
Abstract
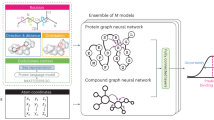
Understanding the interactions formed between a ligand and its molecular target is key to guiding the optimization of molecules. Different experimental and computational methods have been applied to better understanding these intermolecular interactions. Here we report a method based on geometric deep learning that is capable of predicting the binding conformations of ligands to protein targets. The model learns a statistical potential based on the distance likelihood, which is tailor-made for each ligand–target pair. This potential can be coupled with global optimization algorithms to reproduce the experimental binding conformations of ligands. We show that the potential based on distance likelihood, described here, performs similarly or better than well-established scoring functions for docking and screening tasks. Overall, this method represents an example of how artificial intelligence can be used to improve structure-based drug design.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

Data availability
The data that support the findings of this study are available in figshare with identifier https://doi.org/10.6084/m9.figshare.c.540732938. Source data are provided with this paper.
Code availability
The code used to generate the results shown in this study is available under an MIT Licence in the repository https://github.com/OptiMaL-PSE-Lab/DeepDock and https://doi.org/10.5281/zenodo.551020339.
References
Hert, J., Irwin, J. J., Laggner, C., Keiser, M. J. & Shoichet, B. K. Quantifying biogenic bias in screening libraries. Nat. Chem. Biol. 5, 479–483 (2009).
Dobson, C. M. Chemical space and biology. Nature 432, 824–828 (2004).
Congreve, M., Murray, C. W. & Blundell, T. L. Keynote review: structural biology and drug discovery. Drug Discov. Today 10, 895–907 (2005).
Klebe, G. in Drug Design: Methodology, Concepts and Mode-of-Action (ed. Klebe, G.) 61–88 (Springer, 2013).
Renaud, J. P. et al. Cryo-EM in drug discovery: achievements, limitations and prospects. Nat. Rev. Drug Discov. 17, 471–492 (2018).
Gorgulla, C. et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 580, 663–668 (2020).
Lyu, J. et al. Ultra-large library docking for discovering new chemotypes. Nature 566, 224–229 (2019).
De Vivo, M., Masetti, M., Bottegoni, G. & Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 59, 4035–4061 (2016).
Krivák, R. & Hoksza, D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminform. 10, 1–12 (2018).
Pu, L., Govindaraj, R. G., Lemoine, J. M., Wu, H. C. & Brylinski, M. Deepdrug3D: classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 15, e1006718 (2019).
Jiménez, J., Doerr, S., Martínez-Rosell, G., Rose, A. S. & De Fabritiis, G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 33, 3036–3042 (2017).
Ain, Q. U., Aleksandrova, A., Roessler, F. D. & Ballester, P. J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 5, 405–424 (2015).
Li, H., Sze, K. H., Lu, G. & Ballester, P. J. Machine-learning scoring functions for structure-based virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 11, e1478 (2021).
Sanchez-Cruz, N., Medina-Franco, J. L., Mestres, J. & Barril, X. Extended connectivity interaction features: improving binding affinity prediction through chemical description. Bioinformatics 37, 1376–1382 (2020).
Wójcikowski, M., Ballester, P. J. & Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci Rep. 7, 46710 (2017).
Wójcikowski, M., Kukiełka, M., Stepniewska-Dziubinska, M. M. & Siedlecki, P. Development of a protein-ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics 35, 1334–1341 (2019).
Ballester, P. J. & Mitchell, J. B. O. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 26, 1169–1175 (2010).
Ragoza, M., Hochuli, J., Idrobo, E., Sunseri, J. & Koes, D. R. Protein-ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 57, 942–957 (2017).
Stepniewska-Dziubinska, M. M., Zielenkiewicz, P. & Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 34, 3666–3674 (2018).
Hassan-Harrirou, H., Zhang, C. & Lemmin, T. RosENet: improving binding affinity prediction by leveraging molecular mechanics energies with an ensemble of 3D convolutional neural networks. J. Chem. Inf. Model. 60, 2791–2802 (2020).
Jiménez, J., Škalič, M., Martínez-Rosell, G. & De Fabritiis, G. KDEEP: protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model. 58, 287–296 (2018).
Feinberg, E. N. et al. PotentialNet for molecular property prediction. ACS Cent. Sci. 4, 1520–1530 (2018).
Lim, J. et al. Predicting drug-target interaction using a novel graph neural network with 3D structure-embedded graph representation. J. Chem. Inf. Model. 59, 3981–3988 (2019).
Gasteiger, J., Rudolph, C. & Sadowski, J. Automatic generation of 3D-atomic coordinates for organic molecules. Tetrahedron Comput. Methodol. 3, 537–547 (1990).
Velec, H. F. G., Gohlke, H. & Klebe, G. DrugScore CSD knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 48, 6296–6303 (2005).
Fan, H. et al. Statistical potential for modeling and ranking of protein-ligand interactions. J. Chem. Inf. Model. 51, 3078–3092 (2011).
Klebe, G. & Mietzner, T. A fast and efficient method to generate biologically relevant conformations. J. Comput. Aided Mol. Des. 8, 583–606 (1994).
Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020).
Neumaier, A. Complete search in continuous global optimization and constraint satisfaction. Acta Numer. 13, 271–369 (2004).
Liu, Z. et al. PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics 31, 405–412 (2015).
Bishop, C. M. Mixture Density Networks Technical Report. (Aston Univ., 1994).
Li, Y. et al. Assessing protein–ligand interaction scoring functions with the CASF-2013 benchmark. Nat. Protoc. 13, 666–680 (2018).
Su, M. et al. Comparative assessment of scoring functions: the CASF-2016 update. J. Chem. Inf. Model. 59, 895–913 (2019).
Storn, R. & Price, K. Differential evolution—a simple and efficient heuristic for global optimization over continuous spaces. J. Glob. Optim. 11, 341–359 (1997).
Li, H., Leung, K. S., Ballester, P. J. & Wong, M. istar: a web platform for large-scale protein-ligand docking. PLoS ONE 9, e85678 (2014).
Gainza, P. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 17, 184–192 (2020).
Sanner, M. F., Olson, A. J. & Spehner, J. C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 38, 305–320 (1996).
Méndez-Lucio, O., Ahmad, M., del Rio-Chanona, E. A. & Wegner, J. K. A geometric deep learning approach to predict binding conformations of bioactive molecules (dataset). figshare https://doi.org/10.6084/m9.figshare.c.5407329 (2021).
Méndez-Lucio, O., Ahmad, M., del Rio-Chanona, E. A. & Wegner, J. K. OptiMaL-PSE-Lab/DeepDock: DeepDock v1.0.0 (v1.0.0). Zenodo https://doi.org/10.5281/zenodo.5510203 (2021).
Acknowledgements
We thank D. Van Rompaey, J. Verhoeven and N. Dyubankova for supporting this project. We also appreciate comments from W. Heyndrickx that improved the manuscript.
Author information
Authors and Affiliations
Contributions
O.M.-L. conceived the idea, wrote the code, performed the experiments and wrote the manuscript. M.A., E.A.d.R.-C. and J.K.W. helped with the preparation of the manuscript and with insightful discussions. E.A.d.R.-C helped to improve the code.
Corresponding authors
Ethics declarations
Competing interests
O.M.L., M.A. and J.K.W. are employees of Janssen Pharmaceutica NV.
Additional information
Peer review information Nature Machine Intelligence thanks Matteo Aldeghi, Matteo Degiacomi and Hannah E. Bruce Macdonald for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Plot representing the Spearman correlation between RMSD and score for DeepDock and 34 frequently used scoring functions reported by Su et al30.
The x axis represents the ranges [0 to 2 Å], [0 to 3 Å], [0 to 4 Å], etc. Most scoring functions present a high correlation for conformations that are similar to the experimental pose (that is RMSD < 6 Å) but as the RMSD increases Spearman correlation decreases. DeepDock is the only scoring function that presents high Spearman correlation (0.83) taking into account all conformations with and RMSD between 0 and 10 Å.
Extended Data Fig. 2 DeepDock and 34 frequently used scoring functions reported by Su et al30.
Enhancement factor (EF) obtained for The EF measures the number of true binders among the top1% ranked conformations respect to the number of true binders for each of the 57 protein targets during the forward screening task. The red line indicates the mean EF for the scoring function and the bar represents the 90% confidence.
Extended Data Fig. 3 Comparison of real and predicted dihedral angles.
We show the distribution of the12 most common torsions (for example C-C-C-C) using all compounds in the training set predicted with an RMSD < = 1 Å. These plots compare the experimental and predicted dihedral angles for all rotatable bonds used during the optimization step.
Extended Data Fig. 4 Scatter plots summarizing the results of predicting the binding conformation for 1,367 compounds in the validation set.
a-b, show the correlation between the score of the predicted conformation vs the score of the real conformation. c-d, show that predicted conformations for compounds with less rotatable bonds present lower RMSD. e-f, show that compounds with less than 40 atoms usually result in a successful optimization using a differential evolution algorithm. g-h, show that there is no correlation between biological activity and the score obtained using the potential based on distance likelihood.
Extended Data Fig. 5 Scatter plots summarizing the results of predicting the binding conformation for 258 compounds in CASF-2016.
a-b, show the correlation between the score of the predicted conformation vs the score of the real conformation. c-d, show that predicted conformations for compounds with less rotatable bonds present lower RMSD. e-f, show that compounds with less than 40 atoms usually result in a successful optimization using a differential evolution algorithm.
Extended Data Fig. 6 Performance of binding conformation prediction per enzyme type.
Box plots represent the distributions of RMSD between predicted and experimental binding conformations for complexes in the validation set which optimization successfully finished and which target has a valid Enzyme Commission (EC) number.
Supplementary information
Source data
Source Data Fig. 2
Source data for Fig. 2.
Source Data Fig. 3
Source data for Fig. 3g–j.
Source Data Extended Data Fig. 1
Source data for Extended Data Fig. 1.
Source Data Extended Data Fig. 2
Source data for Extended Data Fig. 2.
Source Data Extended Data Fig. 3
Source data for Extended Data Fig. 3.
Source Data Extended Data Fig. 4
Source data for Extended Data Fig. 4.
Source Data Extended Data Fig. 5
Source data for Extended Data Fig. 5.
Source Data Extended Data Fig. 6
Source data for Extended Data Fig. 6.
Rights and permissions
About this article

Cite this article
Méndez-Lucio, O., Ahmad, M., del Rio-Chanona, E.A. et al. A geometric deep learning approach to predict binding conformations of bioactive molecules. Nat Mach Intell 3, 1033–1039 (2021). https://doi.org/10.1038/s42256-021-00409-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-021-00409-9
This article is cited by
-
3D molecular generative framework for interaction-guided drug design
Nature Communications (2024)
-
Applying atomistic neural networks to bias conformer ensembles towards bioactive-like conformations
Journal of Cheminformatics (2023)
-
ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling
Nature Machine Intelligence (2023)
-
SAMPL7 protein-ligand challenge: A community-wide evaluation of computational methods against fragment screening and pose-prediction
Journal of Computer-Aided Molecular Design (2022)
-
Chemoinformatics and artificial intelligence colloquium: progress and challenges in developing bioactive compounds
Journal of Cheminformatics (2022)
