
Abstract
The ability to control translation of endogenous or exogenous RNAs in eukaryotic cells would facilitate a variety of biotechnological applications. Current strategies are limited by low fold changes in transgene output and the size of trigger RNAs (trRNAs). Here we introduce eukaryotic toehold switches (eToeholds) as modular riboregulators. eToeholds contain internal ribosome entry site sequences and form inhibitory loops in the absence of a specific trRNA. When the trRNA is present, eToeholds anneal to it, disrupting the inhibitory loops and allowing translation. Through optimization of RNA annealing, we achieved up to 16-fold induction of transgene expression in mammalian cells. We demonstrate that eToeholds can discriminate among viral infection status, presence or absence of gene expression and cell types based on the presence of exogenous or endogenous RNA transcripts.
Similar content being viewed by others

Main
Synthetic biology techniques for sensing and responding to specific intracellular RNAs are desirable for therapeutic and diagnostic applications, as they provide a means to discriminate and target specific cells, tissues and organisms and can serve as building blocks for sophisticated genetic circuits. We previously developed RNA-based prokaryotic modules, called toehold switches, for detecting specific RNA transcripts1,2. Toehold switches selectively repress translation of an in cis reporter gene by sequestering the ribosome binding site (RBS) upstream of the reporter gene in a stem loop structure in the absence of a trRNA. The RBS is released when a trRNA binds the toehold switch and opens the stem loop structure, thus initiating translation of the reporter gene. Eukaryotic translation, however, is far more complicated and is typically regulated by several factors, including 5′ modified capping recruited by RNA polymerase II, a poly-adenosine (polyA) tail for mRNA stabilization and a Kozak consensus sequence for protein translational regulation. Although the Kozak sequence improves ribosomal binding, it is not as critical to translation as the prokaryotic RBS; previously developed Kozak-based toehold switches have achieved only up to two-fold trRNA-driven induction of eukaryotic translation3. The 5′ cap dominates translational regulation mechanisms and is a major challenge for any eukaryotic RNA-sensing riboswitch that functions at the level of translation.
More complex RNA-based switches have been developed, using Cas9 expression and engineered folding of the guide RNA (gRNA) to hide sequences essential for function4,5. Unfolding of the gRNA by trRNA leads to activation of the Cas9 enzyme and corresponding downstream regulation. However, these mechanisms induce only modest fold changes in both eukaryotes and prokaryotes. A more robust technique involves the use of a ribozyme that cleaves the polyA tail upon small molecule induction6,7. Although recent studies have used ribozymes8,9 to respond to short nucleotide oligomers, these ribozyme-based sensors are not yet compatible with the detection of longer trRNAs, including endogenous transcripts.
Internal ribosome entry sites (IRESs) are RNA elements found in several viruses and endogenous eukaryotic transcripts whose structure has evolved to initiate protein translation independent of mRNA 5′ capping and polyadenylation. In this study, we developed RNA-based eukaryotic modules, called eToeholds, that enable the regulated translation of in cis reporter genes by the presence of specific trRNAs. These eToeholds incorporate modified IRESs that are designed to be inactive until sense–antisense interactions with specific trRNAs cause activation (Fig. 1a). Using this system, we achieve up to 16-fold trRNA-induced translation of transgenes. We demonstrate eToehold functionality in human and yeast cells as well as cell-free lysates. We further show that stable cell lines expressing eToeholds can be used to sense natural viral infection (by Zika virus) and viral transcripts (severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) constructs). We also demonstrate the capability of eToeholds to discriminate different cell states and cell types by selectively activating protein translation based on endogenous RNA levels.

a, eToehold modules are in a locked state in which IRES activity is inhibited, preventing ribosome recruitment and translation. trRNA activates IRES activity through strand invasion and release of the IRES into an activated state, allowing for ribosome binding and protein production. b, Basic screening methodology for eToeholds. Plasmids encoding IRES or eToehold candidates upstream of a reporter protein, polymerase (for example, T7 RNAP) and trRNA sequence (for example, GFP) were co-transfected into HEK293T cells. See Methods and Extended Data Fig. 1 for more detailed information. c, Activities of different IRES modules in HEK293T cells with and without co-transfection with T7 RNAP and GFP trigger sequence. d, Dicistroviridae family IRES structure including three loops critical for translational activity and insertion sites. e, Screen for eToehold modules by inserting two complementary sequences of unequal lengths into insertion sites depicted in d. Numbers denote insertion site of, first, the toehold RNA sequence (~40 base pairs complementary to trRNA) and, second, the smaller fragment (~10–18 base pairs) complementary to the first. f, CrPV IRES structure. Red denotes regions where insertions overlap with stem loops or pseudoknots (BB sites). Examples of sequences for the BB sites needed for the CrPV IRES are included for clarity. g, Effect of choosing insertions with matching base pairs (three base pairs) in the regions depicted in d. See Supplementary Table 1 for construct details. Data are presented as mean values with error bars representing s.d. of three technical replicates. All experiments were repeated at least two times.
Results
Engineering eToeholds from IRES modules
To engineer an RNA-sensing riboswitch that functions in eukaryotic cells, we chose to modify viral IRES modules, which possess structure-dependent translational activity10,11,12,13,14 (Fig. 1b). Although IRESs have been adapted to sense small molecules15, IRES-based systems have not been designed to respond to trRNAs. We first selected IRES modules from viral databases and tested them in a human embryonic kidney 293 (HEK293) cell-based transfection assay (Extended Data Fig. 1a). T7 RNA polymerase (RNAP) was co-transfected into cells and used to produce IRES transcripts, as T7 RNAP does not recruit 5′ capping. The presence of IRES modules resulted in a ~9-fold enhancement in expression of in cis mKate (Fig. 1c and Extended Data Fig. 1b–i). We decided to pursue cricket paralysis virus (CrPV), kashmir bee virus (KBV) and acute bee paralysis virus (ABPV) IRES modules as the basis for further development of eToeholds, with a focus on the CrPV IRES owing to its well-characterized structure and reported functionality in a wide range of eukaryotic systems10,11,12.
Having verified IRES activity, we next hypothesized that inserting short complementary RNA segments into the IRES sequence would disrupt its secondary structure by forming new loops, thereby reducing translational initiation ability, and that breaking of these introduced loops by sense–antisense action of a trRNA would rescue IRES functionality. We tested this hypothesis by inserting complementary sense and antisense segments (two pieces) of nucleic acids into sites within the IRES template, theorizing that base pairing between these sequences in the resulting transcript would distort the functional configuration of the IRES. To enable recovery of IRES function in the presence of trRNA, we designed these complementary introduced segments to bind to a chosen trRNA sequence. Our goal was for base pairing between the trRNA and the new insertion to be sufficient to break the IRES-disrupting loop, so we designed the complementary insertions to be of unequal lengths. The longer piece (40–50 base pairs) was chosen to be the reverse complement of a portion of the trRNA, whereas the shorter piece (6–15 base pairs) was chosen to be the reverse complement of a portion of the longer piece. Based on previous literature11,16, we identified eight sites that were predicted to tolerate insertions and would not on their own erase CrPV IRES activity (Fig. 1d).
We chose green fluorescent protein (GFP) mRNA to act as the trRNA and screened different CrPV IRES sequences with complementary sequences inserted at the eight possible sites. To monitor IRES activity, we used an in cis mKate gene downstream of the modified IRES sequences and co-transfected cells with these constructs as well as a plasmid constitutively expressing GFP (Fig. 1e). Each site combination is named with the format of long piece site number–short piece site number (for example, 1–2). We found that several site combinations (1–2, 1–8, 2–7, 6–7 and 8–6) behaved as expected, producing higher mKate signal when co-expressing GFP. Based on these results, we decided to focus on site combinations 6–7 and 8–6, which reproducibly showed 1.7-fold increases in the percentage of mKate positive cells in the presence of GPF trRNA, which we confirmed via GFP fluorescence.
We observed that our modified IRES appeared to retain substantial translational ability despite the newly introduced insertions. Previous studies suggest that IRES pseudoknots are critical for ribosome recruitment10,17. We reasoned that simultaneously distorting the IRES module, as in our current strategy, and breaking the IRES pseudoknot using the same insertion sequences could further reduce the basal expression of the module. To test this hypothesis, we designed new eToeholds by choosing the shorter insert to be adjacent to sequences present in critical native IRES pseudoknots, with complementary long inserts that included partial sequences of the native IRES pseudoknot. We theorized that, in the absence of trRNA, the annealing between our inserts would impair correct pseudoknot folding. We termed the insert site where this would occur as the base pair breaking site (BB site; Fig. 1f). Designing eToeholds with a BB site led to a reduction of the off (no trigger) state to background levels and increased the on-to-off fold change from 1.7 to 2.5 for site combination 8–6 (Fig. 1g and Extended Data Fig. 4). To further characterize thermodynamic requirements for eToehold switching, we altered the length of the short insert at the 8–6 site, thus changing annealing temperature. Although some eToeholds displayed a correlation between annealing temperature and output, others did not (Extended Data Fig. 2). As the longer eToehold sequence might be of sufficient length to induce an RNA interference response, we assessed RNA levels of IRES constructs designed to bind 40–50-nucleotide-long complementary ‘trigger’ RNA sequences, compared to a non-binding orthogonal sequence (Supplementary Sequences). We observed no significant differences in IRES construct RNA levels in two of the three cell types that we assessed, but we did find ~2-fold reduction in the presence of a ‘trigger’ RNA in primary human fibroblasts (Supplementary Table 2). We also found no significant differences in the production of inflammation-associated cytokines, including CCL2, CCL5 and IL-6, between cells transduced with ‘trigger’ RNA compared to non-binding orthogonal RNA, although transfection alone significantly increased cytokine production (Extended Data Fig. 3)18.
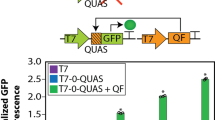
Although T7 promoter (PT7) is generally thought to be highly specific for T7 RNAP, mammalian RNA polymerase II has been shown to bind PT7 and initiate substantial levels of transcription19. We reasoned that part of the unexpected translation from eToeholds in the absence of trRNA might be due to recruitment of endogenous RNA polymerase II and subsequent generation of transcripts with 5′ caps, which independently induces translation and overrides the need for a functional IRES. Accordingly, we screened exogenous promoter sequences for transcription systems orthogonal to PT7 to test whether off-state translation of IRES-controlled reporter would be reduced. We found that the promoter for SP6 (PSP6) resulted in significantly lower basal expression than PT7 and its analogues (Extended Data Fig. 5a). We next tested upstream recruitment of RNA polymerase I, which does not result in 5′ capping and has been shown to decrease RNA polymerase II binding20,21,22, reasoning that this could further decrease basal expression. We identified an upstream activation factor binding DNA sequence from Saccharomyces cerevisiae that successfully reduced basal expression (UAF2; Extended Data Fig. 5b). By combining these components, we minimized basal expression and reached an on-to-off trigger mRNA-based induction of 15.9-fold (Fig. 2a and Extended Data Fig. 6). Similar fold changes upon induction while retaining the use of the T7 RNAP and promoter could be achieved by using a bicistronic design and adding stop codons and stem loops23 before the IRES modules (Extended Data Fig. 7a).

a, Effect of switching promoter-polymerase systems and adding an RNA polymerase I upstream activation sequence on expression of eToehold-gated transgene (mKate). b, eToehold activity, as assessed by mKate expression, in the presence of designed trRNA and unmatched RNA. c, Schematic of bicistronic RNA polymerase II-driven eToehold-gated RNA, including stop codons and stem loops. d, eToehold or IRES activity, assessed by mKate expression, of constructs with and without specific features shown in c. See Supplementary Table 1 for construct details. e, eToeholds were engineered using alternative IRES modules with higher translational activity. These new eToeholds were assessed through creation of stable HEK293T cell lines and subsequent trigger plasmid transfection; nanoluciferase expression and detection outputs are shown (Supplementary Sequences and Supplementary Table 1). Data are presented as mean values with error bars representing s.d. of three technical replicates. All experiments were repeated at least three times. a.u., arbitrary units; EV71, enterovirus 71; HCV, hepatitis C virus; PV, poliovirus.
Having developed an approach to achieve robust fold changes in transgene expression upon trRNA induction, we next tested the specificity of our system to desired trRNA sequences. We designed eToeholds to detect GFP, Azurite and yeast SUMO mRNA and found that our designs specifically sensed their trRNA sequence (Fig. 2b). We further tested the sensitivity of eToeholds to their cognate trRNA by introducing mismatches in the two insertion sequences. We found that eToeholds were sensitive to mismatches in the annealing region (Extended Data Fig. 7b). Additionally, we tested the generalizability of eToehold design to other IRESs. We synthesized eToeholds using both KBV and ABPV IRES modules and observed similar fold changes in trigger-induced translation compared to CrPV IRES module-based eToeholds (Extended Data Fig. 7c). Moreover, we found that RNA sequences that could bind to the short eToehold insertion but not to the longer insertion were not sufficient to activate the eToehold (Extended Data Fig. 7d). Taken together, these findings indicate that eToeholds can be readily designed for different mRNAs with high specificity and that sense–antisense activation is broadly generalizable for IRES-mediated translation.
We next sought to adapt our eToehold system to expression by endogenous eukaryotic polymerases. This would obviate dependency on exogenous polymerases for producing uncapped transcripts, thereby reducing construct size and increasing compatibility of our eToehold system with current cell engineering and gene therapy methods. Although RNA polymerase I24,25 has been shown to produce uncapped transcripts, we found that reliance on RNA polymerase I alone to generate the eToehold transcripts leads to significantly decreased on-to-off ratios and increased basal expression (Extended Data Fig. 5c). Therefore, we decided to explore methods of reducing translational activity in the presence of canonical 5′ capping. By adding stop codons and stem loops23 after a gene controlled by a constitutive promoter, we were able to reduce the basal expression of downstream mRNA despite the reliance on RNA polymerase II (Fig. 2c,d). Furthermore, by inserting an IRES or eToehold module between the stem loops and the coding sequence of a desired gene, thereby creating a bicistronic construct, we retained trRNA-mediated control over translation of the second gene.
To test the applicability of our system in IRES modules that evolved to use mammalian, and specifically human, translational systems, we designed eToehold modules adapted from IRES sequences of hepatitis C virus26, poliovirus27 and enterovirus 71 (refs. 28,29). These new eToeholds also demonstrated an ability to sense specific trRNAs (Fig. 2e). Compared to the CrPV constructs, these new eToeholds, which we call human-optimized Toeholds (hToeholds), produced greater than one order of magnitude more output protein. As a comparison, monocistronic 5′ capped mRNA produced even higher protein output than these hToeholds, consistent with previous findings that monocistronic constructs cause higher levels of mRNA and protein output than bicistronic constructs30.
We next sought to test the functionality of eToehold switches in different eukaryotic systems. To test the eToeholds in a single-celled eukaryote, we created strains of yeast (S. cerevisiae) expressing GFP upon galactose induction, as well as an eToehold designed to produce iRFP670 in the presence of GFP mRNA. We found that, although IRES-mediated expression was low, eToeholds were inducible by galactose-controlled GFP expression, whereas our control constructs (unmodified KBV and CrPV; Extended Data Fig. 8a,b) were not. We next tested eToeholds in plant and mammalian cellular extracts (wheat germ and rabbit reticulocyte lysate) to assess their functionality in cell-free systems. We transcribed different target RNAs and their matching eToeholds and confirmed that eToehold-mediated translation in these extracts was dependent on the presence of trRNA (Extended Data Fig. 8c,d). Furthermore, this induction was dose dependent, suggesting that eToeholds can potentially provide readouts of relative intracellular RNA levels.
eToeholds for detection of endogenous and viral mRNA
As eToeholds have demonstrated an ability to detect exogenously introduced transcripts in mammalian cells, we hypothesized that they could serve as live cell biosensors for viral infection. We generated lentiviral constructs containing eToeholds specific for Zika and SARS-CoV-2 sequences, respectively, that produced either nanoluciferase or an Azurite fluorescent protein as a readout. We transduced these constructs into the Vero E6 cell line, which is commonly used for viral studies and is derived from African green monkey kidney. We found that infection produced up to 9.2-fold increased luminescent signal in cells engineered to express Zika-specific eToeholds (Fig. 3a,b) and, furthermore, demonstrated dose-dependent responsiveness to Zika virus infection at greater sensitivities than existing live cell biosensor approaches31 (Extended Data Fig. 9a). To test the specificity of these eToeholds, we also infected transduced cells with a related Flavivirus virus—dengue virus32—and found that the eToehold response was specific to Zika infection (Fig. 3b). We observed similar results using eToeholds encoding an Azurite fluorescent protein (Extended Data Figs. 9b,c and 10). To test the ability of eToeholds to function as a live cell sensor for SARS-CoV-2, we engineered stable cell lines with eToeholds designed to sense SARS-CoV-2 transcripts. Upon transfection with constructs expressing fragments of SARS-CoV-2, we found that these eToehold-engineered cells could distinguish between the SARS-CoV-2 trRNAs and non-target RNAs (Extended Data Fig. 9d,e).

a, Stable cell lines were created with eToehold modules designed to sense infection with Zika virus. b, Luminescent signal from cells engineered to express nanoluciferase upon Zika infection after mock, Zika or dengue infection. Cells engineered with CrPV-gated nanoluciferase were used as a positive control. c, Stable cell line created with eToehold modules designed to sense exposure to heat by detecting heat shock protein mRNA. d, HeLa cells were transfected with constructs that contained a GFP reporter and eToehold-gated Azurite. e, Constructs designed to translate Azurite protein in the presence of mouse Tyr were transfected into B16, D1 or HEK293T (not shown) cells. f, Expression of eToehold-gated Azurite in B16, D1 or HEK293T cells after transfection. Data are presented as mean values with error bars representing s.d. of three technical replicates (four in Fig. 3f) exposed to the same conditions. All experiments were repeated at least three times. hsp, heat shock protein.
Finally, we explored the potential of the eToehold system for sensing endogenous transcripts, which would enable applications in identifying and targeting specific cell states and cell types. To assess the ability of eToeholds to determine cell state, we designed eToeholds to produce an Azurite protein reporter in response to transcripts of heat shock proteins hsp70 and hsp40, which are upregulated upon exposure to higher temperatures, in HeLa cells. We found that the eToehold constructs increased Azurite production up to 4.8-fold after growth at 42 °C for 24 h, as compared to routine 37 °C culture (Fig. 3c,d). Next, to assess the ability of eToeholds to differentiate between different cell types, we designed eToeholds to sense mouse tyrosinase (Tyr) mRNA, which is abundant in melanin-forming cells. With Azurite as a reporter, we observed a 3.6-fold increase in signal using Tyr-sensing eToeholds transfected into B16-F10 murine melanoma cells, as compared to two control cell lines (HEK293T and D1 marrow stromal cell; Fig. 3e,f). These results indicate that eToeholds can regulate transgene expression based on levels of intracellular, endogenous transcripts, thereby demonstrating their potential for targeting therapies to specific cell types.
Discussion
In this study, we developed eToeholds, an RNA-based eukaryotic sense-and-respond module, based on modified IRES elements that permit the translation of a desired protein in the presence of specific trRNA. Our approach of using sense–antisense interactions to alter the secondary structure of IRES modules for translational control surpasses existing RNA-based sensor systems by expanding the length, and therefore the specificity, of trigger sequences and, furthermore, enables dose responsiveness to triggers of varying quantity. The proven functionality of eToeholds in multiple domains of eukaryotic life, including fungal, plant and mammalian systems, suggests its potential for broad utility in biotechnology.
Although our eToeholds are RNA-based, we relied on DNA transfection and transduction approaches to produce them intracellularly, as these strategies are widely used in biotechnology settings. In doing so, we found that incorporating elements to reduce basal mRNA translation was critical. Endogenous 5′ capping exerts a powerful effect on translation, and we found that using exogenous polymerase systems (T7 or SP6) to transcribe eToeholds in vivo avoided 5′ capping and enabled us to achieve substantial differences between on and off states. Because 5′ capping improves mRNA stability and nuclear export, we also investigated methods to reduce basal translation levels in the presence of 5′ capping. Incorporation of stem loops and stop codons between the 5′ cap and eToehold module retained 5′ capping but still suppressed basal translation. We expect that RNA-only strategies, whereby pre-transcribed eToehold constructs are transfected directly into cells, could enable greater control over the chemistry and configuration of eToeholds. Additionally, we expect that more thorough characterization of thermodynamic interactions between trigger and eToehold constructs will aid in future optimization of eToehold designs.
We found that eToeholds could detect various intracellular RNAs, including those introduced by transfection or infection, and endogenous transcripts, such as those indicative of cell state or cell type. The ability to initiate translation of a desired protein in response to the presence of cell-type-specific or cell-state-specific RNA transcripts, as demonstrated here, has considerable therapeutic potential. For example, the clinical utility of many nucleic acid-targeting therapies is hampered by excessive off-target toxicity. The ability of an eToehold module to translate a protein or protein-based precursor in response to an mRNA signature will help address this challenge by restricting activation of a desired therapy to specific target cells. We expect that future eToehold designs will implement logic gates to allow computation of cell states from multiple trRNAs, which might further enhance the specificity and utility of this system.
Methods
Assembly of DNA constructs
Promoter gene-polyA tail sequences were cloned into pCAG-T7pol (Addgene no. 59926) or pXR1 (plasmid sequence in ‘Example Sequences’). pCAG-T7pol based plasmids were cut with EcoRI and NotI at the 5′ and 3′ ends, respectively, to insert genes for the replacement of T7 RNAP. pXR1 was cut with NotI and NcoI to replace IRES modules. pXR1 was cut with PacI and BglII to replace promoter sequences. pXR1 was cut with NcoI and XhoI to replace the reporter gene. Lentiviral vectors were cloned from pLenti CMV Puro DEST (Addgene no. 17452). pLenti CMV Puro DEST was cut with BspDI and PshAI to change promoters (specifically to PSFFV). pLenti CMV Puro DEST was cut with PshAI and XmaI to change reporter genes. pLenti CMV Puro DEST was cut with XmaI and SalI to add the eToehold-controlled gene construct. All eToehold constructs were created such that the downstream gene was in frame with the non-canonical start codon within the IRES element33. Yeast plasmids were assembled as previously described34.
Qiagen Miniprep or Qiagen Gel Extraction purification kits were used to extract and purify plasmids or fragments. All insertions were either ordered as a gBlock gene fragment from Integrated DNA Technologies or amplified from laboratory plasmids or human genomic DNA through PCR to include homology arms for Gibson isothermal assembly (using a 2× reaction mix purchased from New England Biosciences). All vectors were sequenced using Sanger sequencing from GENEWIZ before experimentation. Tandem repeats were avoided to prevent recombination events.
HEK293T cell transfection and construct testing
Plasmid concentrations were determined using a NanoDrop OneC Microvolume UV-Vis Spectrophotometer. For eToehold construct testing, transfections were performed using Lipofectamine 3000 Transfection Reagent and standard protocols in a 96-well plate of 70% confluent HEK293T cells. For each well, 30 ng of T7/SP6 polymerase-expressing construct, 70 ng of eToehold-mKate construct and 50 ng of trigger RNA-expressing construct were transfected. For samples that did not require T7/SP6 polymerase, 75 ng of eToehold constructs and 75 ng of trRNA-expressing constructs were added. After 60 h of incubation at 37 °C, cells were detached using TrypLE Express and resuspended in 2% FBS in PBS for flow cytometry in a CytoFLEX LX flow cytometer.
Yeast transformation and experimentation
Yeast transformations were carried out as previously described34. EZ-L1 was cut with PmeI and transformed into CEN.PK2–1C to generate strain EZy1. EZy1 was then transformed with EZ-L183, EZ-L184, EZ-L185, EZ-L186 and EZ-L187 to generate strains EZy13, EZy14, EZy15, EZy16 and EZy17, respectively. Liquid yeast cultures were grown in 24-well plates at 30 °C and shaken at 200 r.p.m. in synthetic complete dropout medium supplemented with 2% glucose and 50 ng ml−1 of biliverdin (VWR International, for near-infrared fluorescent protein (iRFP) imaging). Fluorescence and OD600 measurements were taken using the BioTek Synergy H1 plate reader. GFP fluorescence measurements used excitation and emission wavelengths of 485 nm and 535 nm, respectively, and iRFP fluorescence measurements used excitation and emission wavelengths of 640 nm and 675 nm, respectively. Growth and fluorescence normalization were performed as previously described34.
Cell-free lysate testing
RNA was generated using constructs bearing T7 promoters preceding desired RNA (EZ-L212, EZ-L214 and EZ-L366) and HiScribe T7 High Yield RNA Synthesis Kit by New England Biolabs, according to the manufacturer’s instructions (including DNaseI treatment). After purification using a Zymo Research RNA Clean & Concentrator Kit, RNA was added to wheat germ extract from Promega or reticulocyte lysate IVT kit from Thermo Fisher Scientific, according to the manufacturers’ instructions. Changes in fluorescence were measured over 6 h using a BioTek Synergy H1 plate reader.
Lentivirus generation and transduction
Lentiviruses were generated as previously described35, using psPAX2 and pMD2.G as helper plasmids and cloning the transfer vector based on pLenti CMV Puro DEST (w118-1). psPAX2 was a gift from Didier Trono (Addgene plasmid no. 12260; http://n2t.net/addgene:12260; RRID: Addgene_12260). pMD2.G was also a gift from Didier Trono (Addgene plasmid no. 12259; http://n2t.net/addgene:12259; RRID: Addgene_12259). pLenti CMV Puro DEST (w118-1) was a gift from Eric Campeau and Paul Kaufman (Addgene plasmid no. 17452; http://n2t.net/addgene:17452; RRID: Addgene_17452). EZ-L521, EZ-L534 and EZ-L536 were cloned from pLenti CMV Puro DEST (w118-1) through Gibson assembly. After transduction of Vero E6 cells with the lentiviruses, transduced cells were sorted for GFP fluorescence using a Sony SH800 cell sorter.
Zika virus infection testing
Vero E6 cells (American Type Culture Collection (ATCC) CRL-1586; maintained in DMEM 10% FBS), dengue virus serotype 2 (DENV2 strain New Guinea C, accession AAA42941) and Zika virus isolates (Pernambuco isolate 243, accession MF352141) were gifts from Lee Gehrke at the Massachusetts Institute of Technology. Cell lines were plated and grown overnight to 90% confluent monolayers and infected with Zika virus (at multiplicity of infection (MOI) = 0.02 for stable cell line created using EZ-L536 or EZ-L521 and at MOI = 2 for stable cell line created using EZ-L534, in DMEM 2% FBS) or DENV2 (at MOI = 2, in DMEM 2% FBS)32 and fixed at 48 hours post-infection (h.p.i.) using Cytofix from BD Biosciences and subsequently washed twice in DPBS 2% FBS. Replicates of cells were fixed and stained with anti-NS1 antibodies 7724.323 (diluted 1:1,000 anti-dengue NS1 mAb 323) and 7944.644 (diluted 1:2,000; anti-Zika NS1 mAb 644)36,37, obtained from Lee Gehrke. Other replicates of cells were run on a CytoFLEX LX flow cytometer or used for a nanoluciferase assay using Nano-Glo from Promega, according to the manufacturer’s instructions, and measured using a BioTek Synergy H1 plate reader.
Heat shock-sensing eToehold testing
HeLa cells (ATCC CCL-2, a gift from the Ingber lab) were transfected with EZ-L512 (ABPV positive control), EZ-L548 (an eToehold that senses hsp70) or EZ-L554 (an eToehold that sense hsp40). At 36 h.p.i., a portion of the samples was moved to 42 °C for 24 h. Cells were detached using TrypLE Express and resuspended in 2% FBS in PBS for flow cytometry in a CytoFLEX LX flow cytometer.
Mouse Tyr eToehold testing
B16-F10 melanoma cells and D1 marrow stromal cells (ATCC CRL-6475 and CRL-12424, gifts from David Mooney), as well as HEK293T cells (ATCC CRL-3216), were maintained at subconfluency in DMEM 10% FBS. Cells were transfected with plasmids encoding eToeholds or control sequences using TransIT-2020 transfection reagent (Mirus Bio), per the manufacturer’s instructions. At 48 h.p.i., cells were detached, stained for viability (Thermo Fisher Scientific) and analyzed using flow cytometry with an LSRFortessa with High Throughput Sampler (HTS) (BD Biosciences). FACSDiva software (BD Biosciences) was used for analysis of samples. Only viable cells were included for analysis.
Intracellular cytokine staining
HEK293T, human skeletal muscle cells (ATCC) and human dermal fibroblast cells (ATCC) were cultured in DMEM 10% FBS, Primary Skeletal Muscle Growth Kit in Mesenchymal Stem Cell Basal Media (ATCC) and Fibroblast Growth Media 2 (PromoCell), respectively. Cells were transfected or transduced with EZ-L793 to introduce eToehold and either EZ-L1061 or EZ-L1062. Either 24 h after transfection or after antibiotic selection after transduction, cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences); stained with PE/Dazzle 594 anti-human IL-6 (501121), PE anti-human CCL5 (515503) or PE anti-human CCL2 (505903); and analyzed with an LSRFortessa with HTS. FACSDiva software was used for analysis of samples.
qPCR
RNA was isolated after cells were pelleted and frozen by using an RNEasy Plus Mini Kit (Qiagen), following the manufacturer’s instructions, and stored at −80 °C or immediately used. RNA was quantified by NanoDrop spectrophotometer, and reverse transcription and amplication was performed using a Luna Universal One-Step RT–qPCR kit (New England Biolabs) on a CFX96 RT–PCR machine (Bio-Rad). Either PrimePCR primers (Bio-Rad; qHsaCED0038674 for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) or custom primers (5′-TTGACGGAGTCACACCGAAT-3′ and 5′-GTCACTCTGAACAGGAGGCT-3′ for nanoluciferase) were used. Custom primers were validated by calculating primer efficiency from a standard curve of nanoluciferase template RNA. Relative gene expression was computed by the ΔΔCt method, which compared Ct values to a control sample (orthogonal trigger) and reference gene (GAPDH).
Statistics
Statistical significance was determined using a standard t-test for calculating P values. t-scores were calculated by the following formula: \(\frac{{({\mathrm{Mean}}_{{\mathrm{Condition}}\,2} - {\mathrm{Mean}}_{{\mathrm{Condition}}\,1})\sqrt {{\mathrm{Number\,of\,samples}}} }}{{{\mathrm{Standard\,Deviation}}_{{\mathrm{Condition}}\,2}}}\). P values were calculated using two degrees of freedom and a one-sided t-test calculator. For qPCR, statistics were performed using ΔCt values, which are the relative expression normalized to reference gene GAPDH in PRISM using a two-way analysis of variance followed by Sidak multiple comparisons testing.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available in the paper (and in its supplementary information files). Original data that support the findings are available from the corresponding author upon reasonable request. DNA sequences are available in the Supplementary Sequences section of the Supplementary Information.
References
Green, A. A., Silver, P. A., Collins, J. J. & Yin, P. Toehold switches: de-novo-designed regulators of gene expression. Cell 159, 925–939 (2014).
Hanewich-Hollatz, M. H., Chen, Z., Hochrein, L. M., Huang, J. & Pierce, N. A. Conditional guide RNAs: programmable conditional regulation of CRISPR/Cas function in bacterial and mammalian cells via dynamic RNA nanotechnology. ACS Cent. Sci. 5, 1241–1249 (2019).
Siu, K. H. & Chen, W. Riboregulated toehold-gated gRNA for programmable CRISPR–Cas9 function. Nat. Chem. Biol. 15, 217–220 (2019).
Kim, J. et al. De novo-designed translation-repressing riboregulators for multi-input cellular logic. Nat. Chem. Biol. 15, 1173–1182 (2019).
Wang, S., Emery, N. J. & Liu, A. P. A novel synthetic toehold switch for microRNA detection in mammalian cells. ACS Synth. Biol. 8, 1079–1088 (2019).
Litke, J. L. & Jaffrey, S. R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 37, 667–675 (2019).
Felletti, M., Stifel, J., Wurmthaler, L. A., Geiger, S. & Hartig, J. S. Twister ribozymes as highly versatile expression platforms for artificial riboswitches. Nat. Commun. 7, 12834 (2016).
Zhong, G., Wang, H., Bailey, C. C., Gao, G. & Farzan, M. Rational design of aptazyme riboswitches for efficient control of gene expression in mammalian cells. eLife 5, e18858 (2016).
Zhong, G. et al. A reversible RNA on-switch that controls gene expression of AAV-delivered therapeutics in vivo. Nat. Biotechnol. 38, 169–175 (2020).
Ruehle, M. D. et al. A dynamic RNA loop in an IRES affects multiple steps of elongation factor-mediated translation initiation. eLife 4, e08146 (2015).
Kanamori, Y. & Nakashima, N. A tertiary structure model of the internal ribosome entry site (IRES) for methionine-independent initiation of translation. RNA 7, 266–274 (2001).
Zhu, J. et al. Crystal structures of complexes containing domains from two viral internal ribosome entry site (IRES) RNAs bound to the 70S ribosome. Proc. Natl Acad. Sci. USA 108, 1839–1844 (2011).
Yamamoto, H., Nakashima, N., Ikeda, Y. & Uchiumi, T. Binding mode of the first aminoacyl-tRNA in translation initiation mediated by Plautia stali intestine virus internal ribosome entry site. J. Biol. Chem. 282, 7770–7776 (2007).
Colussi, T. M. et al. Initiation of translation in bacteria by a structured eukaryotic IRES RNA. Nature 519, 110–113 (2015).
Ogawa, A., Masuoka, H. & Ota, T. Artificial OFF-riboswitches that downregulate internal ribosome entry without hybridization switches in a eukaryotic cell-free translation system. ACS Synth. Biol. 6, 1656–1662 (2017).
Hodgman, C. E. & Jewett, M. C. Characterizing IGR IRES-mediated translation initiation for use in yeast cell-free protein synthesis. Nat. Biotechnol. 31, 499–505 (2014).
Nishiyama, T. et al. Structural elements in the internal ribosome entry site of Plautia stali intestine virus responsible for binding with ribosomes. Nucleic Acids Res. 31, 2434–2442 (2003).
Wesselhoeft, R. A. et al. RNA circularization diminishes immunogenicity and can extend translation duration in vivo. Mol. Cell 74, 508–520 (2019).
Sandig, V., Lieber, A., Bähring, S. & Strauss, M. A phage T7 class-III promoter functions as a polymerase II promoter in mammalian cells. Gene 131, 255–259 (1993).
Bensaude, O. Inhibiting eukaryotic transcription. Transcription 2, 103–108 (2011).
Keys, D. A. et al. Multiprotein transcription factor UAF interacts with the upstream element of the yeast RNA polymerase I promoter and forms a stable preinitiation complex. Genes Dev. 10, 887–903 (1996).
Vu, L., Siddiqi, I., Lee, B. S., Josaitis, C. A. & Nomura, M. RNA polymerase switch in transcription of yeast rDNA: role of transcription factor UAF (upstream activation factor) in silencing rDNA transcription by RNA polymerase II. Proc. Natl Acad. Sci. USA 96, 4390–4395 (1999).
Bao, C. et al. mRNA stem-loops can pause the ribosome by hindering A-site tRNA binding. eLife 9, e55799 (2020).
Hoffmann, E., Neumann, G., Hobom, G., Webster, R. G. & Kawaoka, Y. ‘Ambisense’ approach for the generation of influenza A virus: vRNA and mRNA synthesis from one template. Virology 267, 310–317 (2000).
Czudai-Matwich, V., Schnare, M. & Pinkenburg, O. A simple and fast system for cloning influenza A virus gene segments into pHW2000- and pCAGGS-based vectors. Arch. Virol. 158, 2049–2058 (2013).
Yokoyama, T. et al. HCV IRES captures an actively translating 80S ribosome. Mol. Cell 74, 1205–1214 (2019).
Ochs, K. et al. Interaction of translation initiation factor eIF4B with the poliovirus internal ribosome entry site. J. Virol. 76, 2113–2122 (2002).
Thompson, S. R. & Sarnow, P. Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology 315, 259–266 (2003).
Davila-Calderon, J. et al. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat. Commun. 11, 4775 (2020).
Sadikoglou, E., Daoutsali, E., Petridou, E., Grigoriou, M. & Skavdis, G. Comparative analysis of internal ribosomal entry sites as molecular tools for bicistronic expression. J. Biotechnol. 181, 31–34 (2014).
McFadden, M. J. et al. A fluorescent cell-based system for imaging Zika virus infection in real-time. Viruses 10, 13–18 (2018).
Medina, F. et al. Dengue virus: isolation, propagation, quantification, and storage. Curr. Protoc. Microbiol. 27, 15D.2.1–15D.2.24 (2012).
Murray, J. et al. Structural characterization of ribosome recruitment and translocation by type IV IRES. eLife 5, e13567 (2016).
Zhao, E. M. et al. Optogenetic regulation of engineered cellular metabolism for microbial chemical production. Nature 555, 683–687 (2018).
Campeau, E. et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529 (2009).
Bosch, I. et al. Serotype-specific detection of dengue viruses in a nonstructural protein 1-based enzyme-linked immunosorbent assay validated with a multi-national cohort. PLoS Negl. Trop. Dis. 14, e0008203 (2020).
Bosch, I. et al. Rapid antigen tests for dengue virus serotypes and Zika virus in patient serum. Sci. Transl. Med. 9, eaan1589 (2017).
Acknowledgements
We thank J. Niemi and N. M. Donghia for their maintenance of laboratory operations. We thank all members of the Collins laboratory for their advice and help throughout the course of this study. We also thank L. Gehrke for providing Zika virus, dengue virus, Vero E6 cells and the 7724.323 and 7944.644 antibodies. We thank D. Inger and G. Goyal for providing HeLa cells. We thank D. Mooney for providing B16-F10 and D1 cells. We thank I. Wickersham for his donation of the pCAG-T7pol plasmid (retrieved from Addgene). We also thank D. Trono, E. Campeau and P. Kaufman for their donations of plasmids necessary for lentiviral synthesis (retrieved from Addgene; see Methods for details). This project was funded by BASF (research grant) and National Institutes of Health grant 1RC2DK120535-01A1. It was also supported by an American Gastroenterological Association Takeda Pharmaceuticals Research Scholar Award in Inflammatory Bowel Disease. E.Z. is supported by the Schmidt Science Fellows program.
Author information
Authors and Affiliations
Contributions
E.M.Z. and A.S.M. planned and performed experiments and analyzed the data. E.M.Z. and A.S.M. wrote the manuscript. E.M.Z., A.S.M., H.P. and X.T. performed experiments and edited the manuscript. H.P., K.Z., N.D.T. and X.T. contributed equally to the manuscript. N.D.T., K.Z., I.H., D.B.T., C.Y.O., E.J.C., F.A.R., T.Y.H., P.R., I.H., D.B.T., C.Y.O. and E.S.Z. performed experiments. P.Q.N., P.R. and M.A.E. helped design schematics. J.J.C. directed the overall research and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
E.M.Z., A.S.M., H.P., X.T., E.J.C., F.A.R. and J.J.C. have filed a patent application (no. PCT/US21/37030) based on the presented work.
Additional information
Peer review information Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Schematic of eToehold screening and gating for flow cytometry experiments.
a, HEK293T cells were plated at 60% confluency and transfected with combinations of plasmids encoding T7-polymerase, T7 promoter driving potential eToehold-gated mKate, and trigger construct (GFP). After incubation at 37 °C for 60 hours, cells were detached and analyzed with flow cytometry. b-e, Representative plots for a negative control. f-i, Representative plots for a positive GFP and mKate control.
Extended Data Fig. 2 eToehold activity dependence on thermodynamics of insertion regions.
a, Constructs were based on CrPV eToehold 8–6 designed for GFP sensing. b, Constructs were based on CrPV eToehold 6–7 designed for GFP sensing. Data are presented as mean values with error bars representing SD of three technical replicates. All experiments were repeated at least three times.
Extended Data Fig. 3 Intracellular cytokine staining in cells expressing CrPV IRES with or without additional RNA binding.
a, Effects of transfection or transduction with a CrPV IRES construct on production of IL-6, CCL5, and CCL2 in primary human fibroblast and muscle skeletal cells. See Supplementary Table 1 for construct specifics. * denotes p = 0.0043 for the column factor (transgene introduction method) in a 2-way ANOVA for fibroblasts, and p = 0.0167 for muscle cells. b, Expression of IL-6, CCL5, and CCL2 in cells transduced to express a CrPV IRES construct and “trigger,” compared to those expressing eToehold and an orthogonal nonbinding sequence. Data are presented as mean values with error bars representing SD of three biologically independent experiments. All experiments were repeated at least three times, and dots represent individual data points.
Extended Data Fig. 4 Mean intensity data for Fig. 1.
a, Intensity data for Fig. 1c. showing activities of different IRES modules with and without co-transfection with T7 RNAP and GFP trigger sequence. b, Intensity data for Fig. 1e, showing eToehold modules screen. c, Intensity data for Fig. 1g, showing effect of using insertions with matching base pairs (three base pairs) in the regions depicted in Fig. 1d. Data are presented as mean values with error bars representing SD of three technical replicates. All experiments were repeated at least two times.
Extended Data Fig. 5 Decreasing basal expression of eToehold modules.
a, Constructs with different promoters driving sfGFP were tested based on Extended Data Fig. 1. b, RNA regions designed to recruit RNA polymerase I factors and decrease 5’ capping were inserted in front of CrPV eToehold 6–7. c, RNA polymerase I responsive promoters were tested based on Extended Data Fig. 1. See Supplementary Table 1 for construct details. Data are presented as mean values with error bars representing SD of three technical replicates. Representative flow cytometry plots were chosen within three experimental replicates showing similar results. All experiments were repeated at least two times.
Extended Data Fig. 6 Mean intensity data for Fig. 2 and Fig. 3.
a, Intensity data for Fig. 2a, showing effects of different promoter-polymerase systems on output transgene (mKate). b, Intensity data for Fig. 2b, showing eToehold activity in the presence of designed trRNA and unmatched RNA. c, Intensity data for Fig. 2d, showing eToehold or IRES activity with or without additional features. d, Intensity data for Fig. 3d, showing function of eToehold modules designed for heat shock protein. e, Intensity data for Fig. 3f, showing function of eToehold modules designed for mouse tyrosinase. Data are presented as mean values with error bars representing SD of three technical replicates. All experiments were repeated at least three times.
Extended Data Fig. 7 Further iterations and testing of eToeholds. a, decrease of T7 promoter basal expression via insertion of stop codons and stem loops in a bicistronic design.
Addition of stop codons and stem loops before the eToehold module tested based on Extended Data Fig. 1. b, Effect of mismatch mutations within or external to the annealing region of inserted RNA sequences on eToehold function. All constructs were based on CrPV eToehold 8–6 designed to sense GFP. c, eToeholds constructed based on other Dicistroviridae IRESs (namely, KBV and ABPV) retain functionality. d, Complements to the smaller fragment insertion do not activate eToeholds. EZ-L287, designed for GFP trigger, was tested using the set up in Extended Data Fig. 1, utilizing different triggers, including a trigger with the reverse complement of the smaller GFP fragment inserted into ySUMO (see Supplementary Table 1). See Supplementary Table 1 for construct specifics. Data are presented as mean values with error bars representing SD of three technical replicates. All experiments were repeated at least two times.
Extended Data Fig. 8 eToeholds function in yeast and cell lysates.
a-b, eToehold modules gating iRFP were integrated into a yeast strain that expressed GFP (trigger RNA) upon switching of carbon source to galactose. iRFP signal (a) and GFP signal (b) were measured after 6 hours of exponential phase growth. Media was supplemented with biliverdin. See Supplementary Table 1 for construct specifics. c-d, eToeholds function in cell-free lysate. c, Wheat germ extract: 50 nM of switch-sfGFP RNA (transcribed from EZ-L214 or EZ-L212 as a control) was added along with different amounts of trigger RNA (transcribed EZ-L366). d, Rabbit reticulocyte lysate: 150 nM of switch-sfGFP RNA (transcribed from EZ-L214 or EZ-L212 as a control) was added along with or without 250 nM of trigger RNA (transcribed EZ-L366). See Supplementary Table 1 for construct details. Data are presented as mean values with error bars representing SD of three technical replicates. All experiments were repeated at least two times.
Extended Data Fig. 9 Further viral infection sensing with eToeholds.
a, Zika virus concentration sensing with eToeholds. Luminescent signal from cells engineered to express nanoluciferase upon Zika infection after mock, Zika, or Dengue infection. Same experiment as in Fig. 3a,b. a, Stable cell line designed for sensing of Zika virus infection contain Zika RNA-responsive eToehold-gated Azurite translation under a T7 promoter. b, Wildtype Vero E6 cells and stable cell line expressing Zika sensing eToehold gated Azurite as depicted in (a) were subjected to infection with Dengue or Zika virus. Sample gates shown in Extended Data Fig. 10. c, Stable cell line designed for sensing of SARS-CoV-2 infection contain SARS-CoV-2 RNA-responsive eToehold-gated Nanoluciferase translation. d, Stable cell lines containing eToeholds that sensed different regions of SARS-CoV-2 were transfected with constructs that expressed GFP and two regions of SARS-CoV-2. Luminescence measurements were then taken after furimazine was added. See Supplementary Table 1 for construct specifics. Data are presented as mean values with error bars representing SD of three technical replicates. All experiments were repeated at least three times.
Extended Data Fig. 10 Gates used for viral sensing applications.
a, Staining results for samples that were infected virus (see Methods) b, Infection level dependence on MOI of infection as measured by staining and subsequent flow cytometry. Representative flow cytometry plots were chosen within two experimental replicates showing similar results. All experiments were repeated at least two times. c, Stable cell lines expressing Zika-sensing eToehold gated Azurite (depicted in Extended Data Fig. 9b) were subjected to infection with Dengue virus and Zika virus. d, Wildtype Vero E6 cells infected with Zika virus do not exhibit increased Azurite fluorescence.
Supplementary information
Supplementary Information
Example sequences and Supplementary Tables 1 and 2.
Rights and permissions
About this article

Cite this article
Zhao, E.M., Mao, A.S., de Puig, H. et al. RNA-responsive elements for eukaryotic translational control. Nat Biotechnol 40, 539–545 (2022). https://doi.org/10.1038/s41587-021-01068-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-021-01068-2
This article is cited by
-
Programmable eukaryotic protein synthesis with RNA sensors by harnessing ADAR
Nature Biotechnology (2023)
-
Targeting cancer with mRNA–lipid nanoparticles: key considerations and future prospects
Nature Reviews Clinical Oncology (2023)
-
Target-dependent RNA polymerase as universal platform for gene expression control in response to intracellular molecules
Nature Communications (2023)
-
Autocatalytic base editing for RNA-responsive translational control
Nature Communications (2023)
-
Modular, programmable RNA sensing using ADAR editing in living cells
Nature Biotechnology (2023)
