
Abstract
Background
To date, certain efforts have been made to investigate the clinical and genetic characteristics of patients with EYS mutations. However, data for Chinese patients are limited.
Objectives
To perform a detailed phenotyping and genetic characterization of 55 Chinese patients with EYS-RD, and to identify risk factors for these clinical data.
Methods
A total of 55 patients with EYS-RD were recruited. Best-corrected visual acuity (BCVA), patient age, age at symptom onset, disease duration, and genetic information were collected.
Results
Thirty-six novel variants, three hot mutations of EYS (30.3%, c.6416G>A, c.6557G>A, c.7492G>C) and one hot region (49.06%, Laminin G domains) were identified. In all, 36.84% of the mutations occurred at base G site, and majority of mutations (56.56%) were missense. Late-truncating mutations are significantly more prevalent (41.30%). The mean age of onset was 15.65 ± 14.67 years old; it had no significant correlation with genotype. The average BCVA was 0.73 ± 0.93 LogMAR, and 61.8% of eyes had a BCVA better than 0.52 logMAR. BCVA was positively correlated with disease duration time. The mean MD was 23.18 ± 7.34 dB, MD showed a significant correlation with genotype and age. Cataract was present in 56.45% of patients, and 42.59% of patients showed an absence of pigmentation in the retina. Cataract and hyperpigmentation both showed a significant correlation with age.
Conclusions
EYS-RD is associated with a moderate phenotype with onset around adolescence, but great variability. Our study largely enhances the current knowledge of phenotypic and genotypic characteristics of EYS-RD, which could pave the way for better management of these patients.
Similar content being viewed by others
Introduction
Eyes shut homolog (EYS; OMIM: 612424), spanning approximately 2 Mb of chr6q12 and consisting of 44 exons, was first reported in 2008 as a disease-causing gene for autosomal recessive retinitis pigmentosa (RP). EYS is the largest gene in the eye and encodes a protein with 3156 amino acids that is predominantly expressed in the retina and plays an essential role in the morphogenesis of photoreceptors [1,2,3,4]. The EYS protein contains 27 epidermal growth factor-like (EGF) domains and five laminin G-like domains, which are highly conserved [1].
Mutations in EYS are associated with several phenotypes, such as RP, cone–rod dystrophy (CRD), and Leber congenital amaurosis (LCA), and we collectively call these phenotypes EYS-associated retinal disease (EYS-RD) here [5,6,7,8]. Our prior study reported that the prevalence of EYS mutations in patients with inherited retinal dystrophies (IRDs) was 7% in the Chinese population, and EYS mutations were ranked as the third most common genetic mutation in patients with IRD [7]. The high prevalence of EYS-RD makes further in-depth research on its clinical and genetic characteristics more urgent and significant.
To date, certain efforts have been made to investigate the clinical and genetic characteristics of patients with EYS mutations. However, most studies were conducted in Japanese cohorts [5, 9,10,11], and data for Chinese patients are limited [12]. In this study, 55 patients with EYS-RD were enrolled, which is the largest cohort to our knowledge. The purpose of this study was to perform a detailed phenotyping and genetic characterization of 55 Chinese patients with EYS-RD, and to identify risk factors for these conditions.
Materials and methods
Subjects and ethical statement
The protocol of this study adhered to the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of the Eye and Ear, Nose and Throat (ENT) Hospital of Fudan University. A total of 55 patients from 46 families with EYS-RD were enrolled from the Eye and ENT Hospital of Fudan University between January 2017 and December 2019; all of the participants were of Chinese descent. Of the patients, 38 were mentioned in our previous report [7]. Written informed consent was obtained from all subjects.
Genetic analysis
Genomic DNA was extracted from the peripheral blood of all affected subjects and the available family members. Molecular testing and data analysis were performed as previously reported [7, 13,14,15,16]. We designed a high-throughput targeted enrichment approach to exon-capture regions of 762 genes involved in common inherited eye diseases. The capture panel was custom designed and produced by the Beijing Genomics Institute (BGI, Shenzhen, China) (Supplementary Table 1). For acquisition of probe sequences, we obtained the exon sequence and its flank ±30 bp of 762 genes from a reference human genome (UCSC hg 38). Each reference sequence begins at one end of a reference sequence and selects the reference sequence of the predetermined length to obtain the probe sequence so that the last total probe can cover the reference sequence at least once. The probe length of the panel is 90 nt; the total target area obtained is 2.3 M. On average, the mean coverage depth was more than 400×, and the coverage of target region was ~99.9% using BGISEQ-2000. Subsequent points for sample quality control were also added to the probe design process. The exon deletion was found by CapCNV analysis followed by CNVkit protocol (https://cnvkit.readthedocs.io/en/stable/pipeline.html). The potential pathogenicity of the variants was interpreted according to the American College of Medical Genetics.
Patients were divided into three groups depending on the number and type of the identified variants, as previously described [17,18,19]. The genotype A group (severe) included patients with ≥2 null variants, the genotype B group included patients with 1 null variant and 1 variant that was a missense or in-frame insertion/deletion, and the genotype C group (moderate) included patients with no null variant but ≥2 variants that were missense or in-frame insertions/deletions. Null variants were those that would be predicted to affect splicing or to introduce a premature truncating codon in the protein, such as nonsense, frameshift, exonic, or intronic variants with significant splice-site alteration [19, 20].
Clinical examination
Clinical information, including medical history, family history, ethnicity, chief complaints of visual symptoms, onset of disease, best Snellen-corrected visual acuity (BCVA, converted to equivalent value of logarithm of minimal angle of resolution (logMAR) unit), slit-lamp biomicroscopy of anterior and posterior segments, wide-field fundus imaging (Optos PLC, Dunfermline, United Kingdom), fundus autofluorescence (FAF, Spectralis HRA COCT; Heidelberg, Germany), visual field (VF, Humphrey Visual Field Analyzer, Carl Zeiss Inc., Dublin, CA, USA), swept-domain optical coherence tomography (Spectralis HRA+OCT, Heidelberg Engineering Inc., Heidelberg, Germany), and full-field electroretinography (according to the standards of the International Society for Clinical Electrophysiology of Vision; www.iscev.org) were assessed. VF was assessed by 30-2 Swedish Interactive Threshold Algorithm (SITA) Fast Programs to measure 30° temporally and nasally and test 76 points. The VF data were excluded if fixation loss and false-positive and false-negative response rates were greater than 20%. The average depression of visual sensitivity was estimated by the mean deviation (MD). To provide numeric values for low BCVAs, the following conversions were performed: light perception, 4 logMAR; hand movements, 3 logMAR; and counting fingers, 2 logMAR.
Statistical analysis
The statistical analysis was carried out using SPSS software version 22.0 (IBM Corp., Armonk, NY). Statistical significance for all statistical tests was set at P < 0.05. Measurement values of the groups were compared using the t-test and one-way ANOVA. Correlations between pairs of measurements were evaluated using the Pearson or Spearman test, as appropriate. Linear mixed models were used to assess the impact of clinical data (age, age of symptom onset, disease duration time, BCVA, MD, retinal hyperpigmentation, and cataract) and genetic data on certain data.
Results
Cohort characteristics
In total, 55 affected subjects (females 21, males 34) from 46 families with EYS-RD were enrolled; all of the participants were of Chinese descent. Pedigrees and mutations of the 46 families are available in Supplementary Fig. 1. The demographic and clinical characteristics are summarized in Table 1. Of the 55 patients, 53 were diagnosed with RP and 2 had LCA. The median age of these subjects was 38.0 years old at the examination (mean, 39.96 ± 14.87; range, 8.0–74.0), and the mean disease duration was 24.13 ± 15.40 years (median 22.5; range, 2.0–66.0).
Genetic analyses
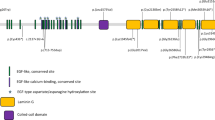
Cosegregation analysis was performed in 46 families. Pedigrees showing the clinical and genetic status of the 46 families are available in Supplementary Fig. 1. A total of 122 alleles representing 56 distinct variants were identified, and of these alleles, 36 were novel (Supplementary Table 2). Sanger verification was performed on all mutations, and Supplementary Fig. 2 shows the partial results of Sanger sequencing. Of the 122 variants identified in these patients, the majority of pathogenic defects (58.20%, n = 71) were missense variants (56.56%, n = 69) or in-frame deletions (1.64%, n = 2), and 41.80% (n = 51) were nonsense (16.39%, n = 20), frameshift (21.31%, n = 26), splice-site (0.82%, n = 1) mutations or copy-number variations (CNVs, 3.28%, n = 4) that severely affected protein function (Fig. 1A). Of the 46 truncating mutations, 13 (28.26%) were located in exon 44 and 6 (13.04%) were located in exon 43 (Fig. 1C), indicating that late-truncating pathogenic variants (exons 43–44) are significantly more prevalent (41.30%). Eleven patients (20.0%) were in the genotype A group, 30 (54.5%) were in the genotype B group, and 14 (25.5%) were in the genotype C group (Fig. 1B). The most prevalent alleles were c.6416G>A p.Cys2139Tyr (11.5%, n = 14), c.6557G>A p.Gly2186Glu (9.8%, n = 12), and c.7492G>C p.Ala2498Pro (9.0%, n = 11), accounting for 30.3% of all alleles (37/122), suggesting that they are hotspot mutations of EYS in the Chinese population. In addition, 15 variants were found in patients presenting with cataracts, 7 were found both in patients with and without cataracts, and 14 were found in patients without cataracts at the time of examinations. Most variants (49.06%) harbored by the cohort were spatially distributed across the laminin G domains toward the carboxy (−COOH) end of the protein (Fig. 2). One striking feature of the point mutations (n = 38) is that 36.84% (n = 14) of them occurred at the base G site.

A The proportion of different mutation types in EYS-associated retinal disease (EYS-RD). A total of 56.56% were missense, 16.39% were nonsense, 21.31% were frameshift, 1.64% were in-frame deletions, 3.28% were copy-number variations (CNVs), and 0.82% were splice-site mutations. B The proportion of different genotypes in EYS-RD patients. C Distribution of the EYS variants (missense, nonsense, and frameshift) on different exons. Late-truncating pathogenic variants (exons 43–44) are significantly more prevalent (41.30%).

Variants identified in patients with and without cataracts are listed in red (n = 15) and black (n = 14), respectively. Variants identified both in patients with and without cataracts are listed in green (n = 7). Variants identified in patients with unknown conditions of the lens are listed in blue (n = 17). The majority of variants (49.06%) harbored by the cohort were spatially distributed across the laminin G domains toward the carboxy (−COOH) end of the protein. Color figure online.
Phenotypic characterization and associated risk factors
Age of symptom onset
Age of symptom onset was available for 43 patients (86 eyes). Disease onset presented a wide spectrum from birth to 62 years of age (mean 15.65 ± 14.67, median 12). In detail, 32.6% (n = 14) of patients had an onset earlier than 5 years of age (infant onset), 37.2% (n = 16) of patients had an onset between 6 and 18 years of age (juvenile onset), 27.9% (n = 12) of patients had an onset between 19 and 49 years of age, and only 1 (2.3%) patient had an onset at age 62 years (Fig. 3A). Spearman analysis revealed that age of onset had no significant correlation with genotype (coefficient: −0.163, P = 0.149).

A Number of patients in different age of onset groups. B Number of eyes in different BCVA logMAR groups. C Correlation of BCVA logMAR with age, age of onset, and disease duration. LogMAR BCVA was positively correlated with disease duration (coefficient: 0.616, P = 0.001) but had no significant associations with age (P = 0.711) or age of symptom onset (P = 0.165). Disease duration is marked in black (font, line, or circle), age is marked in red, and age of symptom onset is marked in blue. D Correlations between BCVA logMAR values and genotype (P = 0.178). BCVA best-corrected visual acuity. Color figure online.
BCVA
BCVA data were available for 38 patients (76 eyes). Measured BCVAs ranged from 0 logMAR to light perception (mean 0.73 ± 0.93), and 17.1% (13/76) of eyes had a BCVA worse than logMAR 1.3, 21.1% (16/76) of eyes had a BCVA worse than 1 logMAR, and 61.8% (47/76) of eyes had a BCVA better than 0.52 logMAR (Fig. 3B and Table 1). To further investigate the impact of clinical data (age, disease duration, age of symptom onset, VF, retinal hyperpigmentation, and genotype) on BCVA, a linear mixed-effects model was used. The results showed that logMAR BCVA was positively correlated with disease duration (coefficient: 0.616, P = 0.001) but had no significant associations with age (P = 0.711), age of symptom onset (P = 0.165), MD (P = 0.065), retinal hyperpigmentation (P = 0.423), or genotype (P = 0.178) (Fig. 3C, D).
Visual field
Severe cataracts could affect the VF test results, so patients with severe cataracts were excluded from the VF analysis. Therefore, VF was available for only 16 patients (32 eyes). The mean MD was 23.18 ± 7.34 dB (range 3.44–32.31, median 25.15). Linear regression analysis showed that MD was not correlated with disease duration (P = 0.755), age of symptom onset (P = 0.148), or BCVA (P = 0.075) but showed a significant correlation with genotype (coefficient: −0.4005, P = 0.001) and age (coefficient: 0.441, P = 0.015) (Fig. 4A, B). Severe genotype and older age were associated with severe VF defects.

A Correlation of MD with age, age of onset, and disease duration. MD showed a significant correlation with age (coefficient: 0.441, P = 0.015) but was not correlated with disease duration (P = 0.755) or age of symptom onset (P = 0.148). B Correlations between MD and genotype (P = 0.178). MD showed a significant correlation with genotype (coefficient: −0.4005, P = 0.001). C Correlation of cataracts with age, age of onset, and disease duration. Cataracts showed a significant correlation with age (coefficient: 0.329, P = 0.002), but there was no significant association of cataracts with age of onset (P = 0.373) or disease duration (P = 0.352). D Correlations between cataract and genotype (P = 0.295). E Correlation of hyperpigmentation with age, age of onset, and disease duration. Hyperpigmentation showed a significant correlation with age (coefficient: 0.334, P = 0.015), but there was no significant association of hyperpigmentation with age of onset (P = 0.197) or disease duration (P = 0.197). F Correlations between cataract and genotype (P = 0.793). Disease duration is marked in black (font, line, or circle), age is marked in red, and age of symptom onset is marked in blue. MD mean deviation. Color figure online.
Cataract
The condition of the lens was available in 31 patients (62 eyes). Cataracts are a common finding in this cohort of patients and were found in all age groups (range 8–74 years old, mean 43.76 ± 16.46 years old). Of the 62 eyes, 35 (56.45%) showed varying degrees of cataracts, with a mean BCVA of 0.48 ± 0.28 logMAR, and the median disease duration was 25.25 ± 13.67 years (Table 1). Patients without cataracts (31.71 ± 14.23 years old) were younger than patients who had cataracts (P = 0.003), and BCVA was worse in patients who had cataracts (1.02 ± 1.07 vs 0.46 ± 0.79 logMAR, P = 0.027). However, age of onset (P = 0.096) and disease duration (P = 0.352) were not significantly different between the two groups. There were no significant associations of cataracts with genotype (P = 0.295), age of onset (P = 0.373) or disease duration (P = 0.352), but cataracts showed a significant correlation with age (coefficient: 0.329, P = 0.002) and BCVA (coefficient: 0.348, P = 0.009) (Fig. 4C, D).
Retinal hyperpigmentation
Data on pigment change in the retina were available for all patients (108 eyes). Of the 108 eyes, 46 (42.59%) showed an absence of pigmentation in the retina, while 62 eyes showed hyperpigmentation (Table 1 and Supplementary Fig. 3). The mean age of the patients who showed hyperpigmentation was 41.11 ± 15.59 years old, the mean BCVA was 0.48 ± 0.28 logMAR, and the median disease duration was 25.25 ± 13.67 years. Patients without pigmentation (31.62 ± 18.06 years old) were younger than those with hyperpigmentation (P = 0.024), and the age of symptom onset was earlier in patients without hyperpigmentation (7.82 ± 7.88 vs 15.00 ± 15.50 years old), but BCVA (P = 0.524) and disease duration (P = 0.705) were not significantly different between the two groups. Correlation analysis revealed that there were no significant associations of hyperpigmentation with genotype (P = 0.793), disease duration (P = 0.197), BCVA (P = 0.446), or MD (P = 0.056), but hyperpigmentation showed a significant correlation with age (coefficient: 0.334, P = 0.015) (Fig. 4E, F). These results indicate that older age was associated with pigmentary changes.
Discussion
EYS-RD is one of the most prevalent IRDs in Japanese and European populations [10, 21,22,23,24,25]. The proportion has been reported to be 5% in the Netherlands and Canada, 7% in Israel, 11% in the UK, 12% in France, 17% in Spain, and 18–23.5% in Japanese cohorts of RP patients. The vast majority of mutations were truncating, and the most prevalent variants were c.4957_4958insA, c.2528G>A, and c.8868C>A in the Japanese population [5, 10, 26]. However, we found that the prevalence of EYS mutations in Chinese patients was 7% in our previous study, ranked as the third most common gene detected in this cohort of patients with IRD (n = 896) [7]; the majority of pathogenic defects (56.56%) were missense variants, and the most prevalent variants were c.6416G>A (11.5%), c.6557G>A (9.8%), and c.7492G>C (9.0%). Only one allele of c.4957_4958insA, c.2528G>A, and c.8868C>A was found in our cohort. These results indicate a different founder mutation of EYS in our population. Of special interest, we found for the first time that late-truncating pathogenic variants are significantly more prevalent (41.30%) in the EYS gene, and 36.84% of point mutations occurred at the base G site. The interpretation of these findings is challenging, which might suggest a site-specific effect, or this variability may be related to underlying genetic variants. Further investigation of these mechanisms should promote better understanding.
Of the 55 patients, 53 (96.36%) were diagnosed with RP, 2 (3.64%) had LCA, and no CRD phenotype was found. We assumed that EYS-associated CRD was rare in the Chinese population. In a recent study of a Japanese cohort, the median age of symptom onset was 19 years (range, 14–62 years) [5], while in a Canadian cohort, the median age of symptom onset was 21 years (range, 1–65 years) [23]. In our cohort, the median age of symptom onset was younger than in the previous two reports (mean 15.65 ± 14.67, median 12, range 0–62 years). Consistent with previous studies, age of symptom onset had no significant correlation with genotype [5, 23], suggesting the involvement of other modifying genes. The mean BCVA was 0.73 ± 0.93, and 61.8% of eyes had a BCVA better than 0.52 logMAR. These results would place EYS-RD in the milder spectrum of disease. To date, limited data have documented factors affecting BCVA. Our study showed that longer duration was associated with poorer BCVA, but BCVA showed no significant associations with age, age of symptom onset, MD, retinal hyperpigmentation, or genotype. Unlike BCVA, MD showed a significant correlation with genotype and age but not with disease duration or age of symptom onset. One possible explanation is that aqueous flare is increased and retinal degeneration progresses in older age, especially in RP patients [27,28,29]. Aqueous flares were negatively correlated with the residual VF area, but no correlation was detected with BCVA [28, 29]. For this reason, severe genotype and older age were associated with severe VF defects but not with BCVA. Although our study did not investigate aqueous flares, understanding the predictive factors associated with visual acuity (VA) and VF may help ophthalmologists or geneticists manage patients’ expectations and provide more accurate genetic counseling. Moreover, these data indicate that gene, stem cell, or drug therapy should be performed as soon as possible in the course of the disease.
Posterior subcapsular cataract occurs in approximately 45% of RP patients [30, 31]. The underlying mechanism is currently unknown, although a possible association with inflammation was proposed [32]. In this study, we found that cataracts were observed in 56.45% of EYS-RD patients, and these patients were distributed in all age groups (range 8–74, mean 43.76 ± 16.46 years). This finding is consistent with prior findings in a Japanese cohort, which revealed that cataracts were observed in seven of the ten affected subjects [9]. These results suggest that the occurrence of cataracts in patients with RP may be associated with genetic predisposition. However, there were no significant associations of cataracts with genotype or disease duration, but cataracts showed a significant correlation with age. This can also be explained by the fact that the inflammatory response increased with age, which is a significant risk factor for cataract formation [32]. One of the characteristic fundoscopic features of RP is intraretinal pigment (bone spicule) migration in regions of photoreceptor degeneration. However, the absence or scarcity of typical RP-related hyperpigmentation has also been described in several RP subtypes [33]. The reason for this is unclear, although myopic degeneration may be a factor in some patients [34]. In this analysis, 42.59% of subjects showed an absence of pigmentation, and they were younger than those who showed hyperpigmentation. Correlation analysis revealed that age was a risk factor for hyperpigmentation, and older age was associated with pigment formation. Takahashi et al. [35] reported that as the age of RP patients increased, there was a higher prevalence of pigment migration, reaching over 95% after 40 years of age [35]. Therefore, as age increases, some patients who do not present pigments may develop pigments. However, there is another possibility that no pigment will be observed in their lifetime due to the relatively milder pathogenicity of EYS.
We are aware that the present study has several limitations. First, our study participants were enrolled from a single tertiary referral center (Eye and ENT Hospital of Fudan University), and this was a cross-sectional study. Multicentre and longitudinal follow-up studies may be conducted in the near future. Second, due to the rare nature of EYS-RD, only 55 individuals were available for inclusion, and this small sample size limits the statistical significance of our results. Additionally, age at symptom onset is a subjective measure, and it is difficult to obtain the exact values.
In conclusion, we explored the clinical and genetic characteristics of EYS-RD in a Chinese cohort. Three hot mutations, 36 novel variants, and two unreported mutational features of EYS were identified. Baseline values and correlations of these clinical data (age, age of onset, disease duration time, BCVA, MD, cataract, and pigment change in the retina) were uncovered. To the best of our knowledge, this is the largest clinical and genetic analysis of EYS-RD in the Chinese population. Our data will serve as a well-founded reference for genetic counseling, better management of these patients, and putative therapeutic approaches.
Summary table
What was known before
-
To date, certain efforts have been made to investigate the clinical and genetic characteristics of patients with EYS mutations. However, data for Chinese patients are limited.
What this study adds
-
Our study largely enhances the current knowledge of phenotypic and genotypic characteristics of EYS-RD in a cohort of Chinese patients, which could pave the way for better management of these patients.
Data availability
All pathogenic variants identified in this study are provided as supplementary materials; further data that support the findings of this study are available from the corresponding author on reasonable request.
References
Abd El-Aziz MM, Barragan I, O’Driscoll CA, Goodstadt L, Prigmore E, Borrego S, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 2008;40:1285–7.
Collin RW, Littink KW, Klevering BJ, van den Born LI, Koenekoop RK, Zonneveld MN, et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet. 2008;83:594–603.
Lu Z, Hu X, Liu F, Soares DC, Liu X, Yu S, et al. Ablation of EYS in zebrafish causes mislocalisation of outer segment proteins, F-actin disruption and cone-rod dystrophy. Sci Rep. 2017;7:46098.
Muriël M, Margo D, Sanne B, Theo AP, Julio CC, Ralph WNS, et al. Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. Plos one. 2018;13:e0200789.
Yang L, Fujinami K, Ueno S, Kuniyoshi K, Hayashi T, Kondo M, et al. Genetic spectrum of EYS-associated retinal disease in a large Japanese cohort: identification of disease-associated variants with relatively high allele frequency. Sci Rep. 2020;10:5497.
Pierrache LHM, Messchaert M, Thiadens A, Haer-Wigman L, de Jong-Hesse Y, van Zelst-Stams WAG, et al. Extending the spectrum of EYS-associated retinal disease to macular dystrophy. Invest Ophthalmol Vis Sci. 2019;60:2049–63.
Gao FJ, Li JK, Chen H, Hu FY, Zhang SH, Qi YH, et al. Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology. 2019;126:1549–56.
Vaclavik V, Munier FL, Schorderet DF, Tran HV. Mutation in EYS gene causes variable expressivity, ranging from Leber Congenital Amaurosis to adult onset Retinitis Pimentosa. Autofluorescence Study. Invest Ophthalmol Vis Sci. 2015;56.
Suto K, Hosono K, Takahashi M, Hirami Y, Arai Y, Nagase Y, et al. Clinical phenotype in ten unrelated Japanese patients with mutations in the EYS gene. Ophthalmic Genet. 2014;35:25–34.
Iwanami M, Oshikawa M, Nishida T, Nakadomari S, Kato S. High prevalence of mutations in the EYS gene in Japanese patients with autosomal recessive retinitis pigmentosa. Investigative Ophthalmol Vis Sci. 2012;53:1033–40.
Hosono K, Ishigami C, Takahashi M, Park DH, Hirami Y, Nakanishi H, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS ONE 2012;7.
Gu S, Tian YY, Chen X, Zhao C. Targeted next-generation sequencing extends the phenotypic and mutational spectrums for EYS mutations. Mol Vis. 2016;22:646–57.
Wang DD, Gao FJ, Li JK, Chen F, Hu FY, Xu GZ, et al. Clinical and genetic characteristics of chinese patients with occult macular dystrophy. Invest. Ophthalmol Vis Sci. 2020;61:10.
Hu FY, Li JK, Gao FJ, Qi YH, Xu P, Zhang YJ, et al. ABCA4 gene screening in a Chinese cohort with Stargardt disease: identification of 37 novel variants. Front Genet. 2019;10:773.
Gao FJ, Qi YH, Hu FY, Wang DD, Xu P, Guo JL, et al. Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy. Br J Ophthalmol. 2019;104:846–51.
Gao FJ, Hu FY, Xu P, Qi YH, Li JK, Zhang YJ, et al. Expanding the clinical and genetic spectrum of Heimler syndrome. Orphanet J Rare Dis. 2019;14:290.
Fujinami K, Lois N, Davidson AE, Mackay DS, Hogg CR, Stone EM, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol. 2013;155:1075–88.
Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology. 2015;122:326–34.
Kong X, Fujinami K, Strauss RW, Munoz B, West SK, Cideciyan AV, et al. Visual acuity change over 24 months and its association with foveal phenotype and genotype in individuals with Stargardt disease: ProgStar Study Report No. 10. JAMA Ophthalmol. 2018;136:920–8.
Georgiou M, Kane T, Tanna P, Bouzia Z, Singh N, Kalitzeos A, et al. Prospective cohort study of childhood-onset Stargardt disease: fundus autofluorescence imaging, progression, comparison with adult-onset disease, and disease symmetry. Am J Ophthalmol. 2020;211:159–75.
Audo I, Sahel JA, Mohand-Said S, Lancelot ME, Antonio A, Moskova-Doumanova V, et al. EYS Is a major gene for rod-cone dystrophies in France. Hum Mutat. 2010;31:E1406–35.
Arai Y, Maeda A, Hirami Y, Ishigami C, Kosugi S, Mandai M, et al. Retinitis pigmentosa with EYS mutations is the most prevalent inherited retinal dystrophy in Japanese populations. J Ophthalmol. 2015;2015:819760.
Littink KW, van den Born LI, Koenekoop RK, Collin RWJ, Zonneveld MN, Blokland EAW, et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology. 2010;117:2026–33.
Barragan I, Borrego S, Pieras JI, Gonzalez-del Pozo M, Santoyo J, Ayuso C, et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Mutat. 2010;31:E1772–E1800.
Bandah-Rozenfeld D, Littink KW, Ben-Yosef T, Strom TM, Chowers I, Collin RWJ, et al. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Invest Ophthalmol Vis Sci. 2010;51:4387–94.
Iwanami M, Oishi A, Ogino K, Seko Y, Nishida-Shimizu T, Yoshimura N, et al. Five major sequence variants and copy number variants in the EYS gene account for one-third of Japanese patients with autosomal recessive and simplex retinitis pigmentosa. Mol Vis. 2019;25:766–79.
Yoshida N, Ikeda Y, Notomi S, Ishikawa K, Murakami Y, Hisatomi T, et al. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology. 2013;120:100–5.
Nishiguchi KM, Yokoyama Y, Kunikata H, Abe T, Nakazawa T. Correlation between aqueous flare and residual visual field area in retinitis pigmentosa. Br J Ophthalmol. 2019;103:475–80.
Murakami Y, Yoshida N, Ikeda Y, Nakatake S, Fujiwara K, Notomi S, et al. Relationship between aqueous flare and visual function in retinitis pigmentosa. Am J Ophthalmol. 2015;159:958–63.
Auffarth GU, Nimsgern C, Tetz MR, Krastel H, Volcker HE. Increased cataract rate and characteristics of Nd:YAG laser capsulotomy in retinitis pigmentosa. Ophthalmologe. 1997;94:791–5.
Fagerholm PP, Philipson BT. Cataract in retinitis pigmentosa. Anal Cataract Surg Acta Ophthalmol (Copenh). 1985;63:50–58.
Fujiwara K, Ikeda Y, Murakami Y, Funatsu J, Nakatake S, Tachibana T, et al. Risk factors for posterior subcapsular cataract in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2017;58:2534–7.
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86.
Bandah-Rozenfeld D, Mizrahi-Meissonnier L, Farhy C, Obolensky A, Chowers I, Pe’er J, et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:382–91.
Takahashi VKL, Takiuti JT, Jauregui R, Mahajan VB, Tsang SH. Rates of bone spicule pigment appearance in patients with retinitis pigmentosa sine pigmento. Am J Ophthalmol. 2018;195:176–80.
Acknowledgements
This study is supported by the National Natural Science Foundation of China (Grant NSFC81770925, 81870670, 82101149), Shanghai Clinical Research Plan of SHDC (SHDC2020CR2041B), Shanghai Municipal Science and Technology major projects (2018SHZDZX05), Outstanding Academic Leaders in Shanghai (20XD1401100), Program for Outstanding Medical Academic Leader (2019LJ01), Aging and Women’s and Children’s Health Special Project of Shanghai Municipal Health Commission (2020YJZX0102), Shanghai Clinical Medical Center of Ocular Disease (2017ZZ01020), Shanghai Committee of Science and Technology (18411965100), and The Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018PT32019).
Author information
Authors and Affiliations
Contributions
J-HW and G-ZX conceived and designed the experiments. WL, QC, PX, J-HW, F-JG, and D-DW collected the clinical samples. F-JG, J-HW, F-YH, J-KL, and D-DW analyzed sequencing data. G-ZX, WL, and F-JG recruited patients, performed clinical examination of patients, and clinical interpretation. F-JG, J-KL, and J-HW analyzed and interpreted the data. F-JG and J-HW drafted and revised the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study adhered to the tenets of the Declaration of Helsinki, and was approved by the Ethics Committee of the Eye and ENT Hospital of Fudan University.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41433_2021_1794_MOESM2_ESM.jpg
Supplementary Figure 2 - Sanger sequencing or polymerase chain reaction results of partial EYS variants identified in this study
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, FJ., Wang, DD., Hu, FY. et al. Genotypic spectrum and phenotype correlations of EYS-associated disease in a Chinese cohort. Eye 36, 2122–2129 (2022). https://doi.org/10.1038/s41433-021-01794-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01794-6

{kind=link}
{kind=link}
{kind=link}