
Abstract
Purpose
Bone marrow-derived, allogeneic, multipotent adult progenitor cells demonstrated safety and efficacy in preclinical models of acute respiratory distress syndrome (ARDS).
Methods
This phase 1/2 trial evaluated the safety and tolerability of intravenous multipotent adult progenitor cells in patients with moderate-to-severe ARDS in 12 UK and USA centres. Cohorts 1 and 2 were open-label, evaluating acute safety in three subjects receiving 300 or 900 million cells, respectively. Cohort 3 was a randomised, double-blind, placebo-controlled parallel trial infusing 900 million cells (n = 20) or placebo (n = 10) within 96 h of ARDS diagnosis. Primary outcomes were safety and tolerability. Secondary endpoints included clinical outcomes, quality of life (QoL) and plasma biomarkers.
Results
No allergic or serious adverse reactions were associated with cell therapy in any cohort. At baseline, the cohort 3 cell group had less severe hypoxia. For cohort 3, 28-day mortality was 25% for cell vs. 45% for placebo recipients. Median 28-day free from intensive care unit (ICU) and ventilator-free days in the cell vs. placebo group were 12.5 (IQR 0,18.5) vs. 4.5 (IQR 0,16.8) and 18.5 (IQR 0,22) vs. 6.5 (IQR 0,18.3), respectively. A prospectively defined severe ARDS subpopulation (PaO2/FiO2 < 150 mmHg (20 kPa); n = 16) showed similar trends in mortality, ICU-free days and ventilator-free days favouring cell therapy. Cell recipients showed greater recovery of QoL through Day 365.
Conclusions
Multipotent adult progenitor cells were safe and well tolerated in ARDS. The clinical outcomes warrant larger trials to evaluate the therapeutic efficacy and optimal patient population.
Similar content being viewed by others

Introduction
Despite multiple clinical studies, no pharmacological treatments have proven effective for acute respiratory distress syndrome (ARDS) [1, 2]. Recently, corticosteroids [3, 4], and possibly IL6 receptor antagonists [5,6,7], have demonstrated favourable results in coronavirus disease 2019 (COVID-19) pneumonia [8]. However, a safe treatment that can reduce mortality of patients who develop ARDS, and improve quality of life for survivors, is still needed.
Mesenchymal stromal cells (MSC) have demonstrated anti-inflammatory properties in animal models of ARDS [9,10,11,12,13,14]. Recently, several clinical studies have investigated MSC treatment for ARDS [15,16,17,18,19]. Multipotent adult progenitor cells are bone marrow-derived, good manufacturing practices (GMP)-manufactured, adherent, allogeneic stromal cells, with immunomodulatory properties similar to MSC. They have been well tolerated in patients with stroke, myocardial infarction, and graft vs host disease [20,21,22]. Pre-clinical studies in a sheep model of ARDS demonstrated that multipotent adult progenitor cells improved blood oxygenation and carbon dioxide clearance, and reduced inflammation, pulmonary vascular pressures, and lung oedema [23, 24].
We hypothesize that multipotent adult progenitor cells are safe in patients with ARDS, and through pleotropic effects, will shift host immune responses from pro-inflammatory towards anti-inflammatory, mitigating lung pathophysiology and promoting repair that could improve important clinical outcomes. The MUST-ARDS study, a multicentre, double-blind, randomised, placebo-controlled trial of multipotent adult progenitor cells in patients with moderate-to-severe ARDS, was presented at the 2019 American Thoracic Society International Conference [25].
Methods
Study design and oversight
This phase 1/2 multicentre, randomised, dose-escalation trial of intravenous multipotent adult progenitor cells was designed and conducted through a collaboration between Athersys, Cell and Gene Therapy Catapult, and senior clinical investigators in critical care, across sites in the UK and USA (see online resource 3 for list of sites involved). An independent Data and Safety Monitoring Board (DSMB) provided oversight of trial governance and safety. The study enrolled 2 open-label, escalating dose tiers (cohorts 1 and 2), then selected the highest well-tolerated dose for a randomised, double-blind, placebo-controlled tier (cohort 3). The study was approved by central and local institutional review boards or ethics committees at the 12 participating sites in the UK and USA.
Between March 2016 and September 2018, with consent from patients or their legally authorised representative, we enrolled adults with moderate-to-severe ARDS (PaO2/FiO2 < 200 mmHg [27 kPa]), consistent with the Berlin definition [26]. We excluded patients with severe interstitial lung disease, chronic obstructive pulmonary disease, chronic liver disease, and those with life expectancy less than 6 months or history of malignancy within the last 2 years. Complete inclusion and exclusion criteria are provided in the protocol (Online Resource 1).
Randomisation and masking
Three patients were assigned to receive open-label intravenous infusion of 300 million multipotent adult progenitor cells (cohort 1). DSMB safety review of cohort 1 patient data included vital sign, ventilator, and vasoactive medication dose changes over the first 4 h, Suspected Unexpected Serious Adverse Reactions (SUSARs) over the first 24 h and laboratory data and Treatment Emergent Adverse Events (TEAEs) over the first 3 days. The next three patients received 900 million cells (cohort 2). Following review of cohort 2 data, the DSMB recommended cohort 3 proceed, evaluating the 900 million cells dose. Thirty patients were randomly assigned, in a 2:1 ratio, to receive intravenous multipotent adult progenitor cells or matched placebo.
Cohort 3, patients were randomised using an interactive web response system (Endpoint—Edinburgh, UK). For each patient randomised, an unblinded staff member of the local cell processing facility received the patient treatment assignment and prepared the cell product or placebo, placing a tinted cover over the intravenous infusion bag and tubing to conceal treatment allocation prior to dispensing. Patients and all trial personnel, including investigators and clinicians, remained blinded to treatment assignment.
Procedures
Cryogenically preserved multipotent adult progenitor cells (Lonza, Walkersville, MD, USA; under contract with Athersys) suspended in PlasmaLyte-A containing dimethyl sulfoxide (DMSO) and human serum albumin (HSA), were thawed, counted, assessed for viability during preparation of each clinical dose. Thawed cells had a median viability of 92% (IQR 83–94%) and were prepared without centrifugation to the appropriate dose of either 300 million or 900 million cells diluted into 300 ml of PlasmaLyte-A. Matching placebo contained PlasmaLyte-A, DMSO, and HSA in the same volume and concentrations (see online resource 3 for further details on cell preparation).
Within the 6 h prior to randomisation, participants were required to achieve a 2-h baseline period of respiratory and haemodynamic stability as well as confirmation of persistent ARDS demonstrated by a PaO2/FiO2 ≤ 300 mmHg (40 kPa). Within 96 h of fulfilling moderate-to-severe ARDS criteria, cells or placebo were administered, through a 200-micron blood filter tubing set, as a single peripheral or central venous infusion over approximately 1 h.
Patients were continuously observed over the first 4 h post-infusion, and further assessed at days 1, 2, 3, 7, 28, 90, and 365 after enrolment. Respiratory mechanics (respiratory rate, tidal volume, and airway pressures [peak and plateau]), mode of ventilation, PaO2/FiO2 ratio, and positive end-expiratory pressure (PEEP) were assessed at each visit, while the patient was on mechanical ventilation. Blood was sampled for inflammatory biomarker multiplex immunoassays (Aeirtec—Newcastle upon Tyne, UK) at baseline, and days 1, 2, 3, and 7. See Online Resource 1 for detailed Protocol including schedule of data collection, pre/post-infusion stability criteria and adverse event definitions.
Outcomes
Primary outcome measures were safety and tolerability of multipotent adult progenitor cells as assessed by (1) physiologic response within 4 h of administration, monitoring vital signs, ventilator volumes and pressures, PaO2 or pulse oximetry, and ventilator setting or vasoactive medication dose adjustment, every 15 min for the first 2 h and at 3 and 4 h after infusion start; and (2) occurrence of SUSARs within 24 h of administration.
Secondary safety outcomes included assessment of vital signs and laboratory parameters through Day 28, and TEAEs through Day 365.
Secondary efficacy outcomes were ventilator-free days, days free from intensive care unit (ICU), and total length of hospital stay through Day 28; changes in PaO2/FiO2 ratio and PEEP requirements from baseline through Days 1, 2, 3, 7 and 28; changes in respiratory physiologic measures (peak and plateau pressures) from baseline through the time the subject is extubated; and all-cause mortality at Days 28, 90 and 365.
Exploratory endpoints included changes in circulating biomarkers of inflammation and lung injury between baseline and Days 1, 2, 3 and 7; and health-related quality of life (EQ-5D-3L) at Days 28, 90 and 365.
Statistical analysis
Descriptive analysis was performed on primary and secondary safety outcomes among all cohorts. Patients randomized in cohort 3 constituted the assessable population for secondary efficacy endpoints, exploratory plasma biomarkers and quality of life assessments. Efficacy assessments were additionally evaluated for a prospectively defined subgroup of patients with baseline PaO2/FiO2 < 150 mmHg (20 kPa). Recent literature suggests patients below this cutoff differ in their anatomical and physiological characteristics, and potentially exhibit differential responses to adjunctive ARDS interventions such as prone positioning and cisatracurium [27, 28].
The DSMB reviewed safety data following cohorts 1 and 2, and after 10 and 20 patients were enrolled to cohort 3. This fully completed study is registered with ClinicalTrials.gov: NCT02611609.
Role of the funding sources
The National Institute on Alcohol Abuse and Alcoholism, and Innovate UK were not involved in the study design, data analysis, data interpretation, or writing of this report. Athersys was involved in study design and in data interpretation. All data collection and analysis were overseen by Synequanon (Norfolk, UK). Two employees of Athersys (AT and EJ) were represented on the writing committee. The corresponding author and the writing group had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Four patients consented and enrolled in cohort 1. One patient withdrew consent prior to receiving cell infusion and was replaced. Baseline characteristics of patients who were dosed in each cohort are presented in Table 1. Three patients enrolled in cohort 1 received 300 million multipotent adult progenitor cells. Vital signs, and any ventilator and vasoactive medication dose adjustments over the first 4 h were carefully evaluated by the DSMB for evidence of deleterious physiologic response to intravenous cell administration. No pattern suggestive of harm was identified. There were no treatment-emergent adverse events causally related cell administration and review of laboratory data revealed no trends concerning for adverse reactions to the therapy through at least the first 3 days following infusion. Reported TEAEs for each cohort are depicted in Table 2.
In cohort 2, 3 patients received 900 million cells. One death occurred in cohort 2, that was determined by the DSMB to be unrelated to cell therapy. Cohort 2 data review revealed no evidence of harm from cell therapy.
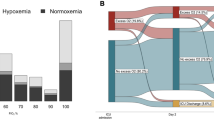
After the DSMB confirmed no acute safety concerns with the 900 million cells dose, 34 patients were consented to cohort 3 with four patients excluded for no longer meeting inclusion criteria at time of randomization. Twenty patients received 900 million cells and 10 patients received placebo (Fig. 1).

Trial Design for Cohort 3
Demographic and baseline clinical information for cohort 3 is presented in Table 1. Notable between group differences included mean modified SOFA (lower in the cells group: 10.9 vs 12.2 for placebo), PaO2/FiO2 (less severe among cell recipients: 173 mmHg [23.1 kPa] than placebo: 128 mmHg [17.1 kPa]) and age (cells: 51 yr vs placebo: 59 year). The cell treatment group had a greater proportion requiring vasopressor support (45%) than the placebo group (30%). A prospectively defined subset for analysis, comprising 16 patients (n = 8 cells; n = 8 placebo) with severe hypoxia (baseline PaO2/FiO2 < 150 mmHg [20 kPa]), had similar group mean baseline PaO2/FiO2 ratios of 121 mmHg (16.1 kPa) vs. 117 mmHg (15.6 kPa), respectively, while a greater proportion of the cell treatment subgroup required vasopressor at baseline (38% vs 25%). Among all cohort 3 patients, 60% in the cell treatment group, and 80% in the placebo group, received corticosteroids at some point between Day minus2 and Day 3.
The primary outcome of the study was acute safety and tolerability of multipotent adult progenitor cells. Close observation during 4 h following administration revealed no evidence of infusional toxicities or protocol specified Adverse Events of Special Interest (AESI), including hypoxemic, haemodynamic, and cardiac events. There were no SUSARs observed within 24 h of administration.
Secondary safety evaluation of vital signs and laboratory parameters through Day 28, and TEAEs through Day 365, revealed no serious TEAEs attributed to the cells. The occurrence of TEAEs was higher in the MultiStem groups compared to placebo (91.3% for patients receiving 900 million cells vs. 60% for placebo), but the occurrence of serious TEAEs was similar (60.9% vs. 60%, respectively). TEAEs reported for 2 or more subjects were those expected in a critically ill population, including pyrexia, sepsis, pneumonia, pleural effusion, venous thrombosis, respiratory and multi-organ failure, hypernatremia, and cardiac arrest. Only 1 possibly related, non-serious TEAE (Common Terminology Criteria for Adverse Events [CTCAE] Grade 1 pyrexia), that resolved without intervention, occurred in the cell treatment group of cohort 3 (further details on Adverse Events see Online Resource 1& 3).
Mortality (Table 3) at Day 28, was lower in the cohort 3 cell treatment group, 5/20 (25%), than in the placebo group, 4/10 (40%). In the prespecified severe hypoxia subpopulation of cohort 3, patients with baseline PaO2/FiO2 < 150 mmHg (20 kPa), Day-28 mortality was 2/8 (25%) in the cell treatment group, and 4/8 (50%) for placebo. Day-365, cohort 3 mortality remained lower in the cell treatment group 8/20 (40%) than the placebo group 5/10 (50%) (Table 2). Reported causes of death included respiratory and multi-organ failure, sepsis, pneumonia, aspiration, pulmonary haemorrhage, and intestinal ischemia.
Through Day 28, the cell treatment group had higher median ventilator-free days, 18.5 (IQR 0, 22), vs 6.5 (IQR 0, 18.3) for the placebo group; and higher median ICU-free days, 12.5 (IQR 0, 18.5), vs to 4.5 (IQR 0, 16.8) for the placebo group (Table 3). For the prespecified severe hypoxia subpopulation, median ventilator-free days was 18.5 (IQR 9, 21.3) in the cell treatment subgroup compared to 3.5 (IQR 0, 16.8) in the placebo subgroup. Median ICU-free days were 12.5 (IQR 6.8, 18) in the cell treatment subgroup vs 1 (IQR 0,9.5) for the placebo subgroup.
With the caveat that ventilator management was not standardized, tidal volumes (mean ± SD) increased 25.4 ± 118.16 mL by Day 3 in the cell treatment group, compared to a decrease of 14.3 ± 98.73 mL in the placebo group. Peak inspiratory pressure and plateau inspiratory pressure also showed an increase at Day 3 in the cell treatment group (1.2 ± 6.54 cmH2O and 1.7 ± 5.33 cmH2O, respectively), compared to a decrease in the placebo group (− 4.5 ± 10.62 cmH2O and − 0.6 ± 6.97 cmH2O, respectively). Increases in PaO2/FiO2 were seen in both groups, indicating improving lung function among ventilated survivors. Notwithstanding the observed baseline differences between groups, the increases in PaO2/FiO2 through Day 3 were greater in the placebo group, at 89.7 ± 109.3 mmHg (12 ± 14.6 kPa), than the cell treatment group at 24 ± 65.7 mmHg (3.2 ± 1.9 kPa) (detailed changes of respiratory physiologic measures over the monitoring period are provided in Online Resource 2).
While no statistical analyses were performed, within cohort 3, several pro-inflammatory biomarkers decreased on average through Day 7 in the cell treatment group and increased through Day 7 among the placebo group (Fig. 2). These include IFN-gamma, IL-1 beta, IL-1R2, IL 6, IL 12, KGF, PD-1, RAGE, and TNF-alpha. Biomarkers that increased through Day 7 in the cell treatment group and decreased in the placebo group included SP-D and TSP-1 (Full biomarker results see Online Resource 2).

Ratio of Day 7 to baseline values of biomarker plasma concentrations. The ratio of the biomarker value at Day 7 compared to the baseline values are presented as medians with upper and lower quartiles. Biomarkers include: angiopoietin 1 (ANG1), angiopoietin 2 (ANG2), C-X-C Motif chemokine ligand 10 (CXCL10), Interferon (IFN) gamma, interleukin 1 beta (IL-1b), interleukin 1 receptor 2 (IL-1R2), interleukin 1 receptor antagonist (IL-1RA), interleukin 6 (IL-6), interleukin 8 (IL-8), interleukin 10 (IL-10), interleukin 12 (IL-12), keratinocyte growth factor (KGF), matrix metalloproteinase 2 (MMP-2), programmed cell death protein (PD-1), receptor for advanced glycation end-products (RAGE), regulated on activation, normal T cell expressed and secreted (RANTES), surfactant protein D (SP-D), tumour necrosis factor alpha (TNF alpha), soluble TNF receptor 1 (sTNFR1) and thrombospondin 1 (TSP-1). Green = patients receiving multipotent adult progenitor cells (n = 19); Blue = patients receiving placebo (n = 7)
Patient well-being during recovery was assessed using the EQ-5D-3L to measure health-related quality of life. Through the course of the study, improvements in the mobility, self-care, usual activities, and anxiety/depression domains of the EQ-5D-3L were seen among survivors in both the cell treatment and placebo groups. The EQ-5D-3L index and visual analogue scale (VAS) scores were more favourable at each timepoint among survivors who had received cells (see Online Resource 2).
Discussion
This study, conducted prior to the COVID-19 pandemic, focused on the results of close observation during the four hours immediately following infusion of multipotent adult progenitor cells, and on the number of reported adverse events in the first 24 h. Neither of the open-label cohorts (300 million or 900 million cells) showed any safety critical events within 24 h. Safety was further supported by the double-blind placebo-controlled cohort 3, wherein no increased rates of 4-h safety events or 24-h adverse events were associated with cell treatment. No serious adverse events related to the cells were observed over the course of the study. One, possibly related, treated-emergent adverse event, Grade-1 pyrexia that resolved quickly without intervention, was reported.
While not powered for efficacy, and with clear baseline imbalances between the study groups, the outcomes from cohort 3 suggest the cells may have beneficial effects in ARDS. At Day 28, cell recipients had greater ventilator-free days, ICU-free days and lower mortality. However, we reiterate the baseline differences between treatment groups. Cell recipients were younger and had higher baseline PaO2/FiO2 and lower SOFA scores than the placebo group. Conversely, a greater proportion of the cell treatment group required vasopressor support at baseline. Although containing only 8 subjects per treatment allocation, the prospectively defined severe hypoxia subgroup (with more comparable baseline PaO2/FiO2 < 150 mmHg [20 kPa]) revealed lower Day-28 mortality among those receiving cells. Overall, results are consistent with therapeutic benefits observed in large animal studies [23, 24] and support conduct of a clinical trial to determine if there is sustained and real clinical benefit.
Several clinical studies have demonstrated adult stem cells are well tolerated in patients with ARDS [15,16,17,18,19]. The largest of these, the START study, infused 10 × 106 MSC/kg in 60 patients with moderate to severe ARDS. Although the START investigators found no significant differences in 28-day mortality, the results revealed numerically higher mortality in the cell group compared to placebo (30% vs 15%). Importantly, in START, MSC viability was variable, ranging from 36 to 85%, compared to a median value of 92% (IQR 83–94%) in this study. In post hoc analysis, Matthay and colleagues reported that outcomes in START participants improved as the cell viability improved [17].
Although this was a small study, with inadequate power to discern significant differences, the cell treatment group had better EQ-5D-3L VAS and index quality of life outcome scores at all three time points through Day 365. The EQ-5D-3L results, numerically favouring cell therapy treated patients, are reassuring, in that greater survival among the cell treatment group does not appear to be accompanied by reduced QoL among those survivors.
There are several potential mechanisms of benefit for multipotent adult progenitor cells in patients with ARDS. Temporal changes in the plasma biomarkers, demonstrating a reduction in pro-inflammatory cytokines, support previously investigated immunomodulatory effects of the multipotent adult progenitor cells. It is notable that the majority of cohort 3 patients (60% cell; 80% placebo), received corticosteroids between Day minus2 and Day 3, likely resulting in corticosteroid pharmacologic effects coinciding with cell biological activity. Trials evaluating specific agents to block selected inflammatory pathways (e.g., statins, ketoconazole, N-acetylcysteine) or to promote resolution (e.g., KGF), have largely failed [1, 29,30,31]. One potential advantage of multipotent adult progenitor cells is that they modulate multiple host responses to tissue injury simultaneously, including down-regulation of hyperinflammatory responses, stimulation of tissue repair, and restoration immune system balance [32]. Similar acute plasma biomarker responses to multipotent adult progenitor cells, including decreases in pro-inflammatory cytokines IL-1beta, TNFα, IL-6, IFN-gamma, and IL-2, have been observed to correlate with improved clinical outcomes following ischemic stroke [20]. Future studies might determine if plasma biomarkers track the efficacy of multipotent adult progenitor cell therapy for ARDS, and whether they might be used to optimize dosing. However, there may be changes within the lung, evidenced by bronchoalveolar lavage but not detectable in circulation [33].
The strengths of this study include evaluation of 2 dose levels of multipotent adult progenitor cells, a double-blind placebo-controlled cohort, performance across multiple centres internationally, and delivery of cells within 96 h of diagnosis. The study confirmed safety and assessed a number of important secondary and exploratory endpoints, including Day-28 clinical outcomes, QoL over one year, and acute changes in plasma biomarkers. The study evaluated an off-the-shelf, standardized, and highly consistent preparation of cryopreserved allogeneic bone marrow-derived stromal cells, demonstrating that delivery without prior centrifugation or washing preserves high cell viability, and that consent, dose preparation, and infusion early in the course of ARDS is logistically feasible and safe. The largest weakness of this study was a sample size designed to confirm the safety and tolerability of multipotent adult progenitor. A larger study will be required to confirm clinical outcome benefits from multipotent adult progenitor cells in ARDS.
Conclusion
This important first study, using multipotent adult progenitor cells in ARDS patients, was a phase 1/2 randomised, blinded, placebo-controlled trial that demonstrated safety and tolerability of intravenous administration of multipotent adult progenitor cells, at doses up to 900 million cells, in patients with ARDS. Our findings support progression to a larger trial to investigate their therapeutic efficacy.
References
Matthay MA, Zemans RL, Zimmerman GA et al (2019) Acute respiratory distress syndrome. Nat Rev Dis Primers 5(1):18. https://doi.org/10.1038/s41572-019-0069-0
Thompson BT, Chambers RC, Liu KD (2017) Acute respiratory distress syndrome. N Engl J Med 377(19):1904–1905. https://doi.org/10.1056/NEJMra1608077
Group RC, Horby P, Lim WS et al (2020) Dexamethasone in hospitalized patients with Covid-19—preliminary report. N Engl J Med. https://doi.org/10.1056/NEJMoa2021436
Angus DC, Derde L, Al-Beidh F et al (2020) Effect of hydrocortisone on mortality and organ support in patients with severe COVID-19: the REMAP-CAP COVID-19 Corticosteroid Domain Randomized Clinical Trial. JAMA 324(13):1317–1329. https://doi.org/10.1001/jama.2020.17022
The REMAP-CAP Investigators (2021) Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med 384:1491–1502. https://doi.org/10.1056/NEJMoa2100433
RECOVERY Collaborative Group (2021) Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet 397:1637–1645. https://doi.org/10.1016/S0140-6736(21)00676-0
Rosas IO, Diaz G, Gottlieb RL et al (2021) Tocilizumab and remdesivir in hospitalized patients with severe COVID-19 pneumonia: a randomized clinical trial. Intensive Care Med. https://doi.org/10.1007/s00134-021-06507-x13
Menk M, Estenssoro E, Sahetya SK et al (2020) Current and evolving standards of care for patients with ARDS. Intensive Care Med 46:2157–2167. https://doi.org/10.1007/s00134-020-06299-6
Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA (2007) Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol 179(3):1855–1863. https://doi.org/10.4049/jimmunol.179.3.1855
Matthay MA, Goolaerts A, Howard JP, Lee JW (2010) Mesenchymal stem cells for acute lung injury: preclinical evidence. Crit Care Med 38(10 Suppl):S569–S573. https://doi.org/10.1097/CCM.0b013e3181f1ff1d
Rojas M, Xu J, Woods CR et al (2005) Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol 33(2):145–152. https://doi.org/10.1165/rcmb.2004-0330OC
Moodley Y, Sturm M, Shaw K et al (2016) Human mesenchymal stem cells attenuate early damage in a ventilated pig model of acute lung injury. Stem cell Res 17(1):25–31. https://doi.org/10.1016/j.scr.2016.05.005
Rojas M, Parker RE, Thorn N et al (2013) Infusion of freshly isolated autologous bone marrow derived mononuclear cells prevents endotoxin-induced lung injury in an ex-vivo perfused swine model. Stem Cell Res Ther 4(2):26. https://doi.org/10.1186/scrt174
Horie S, Curley GF, Laffey JG (2016) What’s new in cell therapies in ARDS? Intensive Care Med 42:779–782. https://doi.org/10.1007/s00134-015-4140-3
Zheng G, Huang L, Tong H et al (2014) Treatment of acute respiratory distress syndrome with allogeneic adipose-derived mesenchymal stem cells: a randomized, placebo-controlled pilot study. Respir Res 15:39. https://doi.org/10.1186/1465-9921-15-39
Wilson JG, Liu KD, Zhuo H et al (2015) Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med 3(1):24–32. https://doi.org/10.1016/S2213-2600(14)70291-7
Matthay MA, Calfee CS, Zhuo H et al (2019) Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respir Med 7(2):154–162. https://doi.org/10.1016/S2213-2600(18)30418-1
Simonson OE, Mougiakakos D, Heldring N et al (2015) In vivo effects of mesenchymal stromal cells in two patients with severe acute respiratory distress syndrome. Stem Cells Transl Med 4(10):1199–1213. https://doi.org/10.5966/sctm.2015-0021
Lv H, Chen W, Xiang AP et al (2020) Mesenchymal stromal cells as a salvage treatment for confirmed acute respiratory distress syndrome: preliminary data from a single-arm study. Intensive Care Med 46:1944–1947. https://doi.org/10.1007/s00134-020-06122-2
Hess DC, Wechsler LR, Clark WM et al (2017) Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 16(5):360–368. https://doi.org/10.1016/S1474-4422(17)30046-7
Maziarz RT, Devos T, Bachier CR et al (2015) Single and multiple dose MultiStem (multipotent adult progenitor cell) therapy prophylaxis of acute graft-versus-host disease in myeloablative allogeneic hematopoietic cell transplantation: a phase 1 trial. Biol Blood Marrow Transplant 21(4):720–728. https://doi.org/10.1016/j.bbmt.2014.12.025
Penn MS, Ellis S, Gandhi S et al (2012) Adventitial delivery of an allogeneic bone marrow-derived adherent stem cell in acute myocardial infarction: phase I clinical study. Circ Res 110(2):304–311. https://doi.org/10.1161/CIRCRESAHA.111.253427
Rojas M, Cardenes N, Kocyildirim E et al (2014) Human adult bone marrow-derived stem cells decrease severity of lipopolysaccharide-induced acute respiratory distress syndrome in sheep. Stem Cell Res Ther 5(2):42. https://doi.org/10.1186/scrt430
Cardenes N, Aranda-Valderrama P, Carney JP et al (2019) Cell therapy for ARDS: efficacy of endobronchial versus intravenous administration and biodistribution of MAPCs in a large animal model. BMJ Open Respir Res 6(1):e000308. https://doi.org/10.1136/bmjresp-2018-000308
Bellingan G, Jacono F, Bannard-Smith J, et al (2019) Primary analysis of a phase 1/2 study to assess MultiStem? Cell therapy, a regenerative advanced therapy medicinal product (ATMP), in acute respiratory distress syndrome (MUST-ARDS). In: B14. LATE BREAKING CLINICAL TRIALS. American Thoracic Society, pp A7353–A7353. https://doi.org/10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A7353
Ranieri VM, Rubenfeld GD, Thompson BT et al (2012) Acute respiratory distress syndrome: the Berlin Definition. JAMA 307(23):2526–2533. https://doi.org/10.1001/jama.2012.5669
Maiolo G, Collino F, Vasques F et al (2018) Reclassifying acute respiratory distress syndrome. Am J Respir Crit Care Med 197(12):1586–1595. https://doi.org/10.1164/rccm.201709-1804OC
Fan E, Brodie D, Slutsky AS (2018) Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA 319(7):698–710. https://doi.org/10.1001/jama.2017.21907
McAuley DF, Cross LM, Hamid U et al (2017) Keratinocyte growth factor for the treatment of the acute respiratory distress syndrome (KARE): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Respir Med 5(6):484–491. https://doi.org/10.1016/S2213-2600(17)30171-6
McAuley DF, Laffey JG, O’Kane CM et al (2014) Simvastatin in the acute respiratory distress syndrome. N Engl J Med 371(18):1695–1703. https://doi.org/10.1056/NEJMoa1403285
Laffey JG, Matthay MA (2017) Fifty years of research in ARDS. Cell-based therapy for acute respiratory distress syndrome. Biology and potential therapeutic value. Am J Respir Crit Care Med 196(3):266–273. https://doi.org/10.1164/rccm.201701-0107CP
Auletta JJ, Cooke KR, Solchaga LA, Deans RJ, van’t Hof W (2010) Regenerative stromal cell therapy in allogeneic hematopoietic stem cell transplantation: current impact and future directions. Biol Blood Marrow Transplant 16(7):891–906. https://doi.org/10.1016/j.bbmt.2009.12.005
Wick KD, Leligdowicz A, Zhuo H et al (2021) Mesenchymal stromal cells reduce evidence of lung injury in patients with ARDS. JCI Insight 6(12):e148983. https://doi.org/10.1172/jci.insight.148983
Acknowledgements
The authors deeply appreciate the contributions of the volunteer study participants as well as the members of the independent Data Safety Monitoring Board: Tim Walsh (chair), Michael Quintel, Sam Janes, and Peter Treasure (Independent Statistician).
Funding
National Institutes of Health, Innovate UK, and Athersys, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Two of the authors, EJ and AT, were employees of Athersys and one, GB, received a travel grant from Athersys to two meetings. No other Conflict of interest are declared
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bellingan, G., Jacono, F., Bannard-Smith, J. et al. Safety and efficacy of multipotent adult progenitor cells in acute respiratory distress syndrome (MUST-ARDS): a multicentre, randomised, double-blind, placebo-controlled phase 1/2 trial. Intensive Care Med 48, 36–44 (2022). https://doi.org/10.1007/s00134-021-06570-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-021-06570-4