2.1. Experimental Procedures
From an experimental perspective, the interaction of the ligand and the macromolecule can be characterized by means of the isothermal titration calorimetry (ITC) technique. Small molecules are bound to macromolecules by four binding modes: Van der Waals, hydrogen bonds, electrostatic, and hydrophobic forces [
25]. According to enthalpy (ΔH°) and entropy (ΔS°) changes, the interaction type can be determined. For ΔH° and ΔS° > 0, the main interaction mode is led by hydrophobic forces; for ΔH° and ΔS° < 0, the main responsible forces are Van der Waals interactions and hydrogen bonds, and for ΔH° < 0 and ΔS° > 0, electrostatic forces [
26].
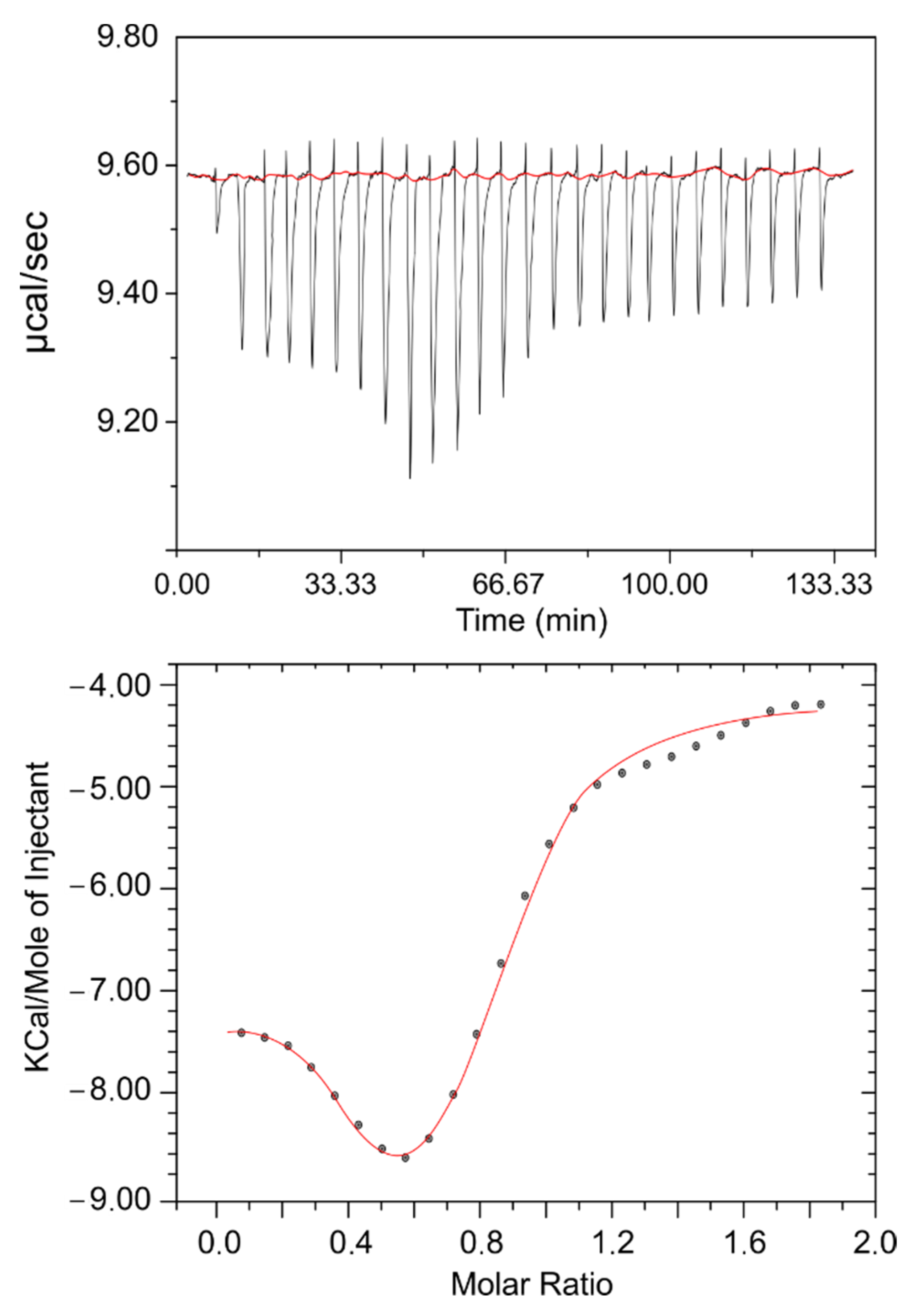
Typically, in this type of test, a protein is inserted in the calorimetric cell and a ligand is injected through a syringe needle at the desired rate. In the present case, the initial concentrations used were 0.02 mM for the protein and 0.5 mM for the drug, assuring a final ratio high enough to reach saturation.
Figure 1 shows the results relating the amount of heat released per injection and the molar ratio (drug/protein). At first glance, a few simple deductions can initially be made: an interaction exists; it is exothermic and gradually decreases as the drug/protein ratio increases; and saturation is found to be reached at molar ratios higher than one. With the designed experimental configuration, equilibrium is roughly reached after 100 min. For further analysis, the thermodynamic parameters were calculated using the Van ‘t Hoff equation:
where R is the molar gas constant, T is the experimental temperature, and K is the association constant.
Data were fitted to a single-site binding model. The binding stoichiometry (n), binding constant (K), enthalpy change (ΔH°), and entropy change (ΔS°) were obtained from the results of the fitting and the values obtained were as follows: n = 0.95 ± 0.14; K = (3.82 ± 0.79) × 10
5 M
−1; ΔH° = −113.8 ± 24.1 kJ mol
−1; and ΔS° = −0.29 ± 0.07 kJ mol
−1 K
−1. The stoichiometry reveals that only one type of binding takes place, while the binding constant denotes that the BTS is a rather moderate binder. For its part, negative values of entropy and enthalpy changes indicate that the hydrogen bonding and Van der Waals forces play a major role in the interaction between the drug and the biomacromolecule, suggesting a loss of conformational freedom related to the formation of the complex [
27].
When BTS binds to some of the proteins under study, it acquires an induced circular dichroism (CD) spectrum through chiral perturbation to its structure or electron rearrangements. The intensity of the CD spectrum is determined by the strength of the interactions and both the geometry of the BTS molecule and the residue in the protein backbone. Thus, the CD can be used to probe the binding of BTS molecules to the proteins under study, providing information about the secondary structure of the protein.
Table 1 shows the changes in the α-helices and β-sheets content of lysozyme at different BTS concentrations [
21]. In general, the presence of BTS does not produce major changes in the protein structure and it can be seen that the β-sheets content is always greater than the α-helices content. α-helices are more sensitive to the presence of BTS, decreasing their content by 37% as the concentration of the drug increases. On the other hand, β-sheets only decrease their content by 17%. These results show that at low concentrations, BTS does not induce important changes in the structure of lysozyme that could alter its physiological behavior.
UV-visible absorption spectroscopy is a very useful tool for assessing conformational alterations in protein-ligand systems. Analyzing the spectral curves under different conditions, the formation of a complex between the biomacromolecule and the drug can be decoded, as well as give insight into the consequential structural changes induced. In the case of lysozyme, the main aromatic amino acid residues that are responsible for the absorption peak at 280 nm are tyrosine, phenylalanine, and tryptophan, with tryptophan being the major contributor in the spectra [
14].
Figure 2A shows the spectral curves of pure lysozyme and different systems of protein-ligand with increasing concentrations of BTS. In this case, the peak at 280 nm slightly decreases with the addition of BTS with no obvious drug shift, suggesting an increase in the hydrophobicity of the microenvironment around the aromatic amino acids of the biomacromolecule [
28].
To obtain further information about the drug-protein interactions, fluorescence measurements were conducted. From previous studies [
29,
30] it is known that lysozyme presents a moderately rigid structure, which includes α-helices, β-sheets, and turns and loops. It is composed of 129 amino acid residues, including six Trp, four disulfide bridges, and three Tyr residues. The fluorophores with a major contribution are Trp-62 and Trp-108, both of them situated at the substrate-binding sites [
31]. Usually, protein fluorescence is excited at wavelengths close to 280 nm or higher. The absorption at this maximum is caused by both Trp and Tyr, while at 300 nm or longer, the protein adsorption is essentially due to Trp. The reduction in the quantum yield of fluorescence from a fluorophore is known as quenching. It can be caused by a series of molecular interactions with a quencher molecule, such as energy transfer, excited-state reactions, ground-state complex formation, molecular rearrangements, and collisional quenching processes [
32]. From the analysis of the spectra, some valuable information on the binding of proteins to simple molecules or even to small particles can be obtained [
33]. In this work, the binding mechanism, constant, binding site, and intermolecular distance were obtained by analyzing the intrinsic fluorescence of pure lysozyme with the addition of different concentrations of the BTS drug. Preliminary experiments were performed to remove any possible imprecision provoked by the existence of inner filter effects. This phenomenon appears when the ligand causes absorption during excitation and emission radiation processes [
28]. In this study, the inner filter effect was corrected following the procedure explained in the Materials and Methods section.
Figure 2B pictures the emission spectra of lysozyme in the presence of increasing concentrations of BTS. As it can be observed, when excited with a wavelength of 280 nm, the protein alone presents a great emission band at 340 nm which is progressively reduced with no shift as the drug is added. This fact signifies the beginning of saturation of the lysozyme binding sites [
30] and, at the same time, indicates that the microenvironment of tryptophan is relatively less hydrophobic upon binding with the drug.
For studying the fluorescence quenching, data is typically analyzed through the well-known Stern–Volmer equation which can classify the type of fluorescence quenching process as static or dynamic:
where F
0 and F are the steady-state fluorescence intensities before and after the addition of the quencher, K
sv is the Stern–Volmer quenching constant, [Q] corresponds to the concentration of the quencher (in this case, BTS), k
q is the quenching rate constant of the protein, and τ
0 is the average lifetime of the biomolecule, taken as 1.8 × 10
−9 s for lyz [
34].
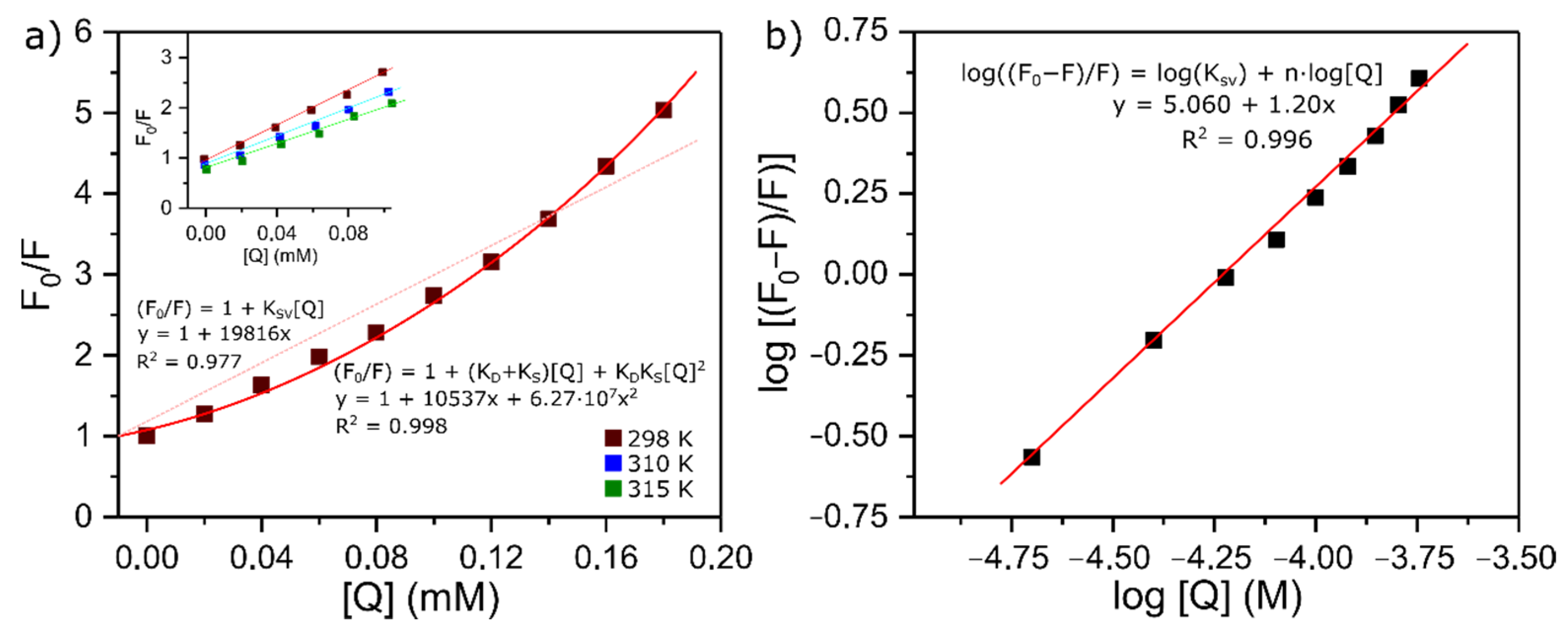
A Stern–Volmer plot of F
0/F against [Q] is linear for purely dynamic (K = 0) or purely static (k = 0) quenching, but curved upwards for combined quenching (eqn (3)). On this basis, a curvature of a Stern–Volmer plot is often taken to indicate combined quenching. The plot obtained for this case is shown in
Figure 3a. It deviates significantly from linearity, which indicates that there is a coexistence of interactions occurring between lysozyme and BTS when the protein is in its ground electronic state and in its first excited single state: static and dynamic quenching, respectively [
34]. Taking into consideration that the intensity ratio F
0/F is given by the product of static and dynamic quenching, Equation (2) would be more accurate as follows:
where K
S and K
D represent the dynamic and static quenching constants. As it can be seen in
Figure 3a, at high concentrations of the quencher, the Stern–Volmer plot follows a continuous ascending curve. In this case, a quadratic least square fit was found to be adequate, with a correlation constant very close to 1 (R
2 = 0.998). However, in the insert are the results for low concentrations of the drug, ranging from 2.0 × 10
−5 to 1.0 × 10
−4 L mol
−1, and it can be observed that they present a good linear relationship (R
2 ≥ 0.98). The K
SV obtained from the slope at a low concentration and 288 K was (1.66 ± 0.01) × 10
−4 L mol
−1 and it descends with an increase in temperature. Therefore, it can be inferred that the interaction between lysozyme and BTS is a static quenching process resulting from the formation of a lysozyme-BTS complex.
This assumption can be further confirmed from the calculated value of k
q. In the present case, this constant has a value of 9.22 × 10
12 L M
−1 s
−1 and is much greater than the value of the highest scatter collision quenching constant (2.0 × 10
10 M
−1 s
−1) for different ligands with proteins [
35]. Gathering this information, it is safe to assume that, at low concentrations, the probable quenching mechanism involves a complex formation, but the upwards curvature shown in
Figure 3a suggests that at higher concentrations the quenching process is a result from a combined one.
Apart from the mechanisms involved, fluorescence measurements are also useful to get quantitative information about the binding. It has been previously demonstrated that if there are independent and similar binding sites in the biomolecule, both the number of binding sites, n, and the binding constant, K
A, can be determined using the following equation [
36]:
The number of binding sites per molecule of lysozyme and the binding constant can be determined by the slope and the intercept of the double logarithm regression curves (
Figure 3b). Linear fitting of experimental data gives values of (1.20 ± 0.14) and (1.15 ± 0.06) × 10
5 M
−1 for n and K
A, respectively. Such results indicate that there is only one independent class of binding sites between the protein and the ligand, and the binder has a reasonable affinity for the protein, reinforcing the conclusions previously drawn from the isothermal titration calorimetry assays.
A more complete description of the interaction can be obtained by the analysis of the energy transfer, which can be used to find the distance between the tryptophan residues in the protein. According to the well-known Förster’s nonradioactive energy transfer theory (FRET), the energy transfer takes place when the fluorescence emission spectrum of the donor and UV-vis absorbance spectrum of the acceptor overlap considerably and the distance between donor and receptor is less than 8 nm [
37,
38]. In the present case, the donor is lysozyme and acceptor is BTS, and there is a significant overlap between emission and absorption spectra. Based on Förster’s theory, the efficiency of energy transfer (E) can be related to the distance between the donor and acceptor (r) as follows:
where F and F
0 are the fluorescence intensities of the protein in the presence and absence of BTS, respectively, and R
0 is the critical distance when the energy transfer efficiency is 50%. This value can be calculated as follows:
where K
2 is the spatial orientation factor of the dipole, n is the refractive index of the medium, Φ is the fluorescence quantum yield of the donor, and J is the effect of the overlap between the emission spectrum of lysozyme and absorption spectrum of BTS. The dipole orientation factor is the most imprecise value when calculating R
0. Its value can theoretically be within the range of 0 to 4, but assuming that both the protein and the ligand are tumbling rapidly and free to take any orientation, then K
2 equals 2/3. J, for its part, can be obtained by the equation:
where F(λ) is the fluorescence intensity of lysozyme at wavelength λ and ε(λ) is the molar absorption coefficient of BTS at wavelength λ. Solving the respective equations with the collected experimental data, the values of E, J, and R
0 can be calculated, and consequentially, the distance between the donor and acceptor (r), can be determined as well.
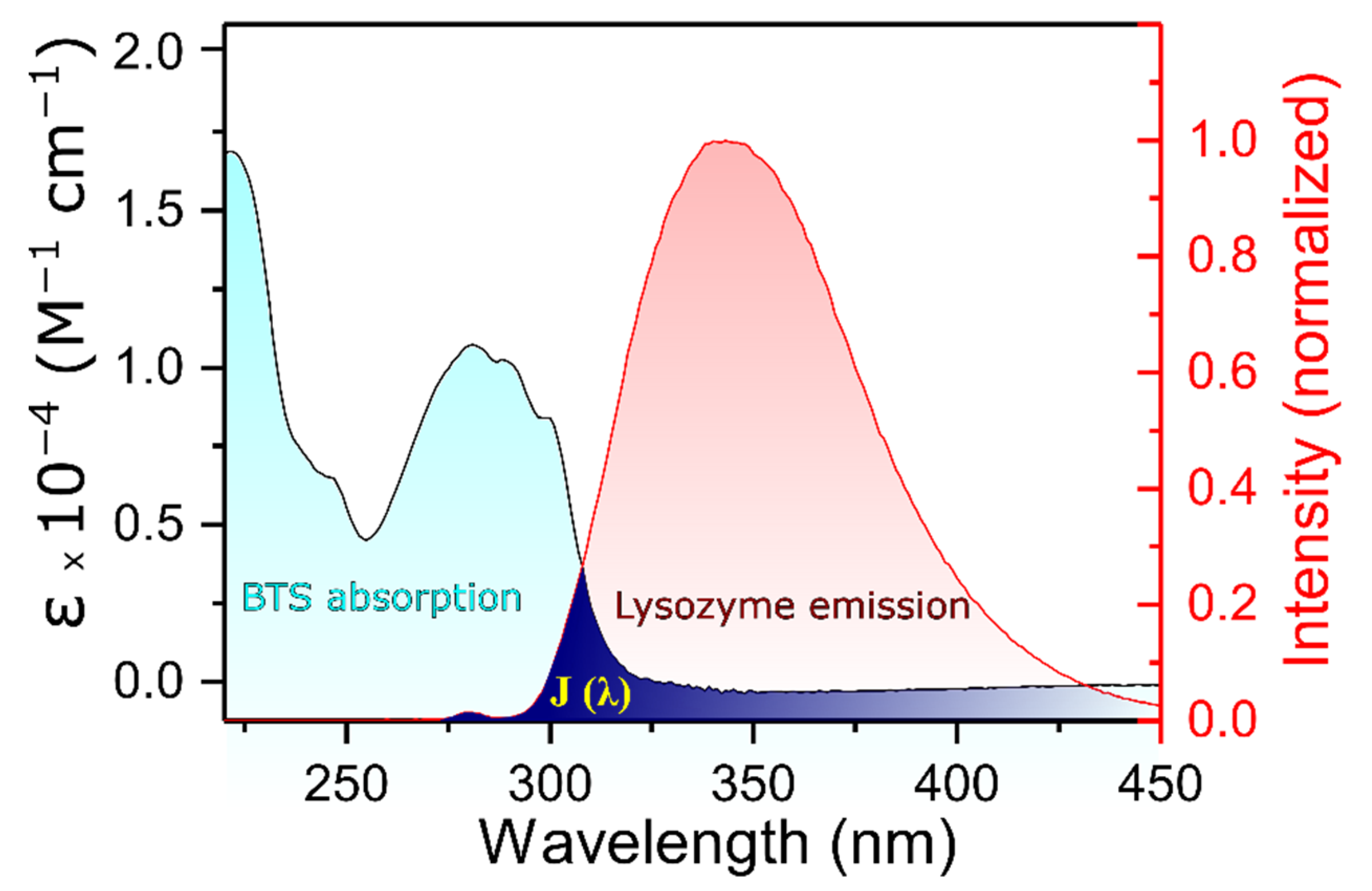
Figure 4 shows the spectral overlap J when the molar ratio of the acceptor and donor is 1:1 and at 298 K. The value obtained for the integral was calculated to be equal to 1.26 ×10
13 M
−1 cm
−1 nm
4. Taking into consideration that the corresponding values of the parameters used were K
2 = 2/3, Φ = 0.15, and n = 1.36 [
39], R
0 and r can be obtained by combining Equations (5) and (6). In this case, E = 0.21, R
0 = 1.78 nm, and r = 2.22 nm, which means that the distance between the donor and acceptor (r) is lower than the maximal academic critical distance for R
0 (5–10 nm) and is within the range of 2–8 nm. Furthermore, 0.5 R
0 < r < 1.5 R
0, clearly suggesting that the energy transfer from lysozyme to the drug occurs with a high probability, again reinforcing the idea that the quenching mechanism involved corresponds to a static one [
30].
2.2. Computational Procedures
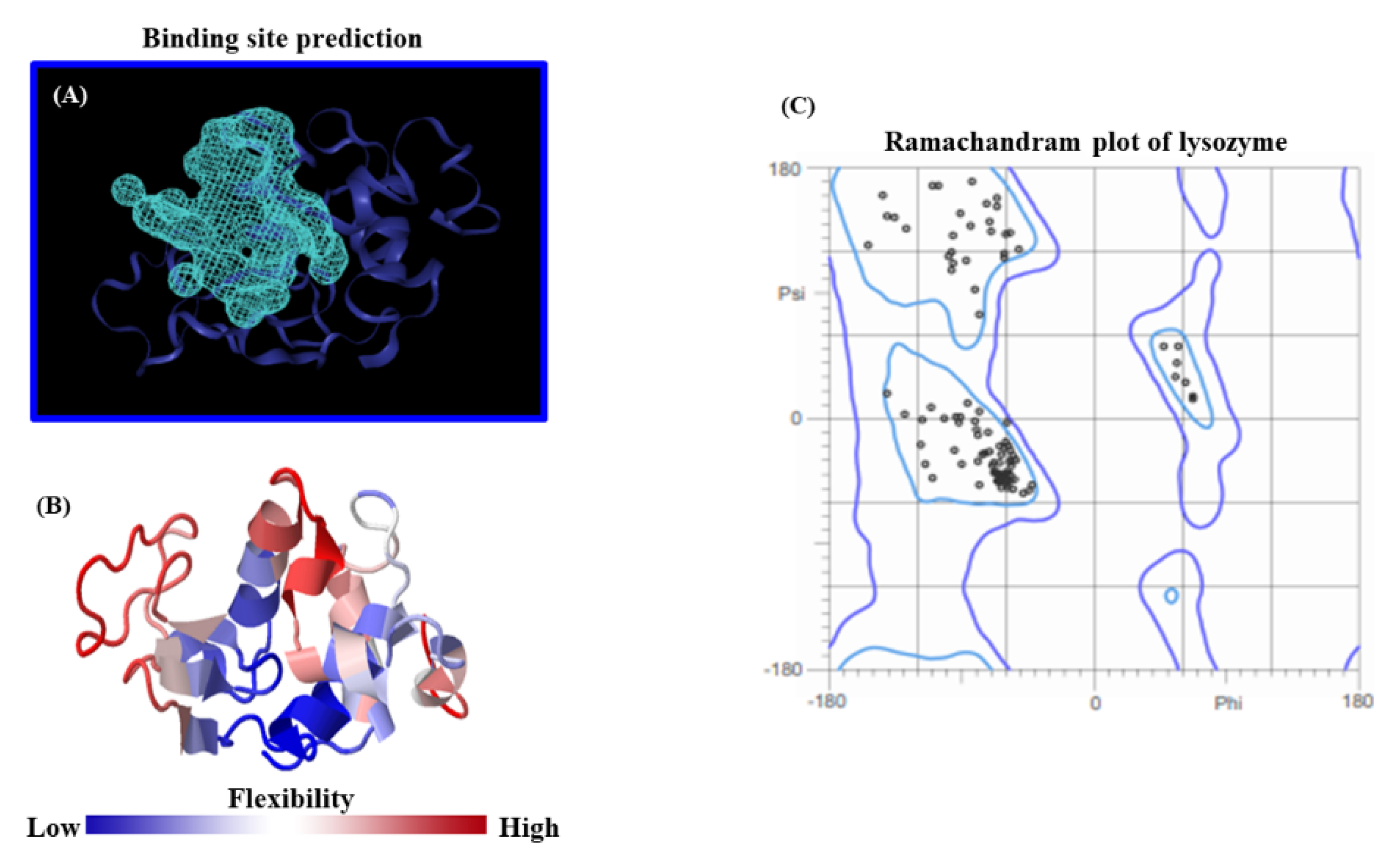
One of the most critical steps to ensuring the quality of modeling results is the accurate prediction of feasible lysozyme binding-sites, coupled with an appropriate crystallographic structural validation of this receptor, and its flexibility properties. Herein, the search space was set up for the lysozyme receptor by using the ezPocket tool with the Deepsite validation procedure [
40,
41,
42], as a cubic grid box with a size of 20 × 20 × 20 Å
3, centered at X = 3.27Å, Y = 24.19Å, Z = 27.15Å, with a volume equal to 431.26 Å, and a discretization of 0.25 Å. The identification of the best-ranked lysozyme binding site was performed according to the details in the Methods section. Furthermore, the lysozyme flexibility properties from the 3D-molecular structure were evaluated from the whole lysozyme 3D-structure together with the corresponding Ramachandran crystallographic validation as depicted in
Figure 5.
As can be seen in
Figure 5, the accuracy of our theoretical docking approach was ensured by performing a proper prediction of the lysozyme binding-site, which suggests the best druggability for the BTS-ligand binding. In the present study, the prediction of the lysozyme-active binding site was performed using an
ezPocket tool with a maximum score equal to 1.0 for the lysozyme. This binding pocket prediction method works with a consensus algorithm [
40,
41,
42] which uses Delaunay triangulation to detect plausible lysozyme binding pockets, typically taking a few seconds and providing accurate Cartesian
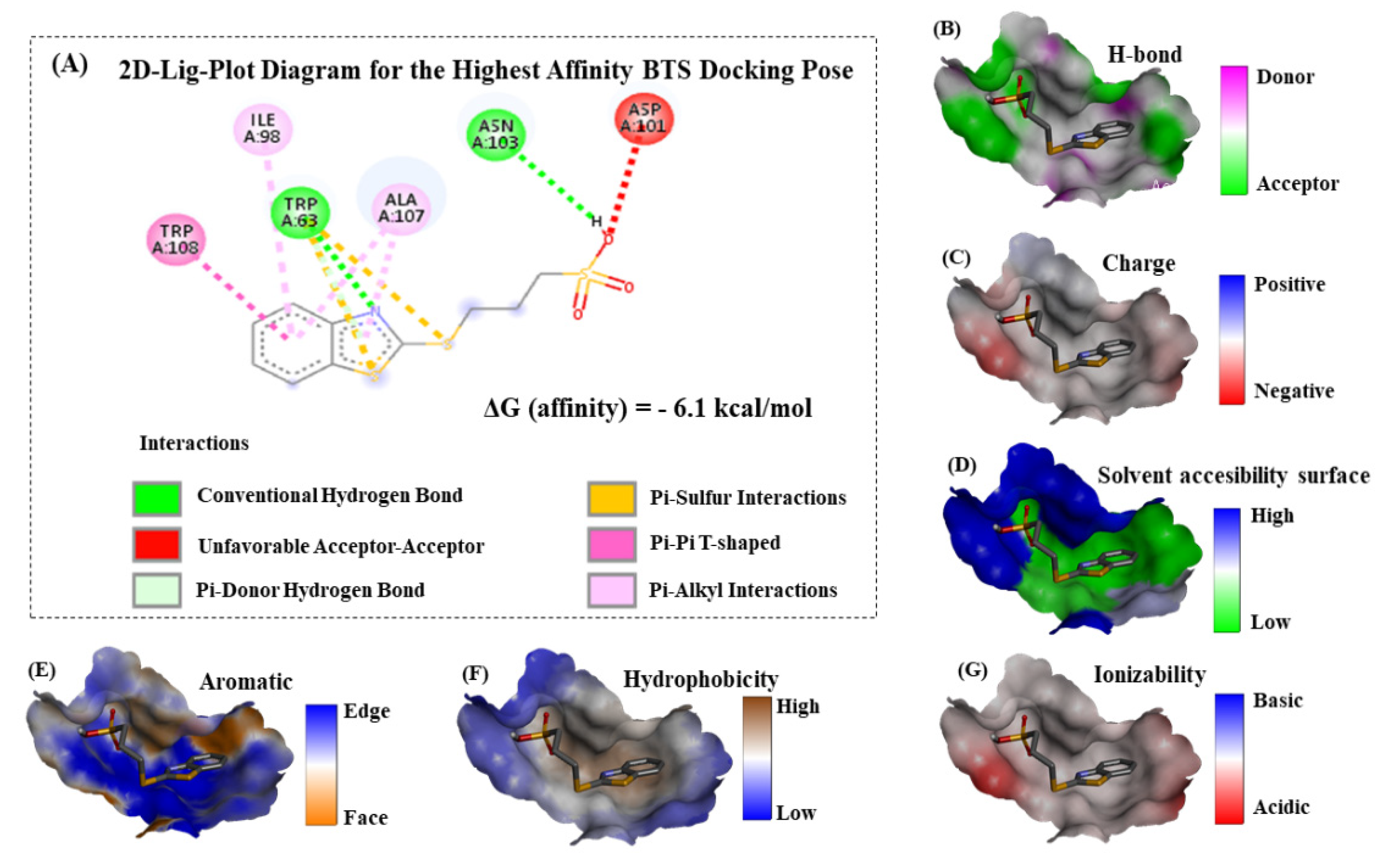
XYZ-coordinates for the docking box simulations. In this context, this computational task acquires a key relevance for exploring the molecular mechanisms of BTS interaction. For this study, the best-ranked binding-conformation that BTS adopts into the lysozyme-binding site was considered the one with the highest affinity according to the more negative values for the Gibbs free energy of binding (FEB) or ΔG (docking affinity). To explain these theoretical results, the 2D-Lig-Plot diagram for the docking complex formed by BTS-lysozyme can provide structural and mechanistic information on the simultaneous modulation of several lysozyme target-residues (i.e., effector and allosteric binding residues). This is accomplished by determining the contribution of the different types of non-covalent docking interactions and thermodynamics factors involved in the binding events, such as hydrophobic, H-bond, electrostatics, and aromatic Π-Π stacking interactions with its corresponding interatomic distances (
dij). For the best-ranked BTS binding-pose obtained from the BTS-lysozyme docking complex [
43], see
Figure 6.
According to the obtained results, we theoretically suggest that the BTS-ligand was able to interact in the predicted binding site placed in the middle of the lysozyme receptor following a spontaneous thermodynamics process according to the negative FEB values of binding ΔG (docking affinity) = −6.1 kcal mol−1 (or expressed in joules as 25.50 kJ mol−1). If we return to the ITC experimental results, we found a stoichiometry of one and a free energy of 26.58 kJ mol−1. It can be seen that both results support the theoretical predictions (in fact, the free energy quantities are really close), which reinforces the validity and quality of the results.
Particularly, we centered our attention on the fact that all the interactions detected correspond to non-covalent binding events. Herein, in order to tackle the different key contributions explaining the molecular mechanisms of interaction with lysozyme (see
Figure 6A), we firstly identified a prevalence of aromatic hydrophobic Van der Waals interactions followed by conventional hydrogen bond interactions between the evaluated BTS-ligand and the lysozyme; namely, we had postulated on the basis of the thermodynamic values obtained by ITC. The aromatic pi-pi stacking edge-to-face interactions (a T-shaped fashion) are between the benzene ring belonging to the 2-(methylthio)-benzothiazole moiety of the BTS and the aromatic amino acid residue TRP108 of the lysozyme receptor. This type of interaction plays an important role in the stabilization of the BTS-lysozyme docking complex and was also verified by UV-vis and fluorescence measurements. Specifically, we identified a slightly displaced face-to-face (displaced pi-pi T-shaped) between the interacting aromatic benzene rings in the docking complex, and this theoretical observation can be corroborated in the Van der Waals map of interaction depicted in
Figure 6E (please, note the presence of orange color locally distributed towards the region of the 2-(methylthio)-benzothiazole moiety in the Van der Waals map of interaction). In addition, these results fit very well with the hydrophobicity Van der Waals interaction map (
Figure 6F) with a trend to express the prevalence of hydrophobic pi-pi T-shaped contacts with the presence of white-to-orange color locally distributed in the hydrophobicity map. Then, it is expected that the presence of the 2-(methylthio)-benzothiazole moiety of BTS in the lysozyme binding site can significantly affect properties such as solvent accessibility surface (
Figure 6D) of the target aromatic amino acid residue TRP108 and its neighboring non-aromatic amino acid residues ALA107 and ILE98. Both interact with the 2-(methylthio)-benzothiazole moiety of BTS through pi-alkyl interactions in the lysozyme binding site (represented as light pink dotted lines in the lig-plot diagram in
Figure 6), which strongly contributes to the stability of the formed docking complex (BTS-lysozyme). Furthermore, note that the pi-alkyl interactions involving the non-aromatic amino acid residue ALA107 is stronger than the ILE98, due to the presence of concomitant pi-donor hydrogen bond interactions with both planar aromatic moieties (benzene ring and heterocyclic 1,3-thiazole) from the 2-(methylthio)-benzothiazole moiety of BTS. In this instance, the pi-alkyl interaction is quite strong over the target residue ALA107 because it is well-known that the presence of concomitant pi-donor hydrogen bond contributes to a larger pi-electron delocalization. Furthermore, we identify the presence of another relevant interaction, as pi-sulfur interaction mixed with a conventional hydrogen bond shared between the lysozyme aromatic target residue (TRP63) with the sulfur group of the 1,3-thiazole moiety from the 2-(methylthio)-benzothiazole moiety of BTS-ligand. In this regard, it is important to note that conventional hydrogen bonds generally contribute to the stabilization of the docking complex (i.e., BTS-lysozyme) and aromatic pi-sulfur interaction buried at a lysozyme’s like TRP63 can affect the interaction energies of ring conformations in the lysozyme and can also modulate specific electron-donating/accepting substitutions on the aromatic ring from both lysozyme and the BTS ligand. Indeed, the presence of two additional interactions in the substitutions of the 2-(methylthio)-benzothiazole moiety of the BTS-ligand were identified in the BTS-lysozyme docking complex which involves electrostatic bonds with neighboring lysozyme target residues such as: (i) a thermodynamically favorable conventional hydrogen bond between the target residue ASN103, and (ii) a thermodynamically unfavorable acceptor-acceptor interaction between the target residue ASP101. Both modulate the electrostatic properties of the oxygen group belonging to the propanesulfonic moiety of the BTS-ligand which at the same time, modulates the binding properties of the 2-(methylthio)-benzothiazole moiety of BTS-ligand in the lysozyme binding site. The coexistence of different interactions could also be guaranteed from an experimental point of view, based on the loss of linearity that we observed in the Stern–Volmer plots (
Figure 3A). In addition, the end result of this balance of interactions is that the complex formed is not excessively stable. The value of the binding constant obtained by ITC defined BTS as a moderate ligand.
The influence of these two last interactions from the electronic point of view can be well corroborated by the presence of a moderate behavior for the analyzed binding properties: acceptor/donor H-bond, charged interactions, and ionizability (as depicted in the
Figure 6B,C,G, respectively), where a predominance of white-to-gray color locally distributed in both is observed, the propanesulfonic moiety of the BTS-ligand and the lysozyme receptor in the corresponding Van der Waals surface maps. Overall, the relevance of these types of interactions and specific orientations between aromatics, electrostatics, and the effect of substitution in the context of a lysozyme’s fold and function under the unbound and bound state is still unexplored and could have paramount significance in rational drug-design targeting lysozyme.
Afterward, we examine how the BTS-ligand could affect the communication efficiency in the inter-residue network of the lysozyme receptor in the unbound and bound state (i.e., in the absence and presence of the BTS-ligand). To this end, we carried out an elastic network approach (ENM models) by regarding the local perturbations induced by the BTS-ligand in the C(α)-residue network that forms the lysozyme binding site [
44,
45,
46]. In this instance, the local perturbations are propagated like an allosteric signal pathway across the communication network composed by C(α)-atom nodes which represent the residues and the network edges representing their mutual contacts connected by the two associated C(α)-residue nodes within a distance of 5.0 Å between C-alpha atoms in the lysozyme receptor structure. Herein, in the ENM model, the lysozyme binding residues composing the allosteric communication network are indexed by a non-ambiguous ordering. Then, using the ENM procedure allows for the visual representation of the strength of allosteric perturbation by generating the matrix of local perturbation response scanning maps from the aforementioned theoretical conditions (unbound and bound state of lysozyme) as depicted in
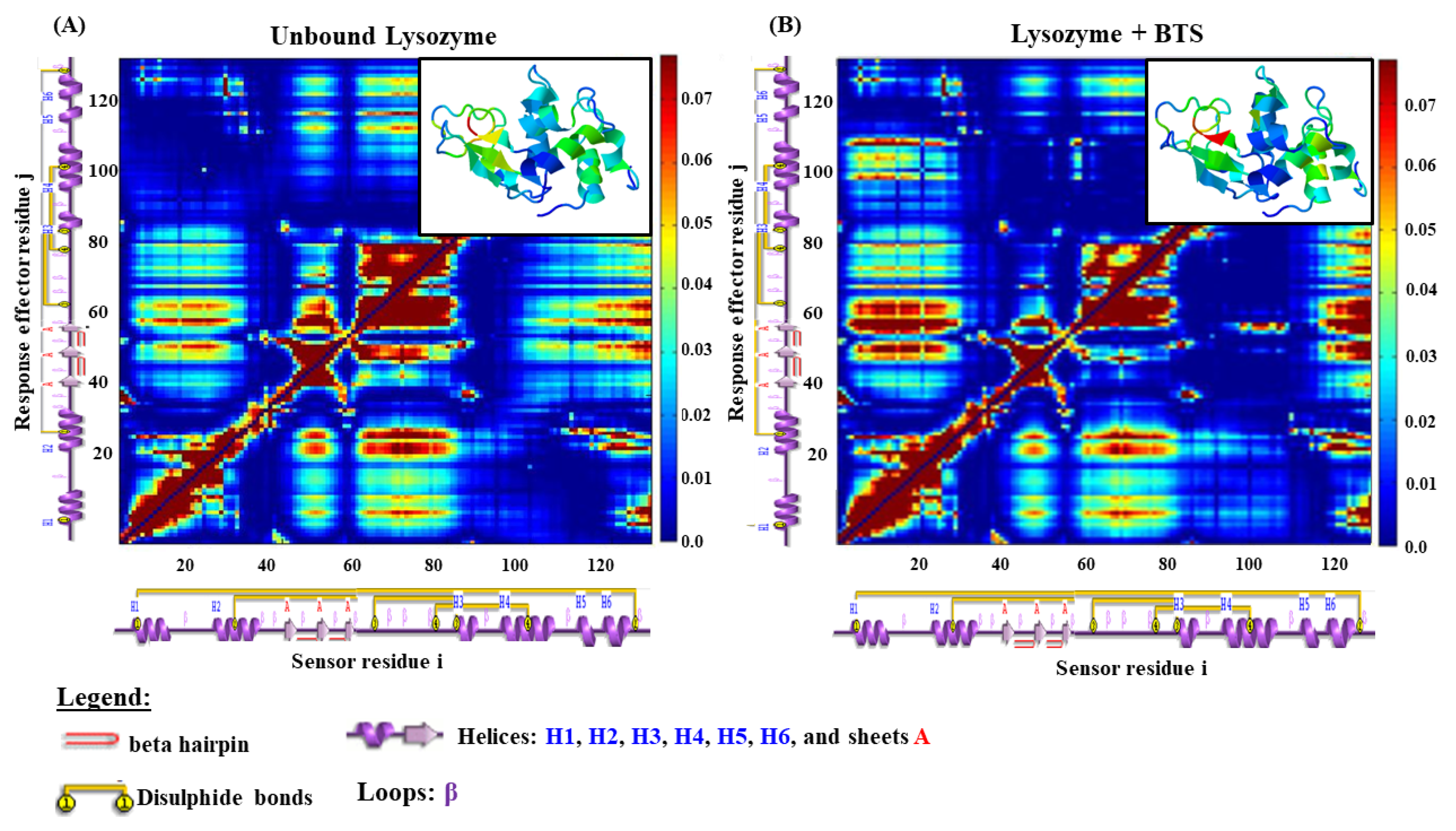
Figure 7.
The obtained LPRS map approaches bring a structural and theoretical vision to the local perturbations induced by the BTS-ligand over the lysozyme receptor in the bound state, which remains unexplored until the present. In this sense, the local perturbations are defined by the collective anisotropic fluctuations generated from the intra-segment C
α-C
α atomic residue distances (labelled by the color intensity in the 2D-perturbation matrix) by the presence of a given ligand (i.e., the best-crystallographic binding pose of the BTS-ligand) which could affect the function and conformational properties of the binding residues of lysozyme. In this context, both the unbound and bound state are fully defined by the local perturbation response 3
N × 3
N Hessian matrix (
Hi,j) which describes the inter-residue allosteric communication pathway in the lysozyme [
44,
45,
46]. Its
N-elements (1 ≤
i, j ≤
N) are the second derivatives of the anisotropic network potential (V
lysozyme or V
lysozyme+ligand) which describes the (3
N)-
XYZ-displacements (
) of all the (
i,
j)-residue pairs sensors (
i) and effectors (
j) in
N-blocks of three dimensions each, and where
γR =
γL = 0.5 is an interaction constant, in the absence (lysozyme unbound state), and upon interactions (or perturbations as lysozyme plus BTS in the bound state) following the Equations (8) and (9):
Then, the anisotropic allosteric perturbations induced by the BTS-ligand in the inter-residue network of the lysozyme were determined using the LPRS maps, mainly focusing on the region of the previously predicted binding site containing around 120 residues. For the case of the control simulation represented by the lysozyme (unbound state), we show the patterns (i.e., theoretical physiological response) generated by coupled pairs of i-sensor residues vs. j-effector residues representing the native structural conformation of the lysozyme like a finely tuned allosteric signal transduction network; this models the catalytic function of the lysozyme binding-site. Herein, labelled-blue regions in the LPRS map denote conformational rigidity for large blocks of consecutive lysozyme i,j-target residues (perturbation direction i-sensor → j-effector residue pair) [
44,
45,
46]. While on the opposite hand, labelled-yellow to dark-red regions in the LPRS map denote conformational flexibilization for large blocks of consecutive lysozyme i,j-target residues (perturbation direction i-sensor → j-effector residue pair).
According to the obtained results, we can clearly identify several differences in the LPRS maps between the unbound (
Figure 7A) and bound state (
Figure 7B) of the lysozyme. Particularly, we note that the first thirty i-sensor residues belonging to the α-helices (H1 and H2 connected by the first β-loop) are structurally and conformationally coupled with j-effector residues which exhibit moderate flexibilization to high conformational rigidity for large blocks of consecutive lysozyme (i-sensor → j-effector residues labelled-yellow to dark-blue color transition regions in the LPRS map of
Figure 7A). This response is released in theoretical physiological conditions (lysozyme unbound state) that can be theoretically explained as physiological modulation or signal allosteric propagation toward j-effector residues belonging to the lysozyme sheets A (i.e., A1, A2, and A3), and also physiological modulation of the lysozyme j-effector residues forming the α-helices (H3, H4, H5, H6, and their corresponding β-loops). When we compare the same block of consecutive lysozyme i-sensor residues (first thirty) under a bound state (i.e.,
Figure 7B as LPRS map under interaction with the BTS-ligand forming the lysozyme-BTS docking complex) we found clear differences in the obtained perturbation response triggered by the lysozyme j-effector residues forming the α-helices (H3, H4, and their corresponding β-loops) which show that the BTS-interaction theoretically induces allosteric signal propagation, which strongly suggests an abnormal conformational flexibilization for large blocks of consecutive lysozyme j-effector residues (perturbation direction i-sensor → j-effector residue pair) at least for the lysozyme j-effector residues belonging to the α-helices (H3, H4, and their corresponding β-loops). Apparently, the BTS interaction does not induce long-distance allosteric perturbation in the lysozyme j-effector residues that form the α-helices H5, H6 and their corresponding β-loops which maintain their conformational properties (i.e., conformational rigidity) like the lysozyme unbound state. Concerning the block of consecutive i-sensor residues placed in the amino-acid sequence position from 60 to 115 (α-helices H3, H4, and their corresponding β-loops), we also detected a presence of conformational allosteric perturbations in the first twenty j-effector residues cluster induced under interaction with the BTS-ligand. These local perturbations were mainly based on moderate conformational flexibilization (labelled-dark yellow region) propagated from the cited i-sensor residues to the first twenty j-effector residues in the α-helices H1, H2, and its corresponding β-loop, in contrast with the LPRS map (unbound state) for the same region of j-effector residues, which presents an intrinsic conformational rigidity (labelled-yellow to dark-blue regions).
This theoretical observation has significant relevance because the block of consecutive lysozyme i-sensor residues placed amino acid sequence position from 60 to 115 of the lysozyme molecular structure includes the key interacting residues previously identified in the 2D-lig-plot analysis (i.e., TRP63, ILE98, ASP101, ASN103, ALA107, and TRP108) which allows for the corroboration and validation of the influence of the non-covalent binding events between the BTS-ligand with the lysozyme receptor.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}