorophyll-loaded liposomal nanoplatform to detect and scavenge excess ROS. Nat. Commun. 11, 534 (2020).
Wallace, J. L. et al. A proof-of-concept, Phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br. J. Pharmacol. 177, 769–777 (2020).
Akinc, A. et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 26, 561–569 (2008).
Khurana, A. et al. Role of nanotechnology behind the success of mRNA vaccines for COVID-19. Nano Today 38, 101142 (2021).
Shao, D. et al. Bioinspired diselenide-bridged mesoporous silica nanoparticles for dual-responsive protein delivery. Adv. Mater. 30, 1801198 (2018).
Shao, D. et al. Biomimetic diselenide-bridged mesoporous organosilica nanoparticles as an X-ray-responsive biodegradable carrier for chemo-immunotherapy. Adv. Mater. 32, 2004385 (2020).
He, W. et al. Circadian expression of migratory factors establishes lineage-specific signatures that guide the homing of leukocyte subsets to tissues. Immunity 49, 1175–1190 (2018).
Westphalen, K. et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 506, 503–506 (2014).
Ghatnekar, G. S. et al. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 4, 205–223 (2009).
Ghatnekar, G. S., Grek, C. L., Armstrong, D. G., Desai, S. C. & Gourdie, R. G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: a multicenter, randomized trial. J. Invest. Dermatol. 135, 289–298 (2015).
Kalluri, R. & LeBleu, V. S. The biology, function, and biomedical applications of exosomes. Science 367, eaau6977 (2020).
Yang, Z. et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 4, 69–83 (2020).
Tay, M. Z., Poh, C. M., Rénia, L., MacAry, P. A. & Ng, L. F. P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20, 363–374 (2020).
Suh, J. S. et al. Periodontitis-induced systemic inflammation exacerbates atherosclerosis partly via endothelial–mesenchymal transition in mice. Int. J. Oral Sci. 11, 21 (2019).
Chen, Z. et al. Real-time observation of leukocyte–endothelium interactions in tissue-engineered blood vessel. Lab Chip 18, 2047–2054 (2018).
Trapecar, M. et al. Gut-Liver physiomimetics reveal paradoxical modulation of IBD-related inflammation by short-chain fatty acids. Cell Syst. 10, 223–239 (2020).
Lee, J. H. et al. Emulating early atherosclerosis in a vascular microphysiological system using branched tissue-engineered blood vessels. Adv. Biol. 5, 2000428 (2021).
Maharjan, S., Cecen, B. & Zhang, Y. S. 3D immunocompetent organ-on-a-chip models. Small Methods 4, 2000235 (2020).
Sharma, A., Sances, S., Workman, M. J. & Svendsen, C. N. Multi-lineage human iPSC-derived platforms for disease modeling and drug discovery. Cell Stem Cell 26, 309–329 (2020).
Irwin, M. R. & Vitiello, M. V. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 18, 296–306 (2019).
Acknowledgements
The authors acknowledge support by the NIH (UG3/UH3TR002151, UG3NS115598, RO1AR073935) and USAMR W81XWH1910463. Support from the National Natural Science Foundation of China (grant no. 82072049), the Fundamental Research Funds for the Central Universities and the Australian Research Council (DE180100736) is also acknowledged. The authors would also like to thank C. Yang and Y. Zhu for scientific discussions.
Author information
Authors and Affiliations
Contributions
Z.T., D.S. and K.W.L. conceived the manuscript; Z.T., Y.Z., D.S. and H.H. wrote the draft; Z.T. and D.S. designed the figures; H.H., R.H., M.S., J.L., B.S. and K.W.L. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Materials thanks Abhay Pandit, Jinjun Shi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tu, Z., Zhong, Y., Hu, H. et al. Design of therapeutic biomaterials to control inflammation. Nat Rev Mater 7, 557–574 (2022). https://doi.org/10.1038/s41578-022-00426-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41578-022-00426-z
This article is cited by
-
Asymmetric adhesive SIS-based wound dressings for therapeutically targeting wound repair
Journal of Nanobiotechnology (2024)
-
Oxidative stress and inflammation: elucidating mechanisms of smoking-attributable pathology for therapeutic targeting
Bulletin of the National Research Centre (2024)
-
Tuning oxidant and antioxidant activities of ceria by anchoring copper single-site for antibacterial application
Nature Communications (2024)
-
Model-based modular hydrogel design
Nature Reviews Bioengineering (2024)
-
The neutrophil-to-Lymphocyte ratio is associated with clinical symptoms in first-episode medication-naïve patients with schizophrenia
Schizophrenia (2024)
