
Abstract
The majority of risk loci identified by genome-wide association studies (GWAS) are in non-coding regions, hampering their functional interpretation. Instead, transcriptome-wide association studies (TWAS) identify gene-trait associations, which can be used to prioritize candidate genes in disease-relevant tissue(s). Here, we aimed to systematically identify susceptibility genes for coronary artery disease (CAD) by TWAS. We trained prediction models of nine CAD-relevant tissues using EpiXcan based on two genetics-of-gene-expression panels, the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) and the Genotype-Tissue Expression (GTEx). Based on these prediction models, we imputed gene expression of respective tissues from individual-level genotype data on 37,997 CAD cases and 42,854 controls for the subsequent gene-trait association analysis. Transcriptome-wide significant association (i.e. P < 3.85e−6) was observed for 114 genes. Of these, 96 resided within previously identified GWAS risk loci and 18 were novel. Stepwise analyses were performed to study their plausibility, biological function, and pathogenicity in CAD, including analyses for colocalization, damaging mutations, pathway enrichment, phenome-wide associations with human data and expression-traits correlations using mouse data. Finally, CRISPR/Cas9-based gene knockdown of two newly identified TWAS genes, RGS19 and KPTN, in a human hepatocyte cell line resulted in reduced secretion of APOB100 and lipids in the cell culture medium. Our CAD TWAS work (i) prioritized candidate causal genes at known GWAS loci, (ii) identified 18 novel genes to be associated with CAD, and iii) suggested potential tissues and pathways of action for these TWAS CAD genes.
Similar content being viewed by others


Introduction
Coronary artery disease (CAD), a leading cause of premature death worldwide, is influenced by interactions of lifestyle, environmental, and genetic risk factors [43]. Genome-wide association studies (GWAS) have identified over 200 risk loci for CAD [11, 17, 35]. Most of them are located in non-coding regions which hampers their functional interpretation. Expression quantitative traits loci (eQTLs) to some extent explain the genomic effects of GWAS signals [19, 61, 64]. By leveraging effects of multiple cis-eQTL variants on gene expression, transcriptome-wide association studies (TWAS) search primarily for gene-trait associations. The approach first builds on prediction models of gene expression from reference panels that correlate genotype patterns with transcript levels in tissues which are relevant for the phenotype. Prediction models are then used to impute tissue-specific gene expression based on genotypes with a given trait in individuals of GWAS cohorts [21]. Since TWAS signals reflect association between transcriptome-wide genetically regulated expression (GReX) and traits or diseases, the approach can be used to prioritize candidate genes across disease-relevant tissues. Thereby, TWAS may point to causal genes at risk loci identified by GWAS and thus provide further insights into biological mechanisms [62, 70]. Moreover, TWAS increase the sensitivity to identify susceptibility genes missed by traditional GWAS analyses. Here we performed a TWAS to identify novel susceptibility genes for CAD comprising more than 80,000 individuals with genotype data along with validation and exploratory analyses for the associated genes.
Materials and methods
Prediction models of nine tissues based on two reference panels
The starting point of this investigation was two large human biobanks with individual-level data on genome-wide genotypes as well as mRNA expression levels in multiple tissues with relevance for CAD. These include atherosclerotic aortic wall (AOR), atherosclerotic-lesion-free internal mammary artery (MAM), liver (LIV), blood (BLD), subcutaneous fat (SF), visceral abdominal fat (VAF), and skeletal muscle (SKLM) in the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) [20], and AOR, LIV, BLD, SF, VAF, and SKLM in the Genotype-Tissue Expression (GTEx) [1] (Supplementary Table 1). Arterial wall coronary (COR) and tibial artery (TIB) datasets were only available in the GTEx. The pipeline used for training prediction models was EpiXcan which was built on the basis of PrediXcan but with improved prediction performance by integrating epigenomic annotation data into model-training process [21, 70]. The samples used for training models were restricted to European ancestry. We adopted the existing expression prediction models established by Zhang except COR and TIB tissues which were not covered yet [70].
We established predictive models for COR and TIB tissues using the same parameters as other tissues [70]. In brief, we first filtered the genotype and expression data of COR and TIB from GTEx v7 [1]. For genotype data, variants with call rate < 0.95, minor allele frequency (MAF) < 0.01, and Hardy Weinberg equilibrium (HWE) < 1e−6 were removed. For expression, we used quality-controlled data and performed sample-level quantile normalization and gene-level inverse quantile normalization using preprocess codes of PredicDB pipeline [21]. We then calculated SNP priors using hierarchical Bayesian model (qtlBHM) [40] that jointly analyzed epigenome annotations of aorta derived from Roadmap Epigenomics Mapping Consortium (REMC) [5], and eQTL statistics. The SNP priors (Supplementary Table 2), genotype data and expression data were jointly applied to tenfold cross-validated weighted elastic-net to train prediction models [70].
Both STARNET- and GTEx-based models were filtered by cross-validated prediction R2 > 0.01 [28, 68]. The summary statistics of sample sizes used for training models and the transcript numbers of genes covered by each predicting model are shown in Supplementary Table 1.
GWAS cohorts
For the discovery cohort, individual-level genotyping data were collected from ten CAD GWAS, a subset of CARDIoGRAMplusC4D, including the German Myocardial Infarction Family Studies (GerMIFS) I–VII [16, 18, 38, 47, 48, 52, 56], Wellcome Trust Case Control Consortium (WTCCC) [7], LURIC [65], and Myocardial Infarction Genetics Consortium (MIGen) [2]. We used a part of individual-level data from UK Biobank (UKB) as the replication cohort [8], by extracting 20,310 CAD cases according to hospital episodes or death registries as reported and randomly selecting 25,000 non-CAD participants as controls. The detailed information about selection criteria of case and control were described at elsewhere [38]. In total, genotypes of 37,997 cases and 42,854 controls were included in our transcriptome-wide association studies (TWAS) of CAD (Supplementary Table 3). The preprocessing steps of genotyping data are as previously described [38].
TWAS analysis
The transcriptome-wide association analysis was performed using prediction models of nine tissues for imputing individual-level GReX from CAD cases and controls of 11 GWAS cohorts and by association of these tissue-specific GReX with CAD risk in each cohort. To test the replicability of TWAS results, we used ten GWAS cohorts as discovery set and UKB as the replication set to test replicability within STARNET- and GTEx-based models, respectively. We compared the consistency of TWAS results between STARNET- and GTEx-based models of the six overlapping tissues using all genotype data. Then, we merged TWAS genes resulted from two reference-based panels as the final list. Finally, we annotated the TWAS genes list by over 200 CAD loci identified by GWASs [17, 35] using MAGMA [37]. Gene resided in the ± 1 Mb regions around known GWAS loci were marked as the known, otherwise genes were marked as the novel.
Colocalization of the eQTL and GWAS signals
Colocalization analysis was performed using COLOC, a Bayesian statistical methodology that takes GWAS and eQTL data as inputs, and tests the posterior probabilities of hypothesis #4 (PP4) that there are shared casual variants for each locus [23]. The summary statistics of GWAS meta-analysis were obtained from CARDIoGRAMplusC4D Consortium [47], and the eQTL data of nine tissues from STARNET [20] and GTEx [1]. The significance threshold is PP4 > 0.55.
Co-expression network for protein coding and lncRNA genes
We used RNA-seq data of STARNET [20] to calculate expression correlations between long non-coding RNA (lncRNA) genes and protein-coding genes in seven tissues. Co-expression pairs with absolute Pearson correlation coefficient larger than 0.4 were considered to be significant. The co-expression network was displayed by cytoscape [34].
Gene set enrichment analyses
Pathway enrichment analysis was carried out using ClueGO (v2.5.2) [6], a plugin of cytoscape [34], based on collated gene sets from public databases including Gene Ontology (GO) [26], KEGG [30], Reactome [12], and WikiPathways [55]. Gene sets with false discovery rate (FDR) by right-sided hypergeometric test less than 0.05 were considered to be significant.
Furthermore, we also studied the diseases or traits associated with risk genes by performing disease enrichment analysis based on DisGeNET [50], the largest publicly available datasets of genes and variants association of human diseases. FDR < 0.05 was used for thresholding.
Rare damaging variants association analysis
To investigate association of damaging variants in TWAS genes with CAD, we used whole-exome sequencing (WES) data of 200,632 participants from UKB [27]. The WES data were processed following the Functional Equivalence (FE) protocol. We performed quality control on the WES data by filtering variants with calling rate < 0.9 and variants with HWE < 1e−6. For the relevant traits, besides CAD, we considered i) three lifestyle factors including body mass index (BMI), diabetes, hypertension; ii) seven categories of blood lipids including low-density lipoproteins cholesterol (LDL-C), high density lipoproteins cholesterol (HDL-C), apolipoprotein A (APOA), apolipoprotein B (APOB), Lipoprotein(a) (LPA), total cholesterol (TC) and triglycerides (TG); iii) four inflammation related factors including C-reactive protein (CRP), lymphocyte count (Lymphocyte), monocyte count (Monocyte) and neutrophil count (Neutrophil). In total, 15 traits were studied.
We defined damaging variants as (i) MAF < 0.01; (ii) annotated into following one of the three classes: loss-of-function (LoF) variants (stop-gained, splice site disrupting, or frameshift variants), pathogenic variants in ClinVar [36], or missense variants predicted to be damaging by one of five computer prediction algorithms (LRT score, MutationTaster, PolyPhen-2 HumDiv, PolyPhen-2 HumVar, and SIFT). The Ensembl Variant Effect Predictor (VEP) [45] and its plugin loftee [31], and annotation databases dbNSFP 4.1a [14] and ClinVar (GRCh38) [36] were used for annotating damaging mutations.
For each analysis, samples were classified into carriers or noncarriers of the gene’s damaging mutations. For binary traits, we used Fisher’s exact test to check if there was incidence difference of mutation carrying between case and controls. For the quantitative traits, we used linear regression model with adjustments of sex, first five principal components, and lipid medication status to investigate the associations between mutation carrying status and traits. We used nominal significance threshold (P < 0.05), given that coding variants are rather rare, and the case–control sample sizes were not balanced which might increase false negative rate.
Association of variants in novel genes with lipid traits
For 18 novel risk genes, we performed association analysis for variants located in respective loci (± 1 Mb) with lipid traits using genotyping data of UKB. The lipid traits include levels of LDL-C, HDL-C, APOA, APOB, LPA, TC, and TG. The variants were filtered by MAF > 0.01, and imputation info score > 0.4. The association test was performed using PLINK2 [10] with adjustment of sex, age, first five principal components, and lipid medication status. The numbers of independent SNPs were estimated using Genetic type 1 error calculator (GEC) tool [39].
Expression-trait association study using mouse data
The hybrid mouse diversity panel (HMDP) is a set of 105 well-characterized inbred mouse strains on a 50% C57BL/6J genetic background [42]. To specifically study atherosclerosis in the HMDP, transgene implementation of human APOE-Leiden and cholesteryl ester transfer protein was performed, promoting distinct atherosclerotic lesion formation [4]. A Western diet containing 1% cholesterol was fed for 16 weeks. Subsequently, gene expression was quantified in aorta and liver of these mice and lesion size was assessed in the proximal aorta using oil red O staining. Fourteen other related traits were measured too, including liver fibrosed area, body weight, TC, VLDL-C (very low-density lipoprotein cholesterol) + LDL-C, HDL-C, TGs, unesterified cholesterol, free fatty acid (FFA), Il-1b, Il-6, Tnfa, Mcp-1, and M-csf. From HMDP, we extracted significant association pairs between TWAS genes and 15 risk traits by applying significance P < 0.05.
Experimental validation of KPTN and RGS19 in human cells
To knock down KPTN and RGS19, two sgRNAs targeting shared exons of all transcription isoforms were delivered by lentivirus into a Cas9-expression huh7, a human hepatoma cell line. Exon 4 of KPTN and exon 5 of RGS19 were targeted by a dual CRISPR strategy to create a 40 bp and 130 bp frame shift deletion, respectively. SgRNAs were carried by Lenti-Guide-Puro vector (addgene, #52963) and infected cells were treated with 10 ug/ml puromycin treatment for 3 days to eliminate the negative cell. Positive targeted cells were expanded in culture and passaged for assays. Cells for measurement of secretive triglycerides, cholesterol, and APOB100 were cultured for 16 h in serum-free medium. Medium triglycerides and cholesterol were enriched for five times by vacuum centrifuge and measured with colorimetric kits, triglyceride (cobas), and CHOL2 (cobas), respectively. The amount of medium APOB100 was measured with an ELISA kit (MABTECH).
RNA isolation and sequencing
Total RNA from huh7 cells was isolated using RNEasy Plus Mini Kit (Qiagen) (control cells, n = 3; knockout cells, n = 3). Quantity and quality of the isolated RNAs were measured by Fragment Analyzer (Agilent). RNA sequencing (RNA-seq) was performed by BGI TECH SOLUTIONS (HONGKONG) using strand specific library preparation with mRNA enrichment, paired-end sequencing with 100 bp read length on the DNBSEQ platform and 20 M clean read pairs per sample. Clean reads were mapped onto the GRCh38.p12. Expression quantifications, differential expression, and gene set enrichment were performed according to BGI RNA-seq pipeline.
Results
Transcriptome-wide significant genes for CAD
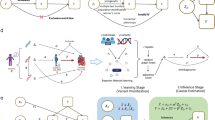
The study design is shown in Fig. 1. Expression prediction models of nine tissues were derived from two reference panels, STARNET [20] and GTEx [1], using EpiXcan pipeline [70] (Materials and methods). We applied these models to impute transcriptome-wide GReX of nine tissues from individual-level genotype data of 11 GWAS cohorts (Supplementary materials; Supplementary Fig. 1–2; Supplementary Tables 1–3) [2, 7, 8, 16, 18, 38, 47, 52, 56, 65]. We next associated the GReX with CAD risk in each cohort (Supplementary materials). The results revealed replicability of TWAS genes when taking ten CARDIoGRAMplusC4D cohorts as discovery and UKB as replication set within the STARNET- and GTEx-based prediction models, respectively (Supplementary Fig. 2I–II; Supplementary Fig. 3). The results also showed consistency and complementarity of TWAS findings in six shared tissues between two reference-based prediction models (Supplementary Fig. 2III–IV; Supplementary Figs. 4–5). Therefore, we combined the results based on the two reference models for the final list of TWAS genes (Supplementary Fig. 2 V).

The study design. Step 1, we trained prediction models using EpiXcan from two eQTL panels, the Stockholm-Tartu Atherosclerosis Reverse Network Engineering Task (STARNET) and the Genotype-Tissue Expression (GTEx) for nine tissues. Step 2, the prediction models were applied to impute genetically regulated expression (GReX) from individual-level genotype data of ten CARDIoGRAMplusC4D sets and UK Biobank (UKB). Step 3, we associated transcriptome-wide GReX with risk of coronary artery disease (CAD) (Supplementary Results) and identified 114 transcriptome-wide significant genes (TWAS genes). Of these, 96 resided within genome-wide significant (GWAS) loci and 18 outside of known GWAS loci (novel genes). Step 4, we tested the plausibility of novel TWAS genes by conducting colocalization analysis and studying effects of damaging mutations, as well as gene set enrichment analyses. Step 5, we explored potential mechanisms of novel genes by testing association with risk traits of CAD in human genotype data of UKB, and association between expressions and risk traits of CAD in atherosclerosis mouse models from the Hybrid Mouse Diversity Panel (HMDP). Lastly, we carried out CRISPR/Cas9-based knockdown experiment for two novel genes RGS19 and KPTN in human hepatocyte cell lines to experimentally validate related functions
From STARNET-based models 129 gene-tissues pairs and from GTEx-based models 106 gene-tissue pairs were significantly associated with CAD (Bonferroni-corrected significance based on 12,995 genes, P < 3.85e−6). Since 42 pairs overlapped between the two panels (Supplementary Fig. 5), the total number of gene-tissue pairs was 193. Given that some genes displayed association in multiple tissues, the final list of significant TWAS genes for CAD was 114 genes (Fig. 2; Supplementary Fig. 6; Supplementary Table 4). Of these, 95 gene-tissue association pairs were confirmed using another commonly used fine-mapping tool (COLOC) [23] with posterior probabilities of shared causal variants in each locus larger than 0.55 (PP4 > 0.55; Materials and methods; Supplementary Table 5; Supplementary Fig. 7).

Manhattan plot of CAD TWAS results. The association results from STARNET- and GTEx-based models were integrated by lowest P values. The blue line marks P = 3.85e−6, i.e. transcriptome-wide significance. Each point corresponds to an association test between gene-tissue pair. 18 novel TWAS genes were highlighted. Supplementary Fig. 6 identifies all genes identified by their genetically-modulated association signals. The color code identifies the tissue in which the genes were differentially expressed by genetic means: AOR aorta, COR coronary artery, MAM mammary artery, BLD blood, LIV liver, SF subcutaneous fat, VAF visceral abdominal fat, SKLM skeletal muscle
Of the 114 TWAS genes, 46 genes displayed genetically mediated differential expression in AOR, 28 in MAM, 25 in LIV, 23 in VAF, 22 in SKLM, 18 in SF, 16 in BLD, 10 in TIB, and 5 in COR (Fig. 3a). Most genes revealed significant associations in only a single tissue; 38 were significant in more than one, almost all having consistent directions of association between predicted expression levels and CAD across tissues (Fig. 3b).

Tissue distribution of 114 TWAS genes of CAD. a Number of transcriptome-wide significant genes across tissues. b Heatmap plot of 38 genes identified in more than one tissues. The color codes indicate direction of effects. Cells marked with * represent significant gene-tissue pairs (P < 3.85e−6). AOR aorta, COR coronary artery, MAM mammary artery, BLD blood, LIV liver, SF subcutaneous fat, VAF visceral abdominal fat, SKLM skeletal muscle, TIB tibial artery
Among the 114 genes, 102 were protein-coding and 12 were lnRNA genes (Supplementary Table 4). The STARNET data showed that most lncRNAs were positively co-expressed with a surrounding gene in affected tissues (Supplementary Fig. 8). LINC00310 was the only exception, which displayed complex co-expression patterns with other genes.
Respective genes were found in 63 genomic regions, thus several regions represented multiple genes with significant associations. Six regions had multiple TWAS genes with shared GWAS and eQTL signals in respective tissues, like 1p13.3 and 2p33.2 (Supplementary Figs. 9–10; Supplementary Table 5). On the other hand, in 39 regions expression of only a single gene was found to be significantly associated, which makes these genes likely candidates for mediating causal effects, particularly, if these genes reside within GWAS risk loci for CAD (these genes are indicated in Supplementary Table 6).
Most TWAS genes (n = 96) could be positionally annotated to the 1 Mb region around one of the over 200 GWAS loci that are currently known to be genome-wide significantly associated with CAD [11, 17, 35]. Therefore, we marked these as known genes (Supplementary Table 6). On the other hand, 18 genes resided outside of these regions and were labeled as novel genes (Table 1). Most novel genes were tissue-specific, except RGS19, FAM114A1 and UFL1 which displayed evidence for differential expression in multiple tissues.
Pathways and diseases enriched by TWAS genes
We carried out two types of gene set enrichment tests to further study the biological relevance of genes giving signals in this TWAS. First, we studied disease-gene sets from the DisGeNET platform which is one of the largest publicly available collections of genes and variants associated with human diseases [50]. The results showed that genes discovered by TWAS were primarily enriched for CAD, coronary atherosclerosis, and hypercholesterolemia (Supplementary Table 7), adding to the plausibility of our TWAS findings.
In line with these results, gene set enrichment analysis based on GO [26], KEGG [30], Reactome [12], and WikiPathways [55] databases showed that the TWAS genes were highly enriched for pathways involved in cholesterol metabolism and regulation of lipoprotein levels. To a lesser extent, risk genes were enriched in regulation of blood pressure as well as development and morphogenesis of the heart and the aortic valve (Supplementary Table 8).
Effects of damaging variants in TWAS genes
We next searched in exome-sequencing data of 200,643 participants from UKB for rare damaging variants in TWAS genes (either loss-of-function mutations or mutations predicted to be adverse by one of five in-silico methods, allele frequency < 0.01) (Materials and methods). In 97 genes we detected such variants. Expectedly these damaging mutations were very rare which limits the power of gene-based burden tests to observe association with risk of CAD or one of 14 CAD-related cardiometabolic traits we tested (15 traits in total). Nevertheless, associations of eight genes with risk traits reached Bonferroni-corrected significance (P < 3.44e−5; 0.05/97genes × 15traits) (Fig. 4; Supplementary Tables 9–10). Mutations of lipoprotein lipase (LPL), a critical regulator of lipid metabolism [29, 60], were evidently associated with lipid traits, including levels of HDL-C (beta = − 0.106; P = 4.54e−68), APOA (beta = − 0.062; P = 6.25e−47), APOB (beta = 0.025; P = 1.38e−12), and TG (beta = 0.241; P = 1.47e−68). ABCG5, encoding a sterol transfer protein [69], was associated with LDL-C (beta = 0.12; P = 3.66e−10), TC (beta = 0.16; P = 8.63e−10). PCSK9, a drug target for cholesterol lowering [13], was associated with LDL-C (beta = − 0.01; P = 4.29e−7) and APOB (beta = − 0.03; P = 4.4e−10). A mutation of SARS was associated with APOB (beta = − 0.02; P = 5.92e−7), MAT2A with lymphocyte counts (beta = 1.34; P = 3.41E−28), and JCAD (odds ratio [OR] = 1.31; 95% confidence interval [CI] 1.18–1.46; P = 5.77e−7) as well as ARHGAP42 (OR = 2.08; 95% CI 1.65–2.59; P = 2.22e−9) were associated with risk of diabetes. We also observed nominally significant associations of several genes with CAD: LPL [29, 60] (OR = 1.168; CI 1.034–1.036; P = 0.016), NOS3 [15] (OR = 1.143; 95% CI 1.109–1.279; P = 0.02), and ADAMTS7 [32] (OR = 1.062; 95% CI 1.011–1.115; P = 0.016) (Supplementary Tables 9–10).

Effects of damaging variants in TWAS genes on CAD and its risk traits. Sign(beta)*−log10(p) displays direction and significance of gene-trait associations. When the Sign(beta)*−log10(P) > 8, they were trimmed to 8. The gene-trait association pairs reached Bonferroni-significance P < 3.44e−5 were highlighted in box. CAD coronary artery disease, LDL-C low-density lipoproteins cholesterol, VLDL-C very low-density lipoprotein cholesterol, HDL-C high density lipoproteins cholesterol, APOA apolipoprotein A, APOB apolipoprotein B, TC total cholesterol, TG triglycerides, CRP C-reactive protein, BMI body mass index
Novel genes associated with risk factors in human and mouse data
We next associated single nucleotide polymorphisms (SNPs) in the regions of ± 1 Mb around the 18 novel TWAS genes to study their associations with a series of lipid traits including LDL-C, HDL-C, APOA, APOB, LPA, TC, and TG in UKB (Materials and methods). There were 883 independent SNPs estimated by GEC. Bonferroni-corrected significance P < 8.09e−6 (0.05/883 × 7 lipid traits) was observed for numerous respective lead variants, of which RGS19, SDCCAG3, EZR, HOMER3, and WWP2 reached genome-wide significant association (P < 5e−8) with multiple lipid traits (Fig. 5a; Supplementary Table 11).

Novel risk genes were associated with lipid traits. a Data from UK Biobank (UKB) indicated that lead variants inside the boundary of risk genes were associated with lipid traits with Bonferroni-corrected significance levels (*P < 8.09e−6), or by genome-wide significance (**P < 5e−8). b Expression levels of novel genes were likewise associated with lipid traits and aortic lesion area in an atherosclerosis mouse model from the hybrid mouse diversity panel (HMDP). *P < 0.05; **FDR < 0.05. LDL-C low-density lipoproteins cholesterol, VLDL-C very low-density lipoprotein cholesterol, HDL-C high density lipoproteins cholesterol, APOA apolipoprotein A, APOB apolipoprotein B, TC total cholesterol, TG triglycerides, FFA free fatty acid
Next, we extracted expression-trait association statistics of TWAS genes from HMDP, which brings together genotypes and expression data from atherosclerosis mouse models [42]. Based on the expression data from mouse aorta and liver tissues, 55 TWAS genes were significantly associated with aortic lesion area and 14 further cardiovascular traits (P < 0.05; Supplementary Table 12). Expression levels of seven novel genes, i.e. Rgs19, Kptn, Ezr, Stx4a, Cand1, Focad, and Wasf1, were associated with aortic lesion area (Fig. 5b), a commonly used measure for atherosclerotic plaque formation in mice. Additionally, we found the novel genes were associated with at least one lipid trait in the mouse model.
Knockdown of RGS19 and KPTN in human liver cells
Potential functional implications of all novel genes, based on the literature, are summarized in Supplementary Table 13. We additionally aimed to validate two exemplary novel TWAS genes by in vitro studies. Based on the above in-silico annotations we focused these studies on novel genes identified in liver with potential effects on lipids, the top risk factor for CAD (Fig. 5). Among the five genes identified in liver including NLRC4, EZR, GAS8, KPTN, and RGS19, the last two were, not only the least studied but also associated with nearly a full spectrum of lipid traits in human or mouse data (Fig. 5). In addition, both KPTN and RGS19 are indeed expressed in hepatocyte (Supplementary Fig. 11a, b). Finally, both KPTN and RGS19 are located within lipid loci identified recently in more than one million individuals [24]. Therefore, we decided to test the influence of KPTN and RGS19 on lipid metabolism of liver cells.
We generated gene knockout (KO) huh7 cell lines by a dual CRISPR strategy (Materials and methods), which substantially reduced expression of the respective genes (Supplementary Fig. 11c, d). We measured secretion levels of TG, cholesterol and APOB in gene-targeted versus control cells. Notably, under normal circumstances, human hepatocytes synthesize cholesterol, assemble TG and APOB100, and secrete these particles in form of VLDL-C [58]. Compared to control huh7 cells, we found reduced APOB and cholesterol levels in culture medium of KPTN-KO cells (Fig. 6a, c). In culture medium of RGS19-KO cells we also detected reduced levels of APOB100, cholesterol, and TG (Fig. 6b, c), in line with strong associations of this gene with an array of lipid traits in both human genotyping and mouse expression data sets (Fig. 5).

Targeting of KPTN and RGS19 reduced lipids and APOB secretion of human liver cells. a Two sgRNAs were used to target the exon4 of KPTN (shared exon among isoforms) in a Cas9-expressing huh7 liver cell line. The dual CRISPR strategy created a 40 bp frame shift deletion in the gene and profound reduction of KPTN at both mRNA and protein levels (Supplementary Fig. 11c, d). The primers (P-Fw and P-Rv) used for analyzing the CRISPR editing as indicated. b The same strategy was used for RGS19 targeting, which resulted in a 130 bp frame shift deletion in the gene, and reduction of mRNA and protein (Supplementary Fig. 11c, d). c Reduced triglyceride and cholesterol levels in knockout (KO) cell lines were detected by colorimetric method and APOB100 secretion was measured by human APOB100 Elisa (n = 6). Triglyceride, cholesterol, and APOB100 levels were normalized to total protein and compared between the KO and control (CTR) cell lines
We further corroborated our experimental results by performing RNA sequencing (RNA-seq) on KPTN-KO and RGS19-KO hepatocytes. In comparison to control cells, dysregulated genes in KPTN-KO and RGS19-KO hepatocytes (P < 0.05; Supplementary Tables 14–15) were indeed enriched for lipid metabolism (Supplementary Fig. 12). For KPTN-KO hepatocytes, the top four significantly enriched pathways plausibly contribute to CAD risk. Pathways ranked 1 and 3, ‘regulation of cholesterol esterification’ and ‘LDL particle remodeling’, strongly suggested that KPTN can affect CAD risk via cholesterol metabolism (Supplementary Fig. 12a, b). For RGS19-KO hepatocytes, the dysregulated genes were enriched for both cholesterol and triglycerides metabolisms (Supplementary Fig. 12c, d) and eight of the top ten significant enriched pathways were related to lipid metabolism, consistent with the reduced secretion of cholesterol and triglyceride of RGS19-KO cells (Fig. 6c).
Discussion
In a stepwise approach, we first generated models which allow to predict gene expression based on genotypes in nine tissues. Next, we applied these models to individual-level genotype data on more than 80,000 CAD cases and controls to perform a transcriptome-wide association analysis. We identified 114 genes with differential expression by genetic means in CAD patients. Many signals were highly plausible as they resided within loci displaying genome-wide significant association with CAD by traditional GWAS. By in-silico analyses, these genes were markedly enriched in established pathways for the disease. Moreover, damaging variants in these genes showed association with CAD risk or its underlying traits in whole exome sequencing data from UKB. Importantly, we also identified 18 genes without prior evidence for their involvement in CAD by GWAS, many of which were found to be associated with lipid metabolism in human and mouse data.
Only a minority of genes residing within published CAD GWAS loci have been validated experimentally for their underlying causal role in atherosclerosis. Our data provide a substantial step towards prioritization of genes at respective GWAS loci [17, 35], because the TWAS association finding is based on expression levels of specific genes in defined tissues. In this respect, 46 genes identified by this TWAS are known for effects in pathophysiological pathways related to CAD, including lipid metabolism, inflammation, angiogenesis, transcriptional regulation, cell proliferation, NO signaling, and high blood pressure, to name a few (Supplementary Table 6), giving credibility to the association findings.
Interestingly, our TWAS uncovered eight novel gene-CAD associations in fat tissue, including MGP and WASF1 in SF, and CAND1, FAM114A1, FOCAD, RGS19, TSPAN11, and TXNRD3 in VAF, representing half of the novel genes. All these genes also showed significant association with multiple lipid traits in a mouse atherosclerosis model (Fig. 5b). Given many CAD patients that are overweight or obese, it will be of great interest to identify how these genes modify cardiometabolic traits leading to cardiovascular disorders. In this respect our TWAS could provide a list of candidate genes and related targetable cardiometabolic traits. In addition, it is of surprise to unveil 22 genes linking SKLM to CAD risk, and eight were unique to this tissue, including HOMER3, SDCCAG3, MTAP, NME9, PSMA4, SLC2A12, UNC119B, and VAMP5, the first two being novel. SDCCAG3 or ENTR1 encodes endosome associated trafficking regulator 1 and involves in recycling of GLUT1 (glucose transporter type 1), supplying the major energy source for muscle contraction. SKLM-based metabolism may have a protective role in CAD as suggested by the many cardioprotective effects of sports [44, 54]. Gene targets enhancing SKLM function in this respect might be effective in CAD prevention, a field relatively unexplored thus far. Here, for the first time, quantitative traits regulated genes in SKLM were associated with CAD by TWAS, providing novel evidence for genes that could modulate CAD risk by their functions in SKLM.
Many novel TWAS genes revealed association with lipid traits in both genotype-trait data of human biobank and expression-trait data of atherosclerosis mouse model. For example, KPTN and RGS19, both novel genes displaying significant TWAS results for CAD—based on their genetically-modulated expression profiles of liver tissue—also showed significant association with various lipid traits as well as aortic lesion area in the atherosclerosis mouse model. Moreover, both gene loci harbor SNPs which are significantly associated with several lipids including LDL-C, HDL-C, TC, and/or TG in human genotype data. Based on these observations, we functionally validated the roles of these two novel genes by studying lipid levels in human liver cells, i.e. the tissue that displayed evidence for differential expression by TWAS. Indeed, we observed that knockout of the two genes lowered secretion of APOB and lipids into culture medium. KPTN, kaptin (actin binding protein), a member of the KPTN, ITFG2, C12orf66, and SZT2 (KICSTOR) protein complex, is a lysosome-associated negative regulator of the mechanistic target of rapamycin complex 1 (mTORC1) signaling [67]. By investigating dysregulated genes of KPTN-KO hepatocytes, we found many genes of mTORC1 pathway to be upregulated (Supplementary Fig. 12b), including a subunit of mTORC1, namely, MLST8 (MTOR associated protein, LST8 Homolog). Interestingly, many lysosome genes were also significantly upregulated including LAMP1 (lysosomal associated membrane protein 1), ACP2 (acid phosphatase 2, lysosomal), AP1B1 (adaptor related protein complex 1 subunit beta 1), ATP6V0C (ATPase H+ transporting V0 subunit c), CTNS (cystinosin, lysosomal cystine transporter), CTSA (cathepsin A), and CLTB (clathrin light chain B). Lysosomes promote lipid catabolism and transport, and maintain cellular lipid homeostasis [57]. Activated mTORC1 located on lysosome membrane [67], acts as a sensor of lysosomal lipids [57], such as cholesterol and phosphatidic acid, which exert as building block for cellular and subcellular membrane system. In fact, cholesterol mediates mTORC1 activation at the lysosome [9]. The interaction of mTORC1 and lysosome may promote lipid-sensing and lipid-trafficking to support the function of other subcellular organelles [46, 57]. These results hint the enhanced cellular usage of cholesterol via mTOR-lysosome axis in KPTN-KO hepatocytes. In addition, several lipoprotein genes were downregulated as well, including APOA1, APOA2, APOA4, and APOB. Both processes might contribute to the reduced cholesterol secretion and the association with CAD.
RGS19 belongs to the RGS (regulators of G-protein signaling) family, who are regulators for G-protein-coupled receptors (GPCRs) [49]. RGS19 inhibits GPCR signal transduction by increasing the GTPase activity of G-protein alpha subunits, thereby transforming them into an inactive GDP-bound form [53, 59]. The targeting GPCR of RGS19 has not been observed before, and how RGS19 regulates lipid metabolism remains unclear. The RGS19 locus was first reported to be associated with TC and TG in 2017 [33, 41]. We observed significant association of this gene with CAD and functionally validated its role in TG and cholesterol secretion. A potential mechanism could be related to PPARα pathway that regulates the expression of genes involving hepatic lipogenesis and lipid storage [63, 66]. PPARα also regulates cholesterol, bile acid homeostasis, and sphingolipid metabolism in the liver [22]. Many genes in PPARα pathway were significantly downregulated in RGS19-KO hepatocytes, including FABP1 (also known as liver fatty acid binding protein), PLTP (phospholipid transfer protein), APOA1, APOA2, and APOC3 (Supplementary Fig. 12d). RGS19 is a regulator for G-protein-coupled receptors (GPCR). Interestingly, we found six dysregulated GPCRs in RGS19-KO hepatocytes, including, ADGRL2, CELSR1, ADGRV1, OXER1, LGR5, and LGR4 (Supplementary Fig. 12d). Furthermore, one of them, OXER1, an activator PPARα [51], was also downregulated in RGS19-KO cells. All in all, one hypothesis could be that RGS19 associated GPCR signaling affects the PPARα pathway, and thereby lipid metabolism and CAD risk. Previous and current studies concordantly suggest from different angles that RGS19 has a role in lipid metabolism and our data further indicate that this function might meditate its effects on CAD risk.
There are certain limitations in our study. First, we observed that about 15% of gene-tissue pairs displayed some degree of heterogeneity in the association findings with CAD risk across the cohorts (Supplementary Table 4). While this number is relatively low and likely result from a play of chance when association findings are being compared across individuals with relatively small case–control samples, it might also indicate some degree of population specific effects within European ancestries from UK, Germany, France, and Italy. Second, since TWAS are strongly dependent on the reference panel linking genetic signatures with gene expression, it had to be expected that STARNET- and GTEx-based predictive models display some differences in gene-CAD associations. STARNET-based TWAS identified 129 gene-tissue pairs, whereas GTEx-based TWAS identified 106 gene-tissue pairs. Yet, 42 gene-tissue pairs were shared between the two analyses, and effect sizes for the shared genes were highly concordant (ρ = 0.97). An average of 62% overlapping genes was observed in the matched tissues of two reference-based models, and the resulting size of expression-CAD associations was linearly consistent with an average ρ = 0.72. The relatively small differences may be due to different sample sizes used for training predictive models [70], different disease states (subjects with and without CAD), intravital (STARNET) or post mortem (GTEx) sample collection, as well as different transcript abundance and genotype coverage leading to differences in expression associated SNPs in our reference panels [20, 25]. Given a fair consistency between the two data sources, we combined results derived from both panels to increase the power for capturing risk genes. Third, although TWAS facilitates candidate risk gene prioritization, co-regulation or co-expression in cis at a given locus limits the precise determination of the culprit gene [62]. Indeed, at 12 loci we observed signals for three or more TWAS genes. For instance, in LIV tissue TWAS identified five genes at 1p13.3, ATXN7L2, CELSR2, PSMA5, PSRC1, SARS, and SORT1 which were co-regulated by same risk variant set, confusing prioritization of the causal gene. While CELSR2, PSRC1, and SORT1 were previously shown to act on lipid metabolism [3], we found that damaging mutations in SARS were also associated with serum levels of HDL-C and APOA. Thus, a combined effects of some or all genes at this locus may contribute to the association signal. In addition, all lncRNA genes identified by our study displayed co-expression with their neighboring coding genes, which makes it difficult to determine their casual effects. Nevertheless, in combining TWAS data with other genetic analyses, e.g. effects of damaging mutations, genetic association with other phenotypes and expression-traits association statistics, we aimed to improve risk gene prioritization, and to provide deeper insights of possible disease-causing mechanisms. For instance, LPL is well-known for its protective role against CAD by lowering lipids [29, 60], and our analyses showed that damaging LPL mutations were associated with higher lipid levels. Last, as with all statistical methods, there are certain limitations and assumptions associated with TWAS. Further evolution and improvement of these methods, as well as functional validation experiments, will assuredly improve the accuracy of these studies.
In summary, our TWAS study based on two genetics-of-gene-expression panels identified 114 gene expression-CAD associations, of which 18 were novel. The extended analyses with multiple datasets supported the reliability of the CAD TWAS signals in prioritizing candidate risk genes and identifying novel associations in a tissue-specific manner. Functional validation of two novel genes, RGS19 and KPTN, lend support to our TWAS findings and provide strong evidence for their role in lipid metabolism. Thus, our study created a set of gene-centered and tissue-annotated associations for CAD, providing insightful guidance for further biological investigation and therapeutic development.
Change history
05 April 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00395-022-00923-w
References
Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park YS, Parsana P, Segrè AV, Strober BJ, Zappala Z, Cummings BB, Gelfand ET, Hadley K, Huang KH, Lek M, Li X, Nedzel JL, Nguyen DY, Noble MS, Sullivan TJ, Tukiainen T, MacArthur DG, Getz G, Addington A, Guan P, Koester S, Little AR, Lockhart NC, Moore HM, Rao A, Struewing JP, Volpi S, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, Kopen G, Leinweber WF, Lonsdale JT, McDonald A, Mestichelli B, Myer K, Roe B, Salvatore M, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Bridge J, Foster BA, Gillard BM, Karasik E, Kumar R, Miklos M, Moser MT, Jewell SD, Montroy RG, Rohrer DC, Valley D, Mash DC, Davis DA, Sobin L, Barcus ME, Branton PA, Abell NS, Balliu B, Delaneau O, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Han B, He AZ, Hormozdiari F, Li X, Liu B, Kang EY, McDowell IC, Ongen H, Palowitch JJ, Peterson CB, Quon G, Ripke S, Saha A, Shabalin AA, Shimko TC, Sul JH, Teran NA, Tsang EK, Zhang H, Zhou YH, Bustamante CD, Cox NJ, Guigó R, Kellis M, McCarthy MI, Conrad DF, Eskin E, Li G, Nobel AB, Sabatti C, Stranger BE, Wen X, Wright FA, Ardlie KG, Dermitzakis ET, Lappalainen T, Battle A, Brown CD, Engelhardt BE, Montgomery SB, Handsaker RE, Kashin S, Karczewski KJ, Nguyen DT, Trowbridge CA, Barshir R, Basha O, Bogu GK, Chen LS, Chiang C, Damani FN, Ferreira PG, Hall IM, Howald C, Im HK, Kim Y, Kim-Hellmuth S, Mangul S, Monlong J, Muñoz-Aguirre M, Ndungu AW, Nicolae DL, Oliva M, Panousis N, Papasaikas P, Payne AJ, Quan J, Reverter F, Sammeth M, Scott AJ, Sodaei R, Stephens M, Urbut S, Van De Bunt M, Wang G, Xi HS, Yeger-Lotem E, Zaugg JB, Akey JM, Bates D, Chan J, Claussnitzer M, Demanelis K, Diegel M, Doherty JA, Feinberg AP, Fernando MS, Halow J, Hansen KD, Haugen E, Hickey PF, Hou L, Jasmine F, Jian R, Jiang L, Johnson A, Kaul R, Kibriya MG, Lee K, Li JB, Li Q, Lin J, Lin S, Linder S, Linke C, Liu Y, Maurano MT, Molinie B, Nelson J, Neri FJ, Park Y, Pierce BL, Rinaldi NJ, Rizzardi LF, Sandstrom R, Skol A, Smith KS, Snyder MP, Stamatoyannopoulos J, Tang H, Wang L, Wang M, Van Wittenberghe N, Wu F, Zhang R, Nierras CR, Carithers LJ, Vaught JB, Gould SE, Lockart NC, Martin C, Addington AM, Koester SE, Undale AH, Smith AM, Tabor DE, Roche NV, McLean JA, Vatanian N, Robinson KL, Valentino KM, Qi L, Hunter S, Hariharan P, Singh S, Um KS, Matose T, Tomaszewski MM, Barker LK, Mosavel M, Siminoff LA, Traino HM, Flicek P, Juettemann T, Ruffier M, Sheppard D, Taylor K, Trevanion SJ, Zerbino DR, Craft B, Goldman M, Haeussler M, Kent WJ, Lee CM, Paten B, Rosenbloom KR, Vivian J, Zhu J (2017) Genetic effects on gene expression across human tissues. Nature 550:204–213. https://doi.org/10.1038/nature24277
Anderson CD, Rosand J, Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles G V, Clark NR, Ma’ayan A, Kim JY, Kim JY, Song KS, Lee YH, Seo JS, Jelinek J, Goldschmidt-Clermont PJ, Issa JP, Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW, Krizsan-Agbas D, Pedchenko T, Smith PG, Li C, Bazzano LA, Rao DC, Hixson JE, He J, Gu D, Gu CC, Shimmin LC, Jaquish CE, Schwander K, Liu DP, Huang J, Lu F, Cao J, Chong S, Lu X, Kelly TN, Sharma P, Kumar J, Garg G, Kumar A, Patowary A, Karthikeyan G, Ramakrishnan L, Brahmachari V, Sengupta S, Pedrinelli R, Ballo P, Fiorentini C, Denti S, Galderisi M, Ganau A, Germanò G, Innelli P, Paini A, Perlini S, Salvetti M, Zacà V, Besingi W, Johansson Å, Cao TH, Quinn PA, Sandhu JK, Voors AA, Lang CC, Parry HM, Mohan M, Jones DJL, Ng LL, Heart N, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups KK, König IR, Cazier J-B, Johansson Å, Hall AS, Lee J-YJ-Y, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikäinen L-P, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, Doney ASF, Mokhtari NEE, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han B-G, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki M-L, Lundmark A, McCarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Müller-Nurasyid M, Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schäfer A, Sivananthan M, Song C, Stewart AFR, Tan S-T, Thorgeirsson G, Schoot CE van der, Wagner PJ, Wells GA, Wild PS, Yang T-P, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrières J, Gauguier D, Go AS, Goodall AH, Gudnason VV, Hazen SL, Holm H, Iribarren C, Jang Y, Kähönen M, Kee F, Kim H-S, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee J-YJ-Y, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Trégouët D-A, Virtamo J, Wallentin L, Wareham NJN, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen TM, Syvänen A-C, Hovingh GK, Dedoussis G, Franks PW, Lehtimäki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O’Donnell CJ, Reilly MP, März W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CNA, Roberts R, Watkins H, Schunkert H, Samani NJ, Willer CJ, Schmidt EM, Al E, Facp AFR, Rodriguez-lopez L, Vargas-ayala G, Mc SH, Serna DC, Lozano-nuevo JJ, Rubio-guerra AF, Studies TIC for BPG-WA, Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Sahota G, Rienstra CM, Rakyan VK, Down TA, Balding DJ, Beck S, Hager J, Kamatani Y, Cazier J-B, Youhanna S, Ghassibe-Sabbagh M, Platt DE, Abchee AB, Romanos J, Khazen G, Othman R, Badro DA, Haber M, Salloum AK, Douaihy B, Shasha N, Kabbani S, Sbeite H, Chammas E, Bayeh H, Rousseau F, Zelenika D, Gut I, Lathrop M, Farrall M, Gauguier D, Zalloua PA, Myocardial Infarction Genetics C, Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand SS, Engert JC, Samani NJ, Schunkert H, Erdmann J, Reilly MP, Rader DJ, Morgan T, Spertus JA, Stoll M, Girelli D, McKeown PP, Patterson CC, Siscovick DS, O’Donnell CJ, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Melander O, Altshuler D, Merlini PA, Berzuini C, Bernardinelli L, Peyvandi F, Tubaro M, Celli P, Ferrario MM, Fetiveau R, Marziliano N, Casari G, Galli M, Ribichini F, Rossi M, Bernardi F, Zonzin P, Piazza A, Yee J, Friedlander Y, Marrugat J, Lucas G, Subirana I, Sala J, Ramos R, Meigs JB, Williams G, Nathan DM, MacRae CA, Havulinna AS, Berglund G, Hirschhorn JN, Asselta R, Duga S, Spreafico M, Daly MJ, Nemesh J, Korn JM, McCarroll SA, Surti A, Guiducci C, Gianniny L, Mirel D, Parkin M, Burtt N, Gabriel SB, Thompson JR, Braund PS, Wright BJ, Balmforth AJ, Ball SG, Hall AS, Wellcome Trust Case Control C, Linsel-Nitschke P, Lieb W, Ziegler A, Konig IR, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann H-EE, Schreiber S, Ouwehand WH, Deloukas P, Scholz M, Cambien F, Li M, Chen Z, Wilensky R, Matthai W, Qasim A, Hakonarson HH, Devaney JM, Burnett MS, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Knouff CW, Waterworth DM, Walker MC, Mooser V, Epstein SE, Scheffold T, Berger K, Huge A, Martinelli N, Olivieri O, Corrocher R, McKeown PP, Erdmann E, Konig IR, Holm H, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Do R, Xie C, Siscovick DS, Liang L, Willis-Owen SAG, Laprise C, Wong KCC, Davies GA, Hudson TJ, Binia A, Hopkin JM, Yang IV, Grundberg E, Busche S, Hudson M, Rönnblom L, Pastinen TM, Schwartz DA, Lathrop GM, Moffatt MF, Cookson WOCM, Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM, Mayer B, Erdmann J, Schunkert H, Myocardial Infarction Genetics C, Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand SS, Engert JC, Samani NJ, Schunkert H, Erdmann J, Reilly MP, Rader DJ, Morgan T, Spertus JA, Stoll M, Girelli D, McKeown PP, Patterson CC, Siscovick DS, O’Donnell CJ, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Melander O, Altshuler D, Merlini PA, Berzuini C, Bernardinelli L, Peyvandi F, Tubaro M, Celli P, Ferrario MM, Fetiveau R, Marziliano N, Casari G, Galli M, Ribichini F, Rossi M, Bernardi F, Zonzin P, Piazza A, Yee J, Friedlander Y, Marrugat J, Lucas G, Subirana I, Sala J, Ramos R, Meigs JB, Williams G, Nathan DM, MacRae CA, Havulinna AS, Berglund G, Hirschhorn JN, Asselta R, Duga S, Spreafico M, Daly MJ, Nemesh J, Korn JM, McCarroll SA, Surti A, Guiducci C, Gianniny L, Mirel D, Parkin M, Burtt N, Gabriel SB, Thompson JR, Braund PS, Wright BJ, Balmforth AJ, Ball SG, Hall AS, Wellcome Trust Case Control C, Linsel-Nitschke P, Lieb W, Ziegler A, Konig IR, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann H-EE, Schreiber S, Ouwehand WH, Deloukas P, Scholz M, Cambien F, Li M, Chen Z, Wilensky R, Matthai W, Qasim A, Hakonarson HH, Devaney JM, Burnett MS, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Knouff CW, Waterworth DM, Walker MC, Mooser V, Epstein SE, Scheffold T, Berger K, Huge A, Martinelli N, Olivieri O, Corrocher R, McKeown PP, Erdmann E, Konig IR, Holm H, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Do R, Xie C, Siscovick DS, The CARDIoGRAMplusC4D Consortium, Studies TIC for BPG-WA, The CARDIoGRAMplusC4D Consortium, Preuss M, König IR, Thompson JR, Erdmann J, Absher D, Assimes TL, Blankenberg SS, Boerwinkle E, Chen L, Cupples LA, Hall AS, Halperin E, Hengstenberg C, Holm H, Laaksonen R, Li M, Marz W, McPherson R, Musunuru K, Nelson CP, Burnett MS, Epstein SE, O’Donnell CJ, Quertermous T, Rader DJ, Roberts R, Schillert A, Stefansson K, Stewart AFR, Thorleifsson G, Voight BF, Wells GA, Ziegler A, Kathiresan S, Reilly MP, Samani NJ, Schunkert H, Avenell A, Broom J, Brown TJ, Poobalan A, Aucott L, Stearns SC, Smith WCS, Jung RT, Campbell MK, Grant AM, Rakyan VK, Down TA, Balding DJ, Beck S, Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AFR, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NEE El, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason VV, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJP, Khaw K-T, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Mühleisen TW, Muhlestein JB, Münzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nöthen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schäfer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups KK, Stoll M, Tang WHW, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJN, Wells GA, Wichmann H-EE, Wild PS, Willenborg C, Witteman JCM, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, März W, Hengstenberg C, Blankenberg SS, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, O’Donnell CJ, McPherson R, Erdmann J, Samani NJ, Sharma P, Garg G, Kumar A, Mohammad F, Ramesh S, Slatkin M, Myocardial T, Genetics I, Investigators C, Udali S, Guarini P, Moruzzi S, Choi S, Friso S, Schübeler D, Setten J Van, Isgum I, Smolonska J, Ripke S, Jong PA De, Oudkerk M, Koning H De, Lammers JJ, Zanen P, Groen HJM, Boezen HM, Postma DS, Wijmenga C, Viergever MA, Th WP, Bakker PIW De (2015) Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 478:103–109. https://doi.org/10.1038/nrg3000.Epigenome-Wide
Arvind P, Nair J, Jambunathan S, Kakkar VV, Shanker J (2014) CELSR2-PSRC1-SORT1 gene expression and association with coronary artery disease and plasma lipid levels in an Asian Indian cohort. J Cardiol 64:339–346. https://doi.org/10.1016/j.jjcc.2014.02.012
Bennett BJ, Davis RC, Civelek M, Orozco L, Wu J, Qi H, Pan C, Packard RRS, Eskin E, Yan M, Kirchgessner T, Wang Z, Li X, Gregory JC, Hazen SL, Gargalovic PS, Lusis AJ (2015) Genetic architecture of atherosclerosis in mice: a systems genetics analysis of common inbred strains. PLoS Genet 11:1005711. https://doi.org/10.1371/journal.pgen.1005711
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA (2010) The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 28:1045–1048. https://doi.org/10.1038/nbt1010-1045
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J (2009) ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093. https://doi.org/10.1093/bioinformatics/btp101
Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, Davison D, Easton D, Evans D, Leung HT, Marchini JL, Morris AP, Spencer CCA, Tobin MD, Attwood AP, Boorman JP, Cant B, Everson U, Hussey JM, Jolley JD, Knight AS, Koch K, Meech E, Nutland S, Prowse CV, Stevens HE, Taylor NC, Walters GR, Walker NM, Watkins NA, Winzer T, Jones RW, McArdle WL, Ring SM, Strachan DP, Pembrey M, Breen G, St. Clair D, Caesar S, Gordon-Smith K, Jones L, Fraser C, Green EK, Grozeva D, Hamshere ML, Holmans PA, Jones IR, Kirov G, Moskvina V, Nikolov I, O’Donovan MC, Owen MJ, Collier DA, Elkin A, Farmer A, Williamson R, McGuffin P, Young AH, Ferrier IN, Ball SG, Balmforth AJ, Barrett JH, Bishop DT, Iles MM, Maqbool A, Yuldasheva N, Hall AS, Braund PS, Dixon RJ, Mangino M, Stevens S, Thompson JR, Bredin F, Tremelling M, Parkes M, Drummond H, Lees CW, Nimmo ER, Satsangi J, Fisher SA, Forbes A, Lewis CM, Onnie CM, Prescott NJ, Sanderson J, Mathew CG, Barbour J, Mohiuddin MK, Todhunter CE, Mansfield JC, Ahmad T, Cummings FR, Jewell DP, Webster J, Brown MJ, Lathrop GM, Connell J, Dominiczak A, Braga Marcano CA, Burke B, Dobson R, Gungadoo J, Lee KL, Munroe PB, Newhouse SJ, Onipinla A, Wallace C, Xue M, Caulfield M, Farrall M, Barton A, Bruce IN, Donovan H, Eyre S, Gilbert PD, Hider SL, Hinks AM, John SL, Potter C, Silman AJ, Symmons DPM, Thomson W, Worthington J, Dunger DB, Widmer B, Frayling TM, Freathy RM, Lango H, Perry JRB, Shields BM, Weedon MN, Hattersley AT, Hitman GA, Walker M, Elliott KS, Groves CJ, Lindgren CM, Rayner NW, Timpson NJ, Zeggini E, Newport M, Sirugo G, Lyons E, Vannberg F, Hill AVS, Bradbury LA, Farrar C, Pointon JJ, Wordsworth P, Brown MA, Franklyn JA, Heward JM, Simmonds MJ, Gough SCL, Seal S, Stratton MR, Rahman N, Ban SM, Goris A, Sawcer SJ, Compston A, Conway D, Jallow M, Rockett KA, Bumpstead SJ, Chaney A, Downes K, Ghori MJR, Gwilliam R, Hunt SE, Inouye M, Keniry A, King E, McGinnis R, Potter S, Ravindrarajah R, Whittaker P, Widden C, Withers D, Cardin NJ, Ferreira T, Pereira-Gale J, Hallgrimsdóttir IB, Howie BN, Spencer CCA, Su Z, Teo YY, Vukcevic D, Bentley D, Compston A (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678.https://doi.org/10.1038/nature05911
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature 562:203–209. https://doi.org/10.1038/s41586-018-0579-z
Castellano BM, Thelen AM, Moldavski O, Feltes M, Van Der Welle REN, Mydock-McGrane L, Jiang X, Van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS, Zoncu R (2017) Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355:1306–1311. https://doi.org/10.1126/SCIENCE.AAG1417
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. https://doi.org/10.1186/s13742-015-0047-8
Chen Z, Schunkert H (2021) Genetics of coronary artery disease in the post-GWAS era. J Int Med. https://doi.org/10.1111/JOIM.13362
Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, Jupe S, Kalatskaya I, MayMahajan S, May B, Ndegwa N, Schmidt E, Shamovsky V, Yung C, Birney E, Hermjakob H, D’Eustachio P, Stein L (2011) Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 39:D691–D697. https://doi.org/10.1093/nar/gkq1018
Dadu RT (2014) Ballantyne CM (2014) Lipid lowering with PCSK9 inhibitors. Nat Rev Cardiol 1110(11):563–575. https://doi.org/10.1038/nrcardio.2014.84
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X (2015) Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24:2125–2137. https://doi.org/10.1093/hmg/ddu733
Emdin CA, Khera AV, Klarin D, Natarajan P, Zekavat SM, Nomura A, Haas M, Aragam K, Ardissino D, Wilson JG, Schunkert H, McPherson R, Watkins H, Elosua R, Bown MJ, Samani NJ, Baber U, Erdmann J, Gormley P, Palotie A, Stitziel NO, Gupta N, Danesh J, Saleheen D, Gabriel S, Kathiresan S (2018) Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 137:222–232. https://doi.org/10.1161/CIRCULATIONAHA.117.028021
Erdmann J, Großhennig A, Braund PS, König IR, Hengstenberg C, Hall AS, Linsel-Nitschke P, Kathiresan S, Wright B, Trégouët DA, Cambien F, Bruse P, Aherrahrou Z, Wagner AK, Stark K, Schwartz SM, Salomaa V, Elosua R, Melander O, Voight BF, O’Donnell CJ, Peltonen L, Siscovick DS, Altshuler D, Merlini PA, Peyvandi F, Bernardinelli L, Ardissino D, Schillert A, Blankenberg S, Zeller T, Wild P, Schwarz DF, Tiret L, Perret C, Schreiber S, El MNE, Schäfer A, März W, Renner W, Bugert P, Klüter H, Schrezenmeir J, Rubin D, Ball SG, Balmforth AJ, Wichmann HE, Meitinger T, Fischer M, Meisinger C, Baumert J, Peters A, Ouwehand WH, Deloukas P, Thompson JR, Ziegler A, Samani NJ, Schunkert H (2009) New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 41:280–282. https://doi.org/10.1038/ng.307
Erdmann J, Kessler T, Munoz Venegas L, Schunkert H (2018) A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res 114:1241–1257. https://doi.org/10.1093/cvr/cvy084
Erdmann J, Willenborg C, Nahrstaedt J, Preuss M, Konig IR, Baumert J, Linsel-Nitschke P, Gieger C, Tennstedt S, Belcredi P, Aherrahrou Z, Klopp N, Loley C, Stark K, Hengstenberg C, Bruse P, Freyer J, Wagner AK, Medack A, Lieb W, Grosshennig A, Sager HB, Reinhardt A, Schafer A, Schreiber S, El Mokhtari NE, Raaz-Schrauder D, Illig T, Garlichs CD, Ekici AB, Reis A, Schrezenmeir J, Rubin D, Ziegler A, Wichmann H-E, Doering A, Meisinger C, Meitinger T, Peters A, Schunkert H (2011) Genome-wide association study identifies a new locus for coronary artery disease on chromosome 10p11.23. Eur Heart J 32:158–168. https://doi.org/10.1093/eurheartj/ehq405
Foroughi Asl H, Talukdar HA, Kindt ASD, Jain RK, Ermel R, Ruusalepp A, Nguyen KDH, Dobrin R, Reilly DF, Schunkert H, Samani NJ, Braenne I, Erdmann J, Melander O, Qi J, Ivert T, Skogsberg J, Schadt EE, Michoel T, Björkegren JLM (2015) Expression quantitative trait loci acting across multiple tissues are enriched in inherited risk for coronary artery disease. Circ Cardiovasc Genet 8:305–315. https://doi.org/10.1161/CIRCGENETICS.114.000640
Franzén O, Ermel R, Cohain A, Akers NK, Di Narzo A, Talukdar HA, Foroughi-Asl H, Giambartolomei C, Fullard JF, Sukhavasi K, Köks S, Gan LM, Giannarelli C, Kovacic JC, Betsholtz C, Losic B, Michoel T, Hao K, Roussos P, Skogsberg J, Ruusalepp A, Schadt EE, Björkegren JLM (2016) Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science (80-) 353:827–830. https://doi.org/10.1126/science.aad6970
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Nicolae DL, Cox NJ, Im HK, Consortium Gte (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47:1091–1098. https://doi.org/10.1038/ng.3367
Ghonem NS, Assis DN, Boyer JL (2015) Fibrates and cholestasis. Hepatology 62:635–643. https://doi.org/10.1002/HEP.27744
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10:e1004383. https://doi.org/10.1371/journal.pgen.1004383
Graham SE, Clarke SL, Wu K-HH, Kanoni S, Zajac GJM, Ramdas S, Surakka I, Ntalla I, Vedantam S, Winkler TW, Locke AE, Marouli E, Hwang MY, Han S, Narita A, Choudhury A, Bentley AR, Ekoru K, Verma A, Trivedi B, Martin HC, Hunt KA, Hui Q, Klarin D, Zhu X, Thorleifsson G, Helgadottir A, Gudbjartsson DF, Holm H, Olafsson I, Akiyama M, Sakaue S, Terao C, Kanai M, Zhou W, Brumpton BM, Rasheed H, Ruotsalainen SE, Havulinna AS, Veturi Y, Feng Q, Rosenthal EA, Lingren T, Pacheco JA, Pendergrass SA, Haessler J, Giulianini F, Bradford Y, Miller JE, Campbell A, Lin K, Millwood IY, Hindy G, Rasheed A, Faul JD, Zhao W, Weir DR, Turman C, Huang H, Graff M, Mahajan A, Brown MR, Zhang W, Yu K, Schmidt EM, Pandit A, Gustafsson S, Yin X, Luan J, Zhao J-H, Matsuda F, Jang H-M, Yoon K, Medina-Gomez C, Pitsillides A, Hottenga JJ, Willemsen G, Wood AR, Ji Y, Gao Z, Haworth S, Mitchell RE, Chai JF, Aadahl M, Yao J, Manichaikul A, Warren HR, Ramirez J, Bork-Jensen J, Kårhus LL, Goel A, Sabater-Lleal M, Noordam R, Sidore C, Fiorillo E, McDaid AF, Marques-Vidal P, Wielscher M, Trompet S, Sattar N, Møllehave LT, Thuesen BH, Munz M, Zeng L, Huang J, Yang B, Poveda A, Kurbasic A, Lamina C, Forer L, Scholz M, Galesloot TE, Bradfield JP, Daw EW, Zmuda JM, Mitchell JS, Fuchsberger C, Christensen H, Brody JA, Feitosa MF, Wojczynski MK, Preuss M, Mangino M, Christofidou P, Verweij N, Benjamins JW, Engmann J, Kember RL, Slieker RC, Lo KS, Zilhao NR, Le P, Kleber ME, Delgado GE, Huo S, Ikeda DD, Iha H, Yang J, Liu J, Leonard HL, Marten J, Schmidt B, Arendt M, Smyth LJ, Cañadas-Garre M, Wang C, Nakatochi M, Wong A, Hutri-Kähönen N, Sim X, Xia R, Huerta-Chagoya A, Fernandez-Lopez JC, Lyssenko V, Ahmed M, Jackson AU, Irvin MR, Oldmeadow C, Kim H-N, Ryu S, Timmers PRHJ, Arbeeva L, Dorajoo R, Lange LA, Chai X, Prasad G, Lorés-Motta L, Pauper M, Long J, Li X, Theusch E, Takeuchi F, Spracklen CN, Loukola A, Bollepalli S, Warner SC, Wang YX, Wei WB, Nutile T, Ruggiero D, Sung YJ, Hung Y-J, Chen S, Liu F, Yang J, Kentistou KA, Gorski M, Brumat M, Meidtner K, Bielak LF, Smith JA, Hebbar P, Farmaki A-E, Hofer E, Lin M, Xue C, Zhang J, Concas MP, Vaccargiu S, van der Most PJ, Pitkänen N, Cade BE, Lee J, van der Laan SW, Chitrala KN, Weiss S, Zimmermann ME, Lee JY, Choi HS, Nethander M, Freitag-Wolf S, Southam L, Rayner NW, Wang CA, Lin S-Y, Wang J-S, Couture C, Lyytikäinen L-P, Nikus K, Cuellar-Partida G, Vestergaard H, Hildalgo B, Giannakopoulou O, Cai Q, Obura MO, van Setten J, Li X, Schwander K, Terzikhan N, Shin JH, Jackson RD, Reiner AP, Martin LW, Chen Z, Li L, Highland HM, Young KL, Kawaguchi T, Thiery J, Bis JC, Nadkarni GN, Launer LJ, Li H, Nalls MA, Raitakari OT, Ichihara S, Wild SH, Nelson CP, Campbell H, Jäger S, Nabika T, Al-Mulla F, Niinikoski H, Braund PS, Kolcic I, Kovacs P, Giardoglou T, Katsuya T, Bhatti KF, de Kleijn D, de Borst GJ, Kim EK, Adams HHH, Ikram MA, Zhu X, Asselbergs FW, Kraaijeveld AO, Beulens JWJ, Shu X-O, Rallidis LS, Pedersen O, Hansen T, Mitchell P, Hewitt AW, Kähönen M, Pérusse L, Bouchard C, Tönjes A, Chen Y-DI, Pennell CE, Mori TA, Lieb W, Franke A, Ohlsson C, Mellström D, Cho YS, Lee H, Yuan J-M, Koh W-P, Rhee SY, Woo J-T, Heid IM, Stark KJ, Völzke H, Homuth G, Evans MK, Zonderman AB, Polasek O, Pasterkamp G, Hoefer IE, Redline S, Pahkala K, Oldehinkel AJ, Snieder H, Biino G, Schmidt R, Schmidt H, Chen YE, Bandinelli S, Dedoussis G, Thanaraj TA, Kardia SLR, Kato N, Schulze MB, Girotto G, Jung B, Böger CA, Joshi PK, Bennett DA, De Jager PL, Lu X, Mamakou V, Brown M, Caulfield MJ, Munroe PB, Guo X, Ciullo M, Jonas JB, Samani NJ, Kaprio J, Pajukanta P, Adair LS, Bechayda SA, de Silva HJ, Wickremasinghe AR, Krauss RM, Wu J-Y, Zheng W, den Hollander AI, Bharadwaj D, Correa A, Wilson JG, Lind L, Heng C-K, Nelson AE, Golightly YM, Wilson JF, Penninx B, Kim H-L, Attia J, Scott RJ, Rao DC, Arnett DK, Walker M, Koistinen HA, Chandak GR, Yajnik CS, Mercader JM, Tusié-Luna T, Aguilar-Salinas CA, Villalpando CG, Orozco L, Fornage M, Tai ES, van Dam RM, Lehtimäki T, Chaturvedi N, Yokota M, Liu J, Reilly DF, McKnight AJ, Kee F, Jöckel K-H, McCarthy MI, Palmer CNA, Vitart V, Hayward C, Simonsick E, van Duijn CM, Lu F, Qu J, Hishigaki H, Lin X, März W, Parra EJ, Cruz M, Gudnason V, Tardif J-C, Lettre G, ’t Hart LM, Elders PJM, Damrauer SM, Kumari M, Kivimaki M, van der Harst P, Spector TD, Loos RJF, Province MA, Psaty BM, Brandslund I, Pramstaller PP, Christensen K, Ripatti S, Widén E, Hakonarson H, Grant SFA, Kiemeney LALM, de Graaf J, Loeffler M, Kronenberg F, Gu D, Erdmann J, Schunkert H, Franks PW, Linneberg A, Jukema JW, Khera AV, Männikkö M, Jarvelin M-R, Kutalik Z, Cucca F, Mook-Kanamori DO, van Dijk KW, Watkins H, Strachan DP, Grarup N, Sever P, Poulter N, Rotter JI, Dantoft TM, Karpe F, Neville MJ, Timpson NJ, Cheng C-Y, Wong T-Y, Khor CC, Sabanayagam C, Peters A, Gieger C, Hattersley AT, Pedersen NL, Magnusson PKE, Boomsma DI, de Geus EJC, Cupples LA, van Meurs JBJ, Ghanbari M, Gordon-Larsen P, Huang W, Kim YJ, Tabara Y, Wareham NJ, Langenberg C, Zeggini E, Kuusisto J, Laakso M, Ingelsson E, Abecasis G, Chambers JC, Kooner JS, de Vries PS, Morrison AC, North KE, Daviglus M, Kraft P, Martin NG, Whitfield JB, Abbas S, Saleheen D, Walters RG, Holmes MV, Black C, Smith BH, Justice AE, Baras A, Buring JE, Ridker PM, Chasman DI, Kooperberg C, Wei W-Q, Jarvik GP, Namjou B, Hayes MG, Ritchie MD, Jousilahti P, Salomaa V, Hveem K, Åsvold BO, Kubo M, Kamatani Y, Okada Y, Murakami Y, Thorsteinsdottir U, Stefansson K, Ho Y-L, Lynch JA, Rader DJ, Tsao PS, Chang K-M, Cho K, O’Donnell CJ, Gaziano JM, Wilson P, Rotimi CN, Hazelhurst S, Ramsay M, Trembath RC, van Heel DA, Tamiya G, Yamamoto M, Kim B-J, Mohlke KL, Frayling TM, Hirschhorn JN, Kathiresan S, Boehnke M, Natarajan P, Peloso GM, Brown CD, Morris AP, Assimes TL, Deloukas P, Sun YV, Willer CJ (2021) The power of genetic diversity in genome-wide association studies of lipids. Nature 600:675–679. https://doi.org/10.1038/s41586-021-04064-3
GTEx Consortium (2013) The genotype-tissue expression (GTEx) project. Nat Genet 45:580–585. https://doi.org/10.1038/ng.2653
Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R (2004) The gene oncology (GO) database and informatics resource. Nucleic Acids Res 32:D258–D261. https://doi.org/10.1093/nar/gkh036
Van Hout CV, Tachmazidou I, Backman JD, Hoffman JD, Liu D, Pandey AK, Gonzaga-Jauregui C, Khalid S, Ye B, Banerjee N, Li AH, O’Dushlaine C, Marcketta A, Staples J, Schurmann C, Hawes A, Maxwell E, Barnard L, Lopez A, Penn J, Habegger L, Blumenfeld AL, Bai X, O’Keeffe S, Yadav A, Praveen K, Jones M, Salerno WJ, Chung WK, Surakka I, Willer CJ, Hveem K, Leader JB, Carey DJ, Ledbetter DH, Cardon L, Yancopoulos GD, Economides A, Coppola G, Shuldiner AR, Balasubramanian S, Cantor M, Nelson MR, Whittaker J, Reid JG, Marchini J, Overton JD, Scott RA, Abecasis GR, Yerges-Armstrong L, Baras A (2020) Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586:749–756. https://doi.org/10.1038/s41586-020-2853-0
Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardiñas AF, Rajagopal VM, Als TD, Nguyen H, Girdhar K, Boocock J, Roussos P, Fromer M, Kramer R, Domenici E, Gamazon ER, Purcell S, Johnson JS, Shah HR, Klein LL, Dang KK, Logsdon BA, Mahajan MC, Mangravite LM, Toyoshiba H, Gur RE, Hahn CG, Schadt E, Lewis DA, Haroutunian V, Peters MA, Lipska BK, Buxbaum JD, Hirai K, Perumal TM, Essioux L, Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, Lee P, Bulik-Sullivan B, Collier DA, Huang H, Pers TH, Agartz I, Agerbo E, Albus M, Alexander M, Amin F, Bacanu SA, Begemann M, Belliveau RA, Bene J, Bergen SE, Bevilacqua E, Bigdeli TB, Black DW, Bruggeman R, Buccola NG, Buckner RL, Byerley W, Cahn W, Cai G, Campion D, Cantor RM, Carr VJ, Carrera N, Catts SV, Chambert KD, Chan RCK, Chen RYL, Chen EYH, Cheng W, Cheung EFC, Chong SA, Cloninger CR, Cohen D, Cohen N, Cormican P, Craddock N, Crowley JJ, Curtis D, Davidson M, Davis KL, Degenhardt F, Del Favero J, Demontis D, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Dudbridge F, Durmishi N, Eichhammer P, Eriksson J, Escott-Price V, Fanous AH, Farrell MS, Frank J, Franke L, Freedman R, Freimer NB, Friedl M, Friedman JI, Fromer M, Genovese G, Georgieva L, Giegling I, Giusti-Rodríguez P, Godard S, Goldstein JI, Golimbet V, Gopal S, Gratten J, de Haan L, Hammer C, Hamshere ML, Hansen M, Hansen T, Hartmann AM, Henskens FA, Herms S, Hirschhorn JN, Hoffmann P, Hofman A, Hollegaard MV, Hougaard DM, Ikeda M, Joa I, Julia A, Kahn RS, Kalaydjieva L, Karachanak-Yankova S, Karjalainen J, Kavanagh D, Keller MC, Kennedy JL, Khrunin A, Kim Y, Klovins J, Knowles JA, Konte B, Kucinskas V, Kucinskiene ZA, Kuzelova-Ptackova H, Kahler AK, Laurent C, Keong JLC, Lee SH, Legge SE, Lerer B, Li M, Li T, Liang KY, Lieberman J, Limborska S, Loughland CM, Lubinski J, Lonnqvist J, Macek M, Magnusson PKE, Maher BS, Maier W, Mallet J, Marsal S, Mattheisen M, Mattingsdal M, McCarley RW, McDonald C, McIntosh AM, Meier S, Meijer CJ, Melegh B, Melle I, Mesholam-Gately RI, Metspalu A, Michie PT, Milani L, Milanova V, Mokrab Y, Morris DW, Mors O, Murphy KC, Murray RM, Myin-Germeys I, Muller-Myhsok B, Nelis M, Nenadic I, Nertney DA, Nestadt G, Nicodemus KK, Nikitina-Zake L, Nisenbaum L, Nordin A, O’Callaghan E, O’Dushlaine C, O’Neill FA, Oh SY, Olincy A, Olsen L, Van Os J, Pantelis C, Papadimitriou GN, Papiol S, Parkhomenko E, Pato MT, Paunio T, Pejovic-Milovancevic M, Perkins DO, Pietiläinen O, Pimm J, Pocklington AJ, Powell J, Price A, Pulver AE, Purcell SM, Quested D, Rasmussen HB, Reichenberg A, Reimers MA, Richards AL, Roffman JL, Salomaa V, Sanders AR, Schall U, Schubert CR, Schulze TG, Schwab SG, Scolnick EM, Scott RJ, Seidman LJ, Shi J, Sigurdsson E, Silagadze T, Silverman JM, Sim K, Slominsky P, Smoller JW, So HC, Spencer CCA, Stahl EA, Stefansson H, Steinberg S, Stogmann E, Straub RE, Strengman E, Strohmaier J, Stroup TS, Subramaniam M, Suvisaari J, Svrakic DM, Szatkiewicz JP, Soderman E, Thirumalai S, Toncheva D, Tosato S, Veijola J, Waddington J, Walsh D, Wang D, Wang Q, Webb BT, Weiser M, Wildenauer DB, Williams NM, Williams S, Witt SH, Wolen AR, Wong EHM, Wormley BK, Xi HS, Zai CC, Zheng X, Zimprich F, Wray NR, Stefansson K, Visscher PM, Adolfsson R, Andreassen OA, Blackwood DHR, Bramon E, Børglum AD, Cichon S, Darvasi A, Ehrenreich H, Esko T, Gejman PV, Gill M, Gurling H, Hultman CM, Iwata N, Jablensky AV, Jonsson EG, Kendler KS, Kirov G, Knight J, Lencz T, Levinson DF, Li QS, Liu J, Malhotra AK, McCarroll SA, McQuillin A, Moran JL, Mortensen PB, Mowry BJ, Nothen MM, Ophoff RA, Owen MJ, Palotie A, Pato CN, Petryshen TL, Posthuma D, Rietschel M, Riley BP, Rujescu D, Sham PC, Sklar P, Clair DS, Weinberger DR, Wendland JR, Werge T, Daly MJ, Sullivan PF, O’Donovan MC, Rajagopal VM, Grove J, Mortensen PB, Pedersen CB, Pedersen MG, Nordentoft M, Bybjerg-Grauholm J, Bækvad-Hansen M, Hansen CS, Sullivan P, Devlin B, Sieberts SK, Cox NJ, Im HK (2019) Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat Genet 51:659–674. https://doi.org/10.1038/s41588-019-0364-4
Investigators MIG and CardiEC (2016) Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med 374:1134. https://doi.org/10.1056/NEJMOA1507652
Kanehisa M (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Aguilar Salinas CA, Ahmad T, Albert CM, Ardissino D, Atzmon G, Barnard J, Beaugerie L, Benjamin EJ, Boehnke M, Bonnycastle LL, Bottinger EP, Bowden DW, Bown MJ, Chambers JC, Chan JC, Chasman D, Cho J, Chung MK, Cohen B, Correa A, Dabelea D, Daly MJ, Darbar D, Duggirala R, Dupuis J, Ellinor PT, Elosua R, Erdmann J, Esko T, Färkkilä M, Florez J, Franke A, Getz G, Glaser B, Glatt SJ, Goldstein D, Gonzalez C, Groop L, Haiman C, Hanis C, Harms M, Hiltunen M, Holi MM, Hultman CM, Kallela M, Kaprio J, Kathiresan S, Kim BJ, Kim YJ, Kirov G, Kooner J, Koskinen S, Krumholz HM, Kugathasan S, Kwak SH, Laakso M, Lehtimäki T, Loos RJF, Lubitz SA, Ma RCW, MacArthur DG, Marrugat J, Mattila KM, McCarroll S, McCarthy MI, McGovern D, McPherson R, Meigs JB, Melander O, Metspalu A, Neale BM, Nilsson PM, O’Donovan MC, Ongur D, Orozco L, Owen MJ, Palmer CNA, Palotie A, Park KS, Pato C, Pulver AE, Rahman N, Remes AM, Rioux JD, Ripatti S, Roden DM, Saleheen D, Salomaa V, Samani NJ, Scharf J, Schunkert H, Shoemaker MB, Sklar P, Soininen H, Sokol H, Spector T, Sullivan PF, Suvisaari J, Tai ES, Teo YY, Tiinamaija T, Tsuang M, Turner D, Tusie-Luna T, Vartiainen E, Ware JS, Watkins H, Weersma RK, Wessman M, Wilson JG, Xavier RJ, Neale BM, Daly MJ, MacArthur DG (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Kessler T, Zhang L, Liu Z, Yin X, Huang Y, Wang Y, Fu Y, Mayr M, Ge Q, Xu Q, Zhu Y, Wang X, Schmidt K, De Wit C, Erdmann J, Schunkert H, Aherrahrou Z, Kong W (2015) ADAMTS-7 inhibits re-endothelialization of injured arteries and promotes vascular remodeling through cleavage of thrombospondin-1. Circulation 131:1191–1201. https://doi.org/10.1161/CIRCULATIONAHA.114.014072
Klimentidis YC, Arora A, Newell M, Zhou J, Ordovas JM, Renquist BJ, Wood AC (2020) Phenotypic and genetic characterization of lower LDL cholesterol and increased type 2 diabetes risk in the UK biobank. Diabetes 69:2194–2205. https://doi.org/10.2337/db19-1134
Kohl M, Wiese S, Warscheid B (2011) Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol 696:291–303. https://doi.org/10.1007/978-1-60761-987-1_18
Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, Momozawa Y, Matsunaga H, Ieki H, Ozaki K, Onouchi Y, Takahashi A, Nomura S, Morita H, Akazawa H, Kim C, Seo J, Higasa K, Iwasaki M, Yamaji T, Sawada N, Tsugane S, Koyama T, Ikezaki H, Takashima N, Tanaka K, Arisawa K, Kuriki K, Naito M, Wakai K, Suna S, Sakata Y, Sato H, Hori M, Sakata Y, Matsuda K, Murakami Y, Aburatani H, Kubo M, Matsuda F, Kamatani Y, Komuro I (2020) Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nat Genet 52:1169–1177. https://doi.org/10.1038/s41588-020-0705-3
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, Hoffman D, Jang W, Kaur K, Liu C, Lyoshin V, Maddipatla Z, Maiti R, Mitchell J, O’leary N, Riley GR, Shi W, Zhou G, Schneider V, Maglott D, Holmes JB, Kattman BL (2019) ClinVar: improvements to accessing data. Nucleic Acids Res 48:835–844.https://doi.org/10.1093/nar/gkz972
De Leeuw CA, Mooij JM, Heskes T, Posthuma D (2015) MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11:1004219. https://doi.org/10.1371/journal.pcbi.1004219
Li L, Pang S, Zeng L, Güldener U, Schunkert H (2021) Genetically determined intelligence and coronary artery disease risk. Clin Res Cardiol 110:211–219. https://doi.org/10.1007/s00392-020-01721-x
Li MX, Yeung JMY, Cherny SS, Sham PC (2012) Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet 131:747–756. https://doi.org/10.1007/S00439-011-1118-2
Li YI, Van De Geijn B, Raj A, Knowles DA, Petti AA, Golan D, Gilad Y, Pritchard JK (2016) RNA splicing is a primary link between genetic variation and disease. Science (80-) 352:600–604. https://doi.org/10.1126/science.aad9417
Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, Amouyel P, Angelantonio E DI, Arveiler D, Assimes TL, Auer PL, Baber U, Ballantyne CM, Bang LE, Benn M, Bis JC, Boehnke M, Boerwinkle E, Bork-Jensen J, Bottinger EP, Brandslund I, Brown M, Busonero F, Caulfield MJ, Chambers JC, Chasman DI, Chen YE, Chen YDI, Chowdhury R, Christensen C, Chu AY, Connell JM, Cucca F, Cupples LA, Damrauer SM, Davies G, Deary IJ, Dedoussis G, Denny JC, Dominiczak A, Dubé MP, Ebeling T, Eiriksdottir G, Esko T, Farmaki AE, Feitosa MF, Ferrario M, Ferrieres J, Ford I, Fornage M, Franks PW, Frayling TM, Frikke-Schmidt R, Fritsche LG, Frossard P, Fuster V, Ganesh SK, Gao W, Garcia ME, Gieger C, Giulianini F, Goodarzi MO, Grallert H, Grarup N, Groop L, Grove ML, Gudnason V, Hansen T, Harris TB, Hayward C, Hirschhorn JN, Holmen OL, Huffman J, Huo Y, Hveem K, Jabeen S, Jackson AU, Jakobsdottir J, Jarvelin MR, Jensen GB, Jørgensen ME, Jukema JW, Justesen JM, Kamstrup PR, Kanoni S, Karpe F, Kee F, Khera AV, Klarin D, Koistinen HA, Kooner JS, Kooperberg C, Kuulasmaa K, Kuusisto J, Laakso M, Lakka T, Langenberg C, Langsted A, Launer LJ, Lauritzen T, Mliewald DC, Lin LA, Linneberg A, Loos RJF, Lu Y, Lu X, Mägi R, Malarstig A, Manichaikul A, Manning AK, Mäntyselkä P, Marouli E, Masca NGD, Maschio A, Meigs JB, Melander O, Metspalu A, Morris AP, Morrison AC, Mulas A, Müller-Nurasyid M, Munroe PB, Neville MJ, Nielsen SF, Nielsen JB, Nordestgaard BG, Ordovas JM, Mehran R, O’Donnell CJ, Orho-Melander M, Molony CM, Muntendam P, Padmanabhan S, Palmer CNA, Pasko D, Patel AP, Pedersen O, Perola M, Peters A, Pisinger C, Pistis G, Polasek O, Poulter N, Psaty BM, Rader DJ, Rasheed A, Rauramaa R, Reilly DF, Reiner AP, Renström F, Rich SS, Ridker PM, Rioux JD, Robertson NR, Roden DM, Rotter JI, Rudan I, Salomaa V, Samani NJ, Sanna S, Sattar N, Schmidt EM, Scott RA, Sever P, Sevilla RS, Shaffer CM, Sim X, Sivapalaratnam S, Small KS, Smith AV, Smith BH, Somayajula S, Southam L, Spector TD, Speliotes EK, Starr JM, Stirrups KE, Stitziel N, Strauch K, Stringham HM, Surendran P, Tada H, Tall AR, Tang H, Tardif JC, Taylor KD, Trompet S, Tsao PS, Tuomilehto J, Tybjaerg-Hansen A, Zuydam NRV, Varbo A, Varga TV, Virtamo J, Waldenberger M, Wang N, Wareham NJ, Warren HR, Weeke PE, Weinstock J, Wessel J, Wilson JG, Wilson PWF, Xu M, Yaghootkar H, Young R, Zeggini E, Zhang H, Zheng NS, Zhang W, Zhang Y, Zhou W, Zhou Y, Zoledziewska M, Howson JMM, Danesh J, McCarthy MI, Cowan CA, Abecasis G, Deloukas P, Musunuru K, Willer CJ, Kathiresan S (2017) Exome-wide association study of plasma lipids in > 300,000 individuals. Nat Genet 49:1758–1766.https://doi.org/10.1038/NG.3977
Lusis AJ, Seldin MM, Allayee H, Bennett BJ, Civelek M, Davis RC, Eskin E, Farber CR, Hui S, Mehrabian M, Norheim F, Pan C, Parks B, Rau CD, Smith DJ, Vallim T, Wang Y, Wang J (2016) The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. J Lipid Res 57:925–942. https://doi.org/10.1194/jlr.R066944
Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, Chakraborty S (2019) A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol 234:16812–16823. https://doi.org/10.1002/jcp.28350
McGough IJ, Steinberg F, Gallon M, Yatsu A, Ohbayashi N, Heesom KJ, Fukuda M, Cullen PJ (2014) Identification of molecular heterogeneity in SNX27-retromermediated endosome-to-plasma-membrane recycling. J Cell Sci 127:4940–4953. https://doi.org/10.1242/jcs.156299
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F (2016) The ensembl variant effect predictor. Genome Biol. https://doi.org/10.1186/s13059-016-0974-4
Meng Y, Heybrock S, Neculai D, Saftig P (2020) Cholesterol handling in lysosomes and beyond. Trends Cell Biol 30:452–466. https://doi.org/10.1016/J.TCB.2020.02.007
Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, Giannakopoulou O, Jiang T, Hamby SE, Di Angelantonio E, Assimes TL, Bottinger EP, Chambers JC, Clarke R, Palmer CNA, Cubbon RM, Ellinor P, Ermel R, Evangelou E, Franks PW, Grace C, Gu D, Hingorani AD, Howson JMM, Ingelsson E, Kastrati A, Kessler T, Kyriakou T, Lehtimäki T, Lu X, Lu Y, März W, McPherson R, Metspalu A, Pujades-Rodriguez M, Ruusalepp A, Schadt EE, Schmidt AF, Sweeting MJ, Zalloua PA, Alghalayini K, Keavney BD, Kooner JS, Loos RJF, Patel RS, Rutter MK, Tomaszewski M, Tzoulaki I, Zeggini E, Erdmann J, Dedoussis G, Björkegren JLM, Schunkert H, Farrall M, Danesh J, Samani NJ, Watkins H, Deloukas P (2017) Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 49:1385–1391. https://doi.org/10.1038/ng.3913
Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, CHopewell J, Webb TR, Zeng L, Dehghan A, Alver M, Marmasu S, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, De Andrade M, De Vries PS, Van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, MLindgren C, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FUR, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, EStirrups K, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples L, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, SHall A, Hamsten A, Harris TB, LHazen S, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJF, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O’Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M (2015) A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47:1121–1130.https://doi.org/10.1038/ng.3396
Stewart A, Fisher RA (2015) Introduction: G protein-coupled receptors and RGS proteins. Prog Mol Biol Transl Sci 133:1–11. https://doi.org/10.1016/BS.PMBTS.2015.03.002
Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, Ronzano F, Centeno E, Sanz F, Furlong LI (2020) The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res 48:D845–D855. https://doi.org/10.1093/nar/gkz1021
Powell WS, Rokach J (2015) Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim Biophys Acta 1851:340–355. https://doi.org/10.1016/J.BBALIP.2014.10.008
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann H-E, Barrett JH, König IR, Stevens SE, Szymczak S, Tregouet D-A, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Brænne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H (2007) Genomewide association analysis of coronary artery disease. N Engl J Med 357:443–453. https://doi.org/10.1056/NEJMoa072366
Sangphech N, Osborne BA, Palaga T (2014) Notch signaling regulates the phosphorylation of Akt and survival of lipopolysaccharide-activated macrophages via regulator of G protein signaling 19 (RGS19). Immunobiology 219:653–660. https://doi.org/10.1016/j.imbio.2014.03.020
Sixt S, Beer S, Blüher M, Korff N, Peschel T, Sonnabend M, Teupser D, Thiery J, Adams V, Schuler G, Niebauer J (2010) Long- but not short-term multifactorial intervention with focus on exercise training improves coronary endothelial dysfunction in diabetes mellitus type 2 and coronary artery disease. Eur Heart J 31:112–119. https://doi.org/10.1093/eurheartj/ehp398
Slenter DN, Kutmon M, Hanspers K, Riutta A, Windsor J, Nunes N, Mélius J, Cirillo E, Coort SL, Digles D, Ehrhart F, Giesbertz P, Kalafati M, Martens M, Miller R, Nishida K, Rieswijk L, Waagmeester A, Eijssen LMT, Evelo CT, Pico AR, Willighagen EL (2018) WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 46:D661–D667. https://doi.org/10.1093/nar/gkx1064
Stitziel NO, Won HH, Morrison AC, Peloso GM, Do R, Lange LA, Fontanillas P, Gupta N, Duga S, Goel A, Farrall M, Saleheen D, Ferrario P, König I, Asselta R, Merlini PA, Marziliano N, Notarangelo MF, Schick U, Auer P, Assimes TL, Reilly M, Wilensky R, Rader DJ, Kees Hovingh G, Meitinger T, Kessler T, Kastrati A, Laugwitz KL, Siscovick D, Rotter JI, Hazen SL, Tracy R, Cresci S, Spertus J, Jackson R, Schwartz SM, Natarajan P, Crosby J, Muzny D, Ballantyne C, Rich SS, O’Donnell CJ, Abecasis G, Sunyaev S, Nickerson DA, Buring JE, Ridker PM, Chasman DI, Austin E, Ye Z, Kullo IJ, Weeke PE, Shaffer CM, Bastarache LA, Denny JC, Roden DM, Palmer C, Deloukas P, Lin DY, Tang ZZ, Erdmann J, Schunkert H, Danesh J, Marrugat J, Elosua R, Ardissino D, McPherson R, Watkins H, Reiner AP, Wilson JG, Altshuler D, Gibbs RA, Lander ES, Boerwinkle E, Gabriel S, Kathiresan S (2014) Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med 371:2072–2082. https://doi.org/10.1056/NEJMoa1405386
Thelen AM, Zoncu R (2017) Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol 27:833–850. https://doi.org/10.1016/J.TCB.2017.07.006
Tiwari S, Siddiqi SA (2012) Intracellular trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol 32:1079–1086. https://doi.org/10.1161/ATVBAHA.111.241471
Tso PH, Yung LY, Wang Y, Wong YH (2011) RGS19 stimulates cell proliferation by deregulating cell cycle control and enhancing Akt signaling. Cancer Lett 309:199–208. https://doi.org/10.1016/j.canlet.2011.06.002
Tsutsumi K (2003) Lipoprotein lipase and atherosclerosis. Curr Vasc Pharmacol 1:11–17. https://doi.org/10.2174/1570161033386673
Vilne B, Schunkert H (2018) Integrating genes affecting coronary artery disease in functional networks by multi-OMICs approach. Front Cardiovasc Med 5:89. https://doi.org/10.3389/fcvm.2018.00089
Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, Ermel R, Ruusalepp A, Quertermous T, Hao K, Björkegren JLM, Im HK, Pasaniuc B, Rivas MA, Kundaje A (2019) Opportunities and challenges for transcriptome-wide association studies. Nat Genet 51:592–599. https://doi.org/10.1038/s41588-019-0385-z
Wang Y, Nakajima T, Gonzalez FJ, Tanaka N (2020) PPARs as metabolic regulators in the liver: lessons from liver-specific PPAR-null mice. Int J Mol Sci. https://doi.org/10.3390/IJMS21062061
Wild PS, Zeller T, Schillert A, Szymczak S, Sinning CR, Deiseroth A, Schnabel RB, Lubos E, Keller T, Eleftheriadis MS, Bickel C, Rupprecht HJ, Wilde S, Rossmann H, Diemert P, Cupples LA, Perret C, Erdmann J, Stark K, Kleber ME, Epstein SE, Voight BF, Kuulasmaa K, Li M, Schäfer AS, Klopp N, Braund PS, Sager HB, Demissie S, Proust C, König IR, Wichmann H-E, Reinhard W, Hoffmann MM, Virtamo J, Burnett MS, Siscovick D, Wiklund PG, Qu L, El Mokthari NE, Thompson JR, Peters A, Smith AV, Yon E, Baumert J, Hengstenberg C, März W, Amouyel P, Devaney J, Schwartz SM, Saarela O, Mehta NN, Rubin D, Silander K, Hall AS, Ferrieres J, Harris TB, Melander O, Kee F, Hakonarson H, Schrezenmeir J, Gudnason V, Elosua R, Arveiler D, Evans A, Rader DJ, Illig T, Schreiber S, Bis JC, Altshuler D, Kavousi M, Witteman JCM, Uitterlinden AG, Hofman A, Folsom AR, Barbalic M, Boerwinkle E, Kathiresan S, Reilly MP, O’Donnell CJ, Samani NJ, Schunkert H, Cambien F, Lackner KJ, Tiret L, Salomaa V, Munzel T, Ziegler A, Blankenberg S (2011) A genome-wide association study identifies LIPA as a susceptibility gene for coronary artery disease. Circ Cardiovasc Genet 4:403–412. https://doi.org/10.1161/CIRCGENETICS.110.958728
Winkelmann BR, März W, Boehm BO, Zotz R, Hager J, Hellstern P, Senges J (2001) Rationale and design of the LURIC study—a resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics. https://doi.org/10.1517/14622416.2.1.s1
Wolfrum C, Borrmann CM, Börchers T, Spener F (2001) Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci USA 98:2323–2328. https://doi.org/10.1073/PNAS.051619898
Wolfson RL, Chantranupong L, Wyant GA, Gu X, Orozco JM, Shen K, Condon KJ, Petri S, Kedir J, Scaria SM, Abu-Remaileh M, Frankel WN, Sabatini DM (2017) KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 543:438–442. https://doi.org/10.1038/nature21423
Wu L, Shi W, Long J, Guo X, Michailidou K, Beesley J, Bolla MK, Shu XO, Lu Y, Cai Q, Al-Ejeh F, Rozali E, Wang Q, Dennis J, Li B, Zeng C, Feng H, Gusev A, Barfield RT, Andrulis IL, Anton-Culver H, Arndt V, Aronson KJ, Auer PL, Barrdahl M, Baynes C, Beckmann MW, Benitez J, Bermisheva M, Blomqvist C, Bogdanova NV, Bojesen SE, Brauch H, Brenner H, Brinton L, Broberg P, Brucker SY, Burwinkel B, Caldés T, Canzian F, Carter BD, Castelao JE, Chang-Claude J, Chen X, Cheng TYD, Christiansen H, Clarke CL, Collée M, Cornelissen S, Couch FJ, Cox D, Cox A, Cross SS, Cunningham JM, Czene K, Daly MB, Devilee P, Doheny KF, Dörk T, Dos-Santos-Silva I, Dumont M, Dwek M, Eccles DM, Eilber U, Eliassen AH, Engel C, Eriksson M, Fachal L, Fasching PA, Figueroa J, Flesch-Janys D, Fletcher O, Flyger H, Fritschi L, Gabrielson M, Gago-Dominguez M, Gapstur SM, García-Closas M, Gaudet MM, Ghoussaini M, Giles GG, Goldberg MS, Goldgar DE, González-Neira A, Guénel P, Hahnen E, Haiman CA, Håkansson N, Hall P, Hallberg E, Hamann U, Harrington P, Hein A, Hicks B, Hillemanns P, Hollestelle A, Hoover RN, Hopper JL, Huang G, Humphreys K, Hunter DJ, Jakubowska A, Janni W, John EM, Johnson N, Jones K, Jones ME, Jung A, Kaaks R, Kerin MJ, Khusnutdinova E, Kosma VM, Kristensen VN, Lambrechts D, Le Marchand L, Li J, Lindström S, Lissowska J, Lo WY, Loibl S, Lubinski J, Luccarini C, Lux MP, MacInnis RJ, Maishman T, Kostovska IM, Mannermaa A, Manson JAE, Margolin S, Mavroudis D, Meijers-Heijboer H, Meindl A, Menon U, Meyer J, Mulligan AM, Neuhausen SL, Nevanlinna H, Neven P, Nielsen SF, Nordestgaard BG, Olopade OI, Olson JE, Olsson H, Peterlongo P, Peto J, Plaseska-Karanfilska D, Prentice R, Presneau N, Pylkäs K, Rack B, Radice P, Rahman N, Rennert G, Rennert HS, Rhenius V, Romero A, Romm J, Rudolph A, Saloustros E, Sandler DP, Sawyer EJ, Schmidt MK, Schmutzler RK, Schneeweiss A, Scott RJ, Scott CG, Seal S, Shah M, Shrubsole MJ, Smeets A, Southey MC, Spinelli JJ, Stone J, Surowy H, Swerdlow AJ, Tamimi RM, Tapper W, Taylor JA, Terry MB, Tessier DC, Thomas A, Thöne K, Tollenaar RAEM, Torres D, Truong T, Untch M, Vachon C, Van Den Berg D, Vincent D, Waisfisz Q, Weinberg CR, Wendt C, Whittemore AS, Wildiers H, Willett WC, Winqvist R, Wolk A, Xia L, Yang XR, Ziogas A, Ziv E, Dunning AM, Pharoah PDP, Simard J, Milne RL, Edwards SL, Kraft P, Easton DF, Chenevix-Trench G, Zheng W (2018) A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat Genet 50:968–978. https://doi.org/10.1038/s41588-018-0132-x
Zein AA, Kaur R, Hussein TOK, Graf GA, Lee J-Y (2019) ABCG5/G8: a structural view to pathophysiology of the hepatobiliary cholesterol secretion. Biochem Soc Trans 47:1259. https://doi.org/10.1042/BST20190130
Zhang W, Voloudakis G, Rajagopal VM, Readhead B, Dudley JT, Schadt EE, Björkegren JLM, Kim Y, Fullard JF, Hoffman GE, Roussos P (2019) Integrative transcriptome imputation reveals tissue-specific and shared biological mechanisms mediating susceptibility to complex traits. Nat Commun 10:1–13. https://doi.org/10.1038/s41467-019-11874-7
Acknowledgements
We’d like to thank Dr. Wen Zhang, Pro. Panos Roussos for sharing the prediction models of seven tissues trained from either STARNET or GTEx panels, as well as the EpiXcan pipeline.
Funding
Open Access funding enabled and organized by Projekt DEAL. The work was funded by the German Federal Ministry of Education and Research (BMBF) within the framework of ERA-NET on Cardiovascular Disease (Druggable-MI-genes: 01KL1802), within the scheme of target validation (BlockCAD: 16GW0198K), COMMITMENT (01ZX1904A), and within the scheme of e:Med research and funding concept (AbCD-Net: 01ZX1706C). As a Co-applicant of the British Heart Foundation (BHF)/German Centre of Cardiovascular Research (DZHK)-collaboration (DZHK-BHF: 81X2600522) and the Leducq Foundation for Cardiovascular Research (PlaqOmics: 18CVD02), we gratefully acknowledge their funding. Additional support has been received from the German Research Foundation (DFG) as part of the Sonderforschungsbereich SFB 1123 (B02) and the Sonderforschungsbereich SFB TRR 267 (B05). Further, we kindly acknowledge the support of the Bavarian State Ministry of Health and Care who funded this work with DigiMed Bayern (grant No: DMB-1805–0001) within its Masterplan “Bayern Digital II” and of the German Federal Ministry of Economics and Energy in its scheme of ModulMax (grant No: ZF4590201BA8). Finally, we acknowledge the support of DZHK with Postdoc Start-up Grant (“Förderkennzeichen”).
Author information
Authors and Affiliations
Contributions
HS, LL, ZC designed the study and wrote the manuscript. LL ran analyses. ZC, SL and AS performed experiment. MS, UG, SK, AM, KH, CP, AJL, SP, TK, RE, KS, AR, JG, JE, JCK, JLMB provided research data, technical support and gave conceptual advice.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Tools and data
EpiXcan pipeline: https://bitbucket.org/roussoslab/epixcan/src/master/, and predictive models based on STARNET and GTEx databases: http://predictdb.org/.
PrediXcan pipeline: https://github.com/hakyim/PrediXcan.
qtlBHM: https://github.com/rajanil/qtlBHM.
STARNET database: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001203.v1.p1. Project ID: 13585.
GTEx database: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v8.p2. Project ID: 20848.
UK Biobank: https://www.ukbiobank.ac.uk/. Project ID: 25214.
MAGMA: https://ctg.cncr.nl/software/magma.
R package for colocalization analysis, coloc: https://cran.r-project.org/web/packages/coloc/vignettes/vignette.html.
DisGeNET: https://www.disgenet.org/.
CARDIoGRAMplusC4D Consortium: http://www.cardiogramplusc4d.org/.
Additional information
The original online version of this article was revised: Figure 6 has been corrected.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Chen, Z., von Scheidt, M. et al. Transcriptome-wide association study of coronary artery disease identifies novel susceptibility genes. Basic Res Cardiol 117, 6 (2022). https://doi.org/10.1007/s00395-022-00917-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-022-00917-8