
C,C- and C,N-Chelated Organocopper Compounds


1
Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China
2
Henan Institute of Chemistry Co., Ltd., Henan Academy of Sciences, Zhengzhou 450002, China
3
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(19), 5806; https://doi.org/10.3390/molecules26195806
Submission received: 1 September 2021
/
Revised: 22 September 2021
/
Accepted: 23 September 2021
/
Published: 25 September 2021
(This article belongs to the Special Issue Catalysis and Its Contributions to Society: Theme Issue Honoring Professor Gerard van Koten on His 80th Birthday)
Abstract
:Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure-function relationship of organocopper compounds could advance the rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono-carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize unstable organocopper compounds. Bidentate ligands can chelate to the same copper atom via η2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via η1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. The anionic coordinating sites of the bidentate ligand can also bridge two metals via μ2-mode, forming organocopper aggregates with Cu-Cu interactions and organocuprates with contact ion pair structures. The reaction chemistry of some selected organocopper compounds is highlighted, showing their unique structure–reactivity relationships.
1. Introduction
Copper-mediated or -catalyzed reactions have been upgrading the toolbox of organic synthesis with a variety of cheap, efficient transformations, which benefit scientific research in a broad range of areas, such as in polymer chemistry and biochemistry [1,2,3,4,5,6,7,8,9,10,11,12]. To have a deep understanding of the reaction mechanisms, organocopper species, as proposed intermediates, are sought to be isolated. On the other hand, organocopper compounds can be easily prepared in situ and utilized as organometallic synthons to construct various small organic molecules [13,14,15]. A well-recognized example is that “hard” nucleophiles, e.g., organolithium or Grignard reagents, react with α,β-unsaturated carbonyl compounds via 1,2-addition, whereas the “soft” organocuprates will end up with 1,4-addition products with the same substrates [16,17]. In a catalytic version, in the presence of copper salts, Grignard reagents can behave similarly to organocuprates [18]. It is known that reactivities are closely connected to structural configurations. Organocopper chemistry focuses on establishing the structure–reactivity relationship of well-defined organocopper compounds, rationalizing the dissimilar reaction patterns of different organometallic copper reagents.
Transmetalation provides the most straightforward and reliable method f preparing organocopper compounds from readily available organometallic reagents [19,20], though many other synthetic strategies are also known, such as copper-halide exchange reactions [21], direct cupration [22], and the oxidative addition of reactive Rieke Cu* [23]. It should be emphasized that the identity of forming organocopper compounds is very much dependent on the ratio of starting materials. For example, in the early 19th century, Gilman et al. found that the reaction of methyl lithium and 1.0 equiv. of copper(I) salts formed insoluble methylcopper polymers as yellow solids in diethyl ether [24]. When 0.5 equiv. of copper(I) salts was used in the above condition, a clear solution of dimethyl cuprate (Gilman reagent) was created. The initial isolation and characterizations of organocopper compounds were very slow since the reactive species were subjected to thermally homolytic, oxidative, and hydrolytic decompositions [25]. A general trend is that the stability increases from alkyl, alkenyl, and aryl to alkynyl copper compounds. For example, methylcopper [24] and phenylcopper [26] clusters display rapid and slow decompositions at room temperature, respectively, while (phenylethynyl)copper [27] looks stable under ambient conditions. This stability trend could also be reflected by the numbers of single-crystal structures in the Cambridge Structural Database (CSD 5.42, 2020 November), in which roughly 185 alkyl, 288 aryl, and 326 alkynyl copper compounds have been collected. Moreover, the aggregate state, which is favorable in charge-neutral organocopper compounds, makes these polymeric compounds poorly soluble in common organic solvents. To solve the above inherent problems of organocopper compounds, coordinating heteroatom-containing moieties, such as dimethyl amino groups, were rationally introduced into the ligand skeleton by van Koten and other researches to enhance stability and solubility [28,29]. The hybrid C,N-bidentate ligands can bind to copper atoms with an enhanced chelating effect. Following a different path, Xi’s group discovered that 1,4-dilithio 1,3-butadienes (C,C-bidentate ligand) can stabilize reactive organocopper species to a great extent via a cooperative effect [30,31]. In addition, this C,C-bidentate ligand can also be regarded as an analogue of 1,2-diketimine, which turns out to be a special Z-type non-innocent ligand, producing an unprecedented aromatic dicupro-annulenes [32].
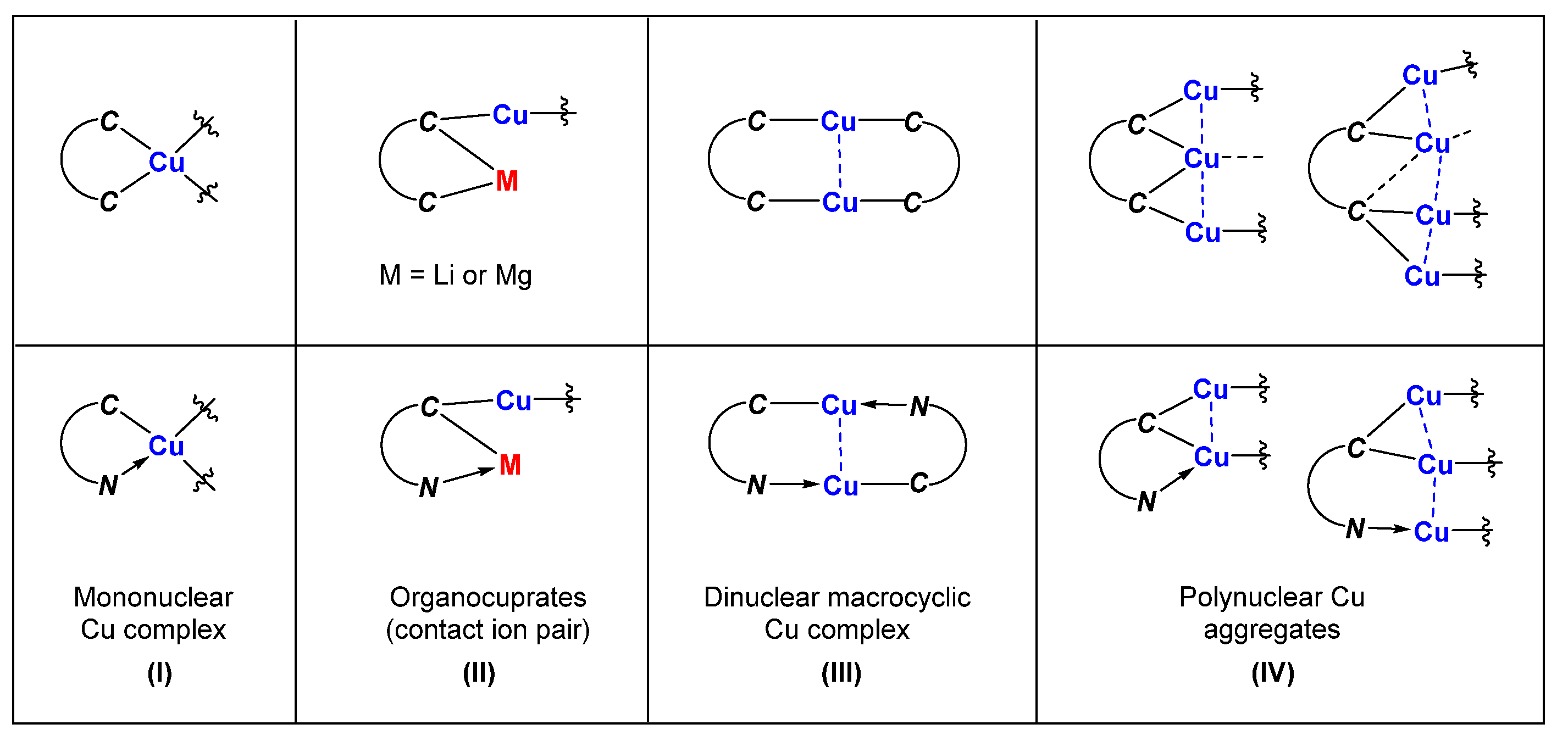
Here, in this review, we are interested in summarizing organocopper compounds with at least one Cu-C σ-bond in which the organic fragments behave as actor ligands rather than spectator ligands (e.g., Cp, NHC). For mono-dentate carbonic ligands, the binding between copper and carbon atoms normally consists of the regular two-center two-electron (2c-2e) σ-bonds and three-center two-electron (3c-2e) bonds [33] when the carbon atom bridges two copper atoms. In cases where the bridging carbon atom is linked with a copper and a lithium (or magnesium) atom, the binding is more likely between the 2c-2e and 3c-2e bonds. For neutral nitrogen-containing groups, the binding of Cu-N is a traditional dative bond. When extending from one coordinating site to C,N- or C,C-chelating ligands, the binding in the corresponding organocopper compounds becomes more diverse. Based on the characteristic connectivity of the fragments in these structures, the C,N- and C,C-chelated organocopper compounds can be put into the following four categories:
- (1)
- Mononuclear organocopper complexes:
The two coordinating sites of the C,C- and C,N-bidentate ligand bind to the same Cu center together, forming a 5-membered chelate ring, as shown in (I) of Scheme 1. The two chelate rings are connected with a copper atom and will generate a spiro complex.
- (2)
- Organocuprates with contact ion pair structures:
The two coordinating sites can be attached with different metals, with one site bound to the Cu/M atoms and the other one bound to the main group metal atoms M only. The resulting product can be regarded as being organocuprates with contact ion pair structures (II).
- (3)
- Binuclear macrocyclic copper complexes:
Each coordinating site of the C,C- and C,N-bidentate ligand can also bind an individual Cu atom with 2c-2e bonds, forming a macrocyclic dinuclear complex. As a result, each Cu atom forms a linear geometry with two units of bidentate ligands that provide only one coordinating site to each Cu atom (III).
- (4)
- Polynuclear organocopper aggregates:
If the carbanion bridges two copper atoms, the structural feature will likely be polynuclear copper aggregates with Cu-Cu interactions (IV).
2. Mononuclear Organocopper Complexes
Although neutral N,N-chelated spiro copper complexes are relatively common, the anionic C,N- or C,C-cheated spiro counterparts are sporadic. As mentioned above, Xi’s group found that the C,C-bidentate ligand is an excellent platform to build up diversified coordination complexes across the periodic table [32,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. The chelating effect increases the thermal stability of these organometallic species, facilitating isolation and following characterization. Additionally, their metal–carbon bonds remain reactive toward electrophiles. As a result, a good balance between stability and reactivity is achieved.
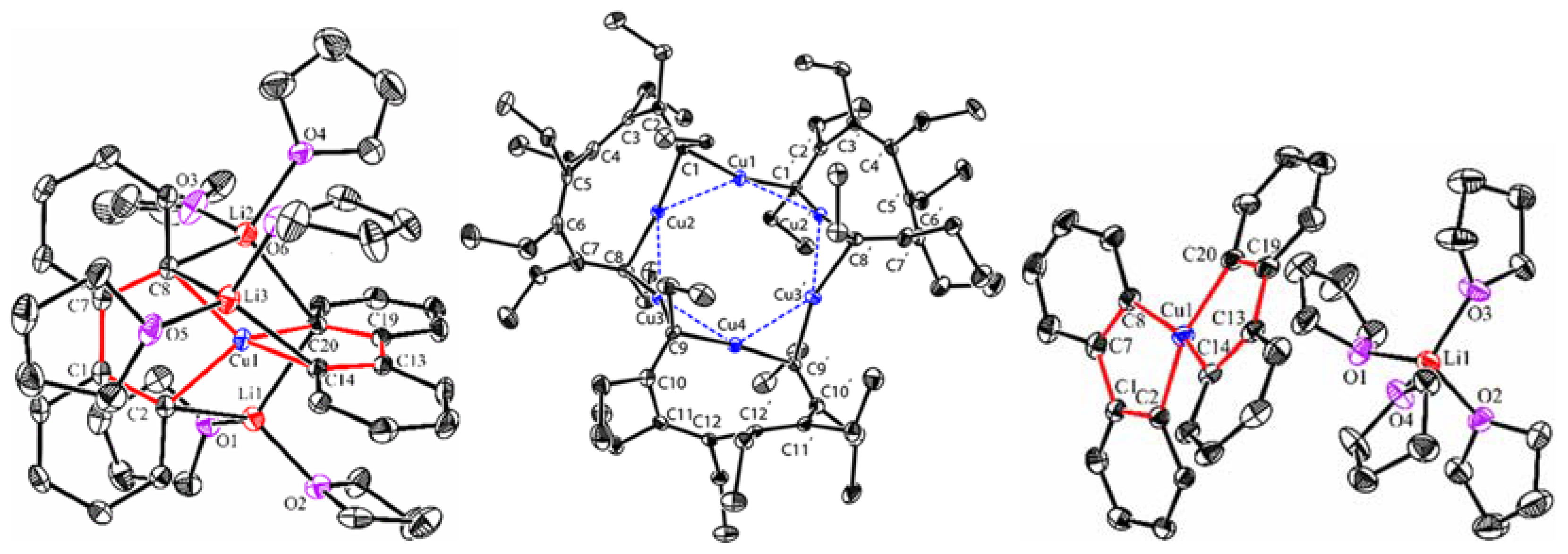
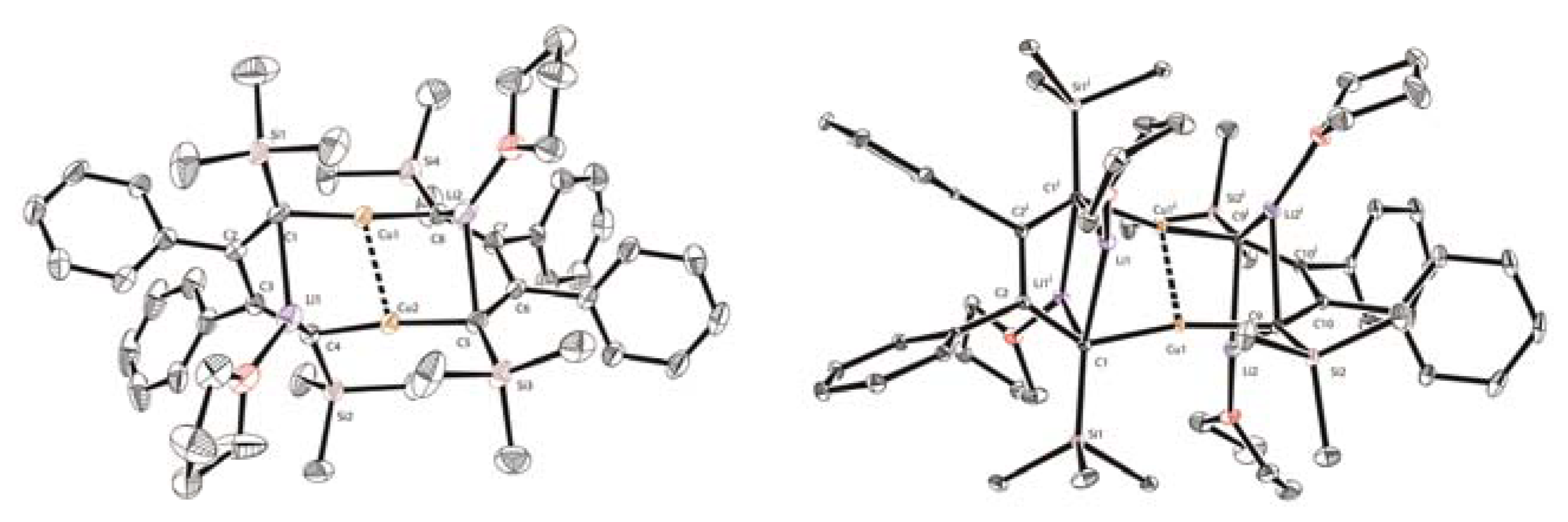
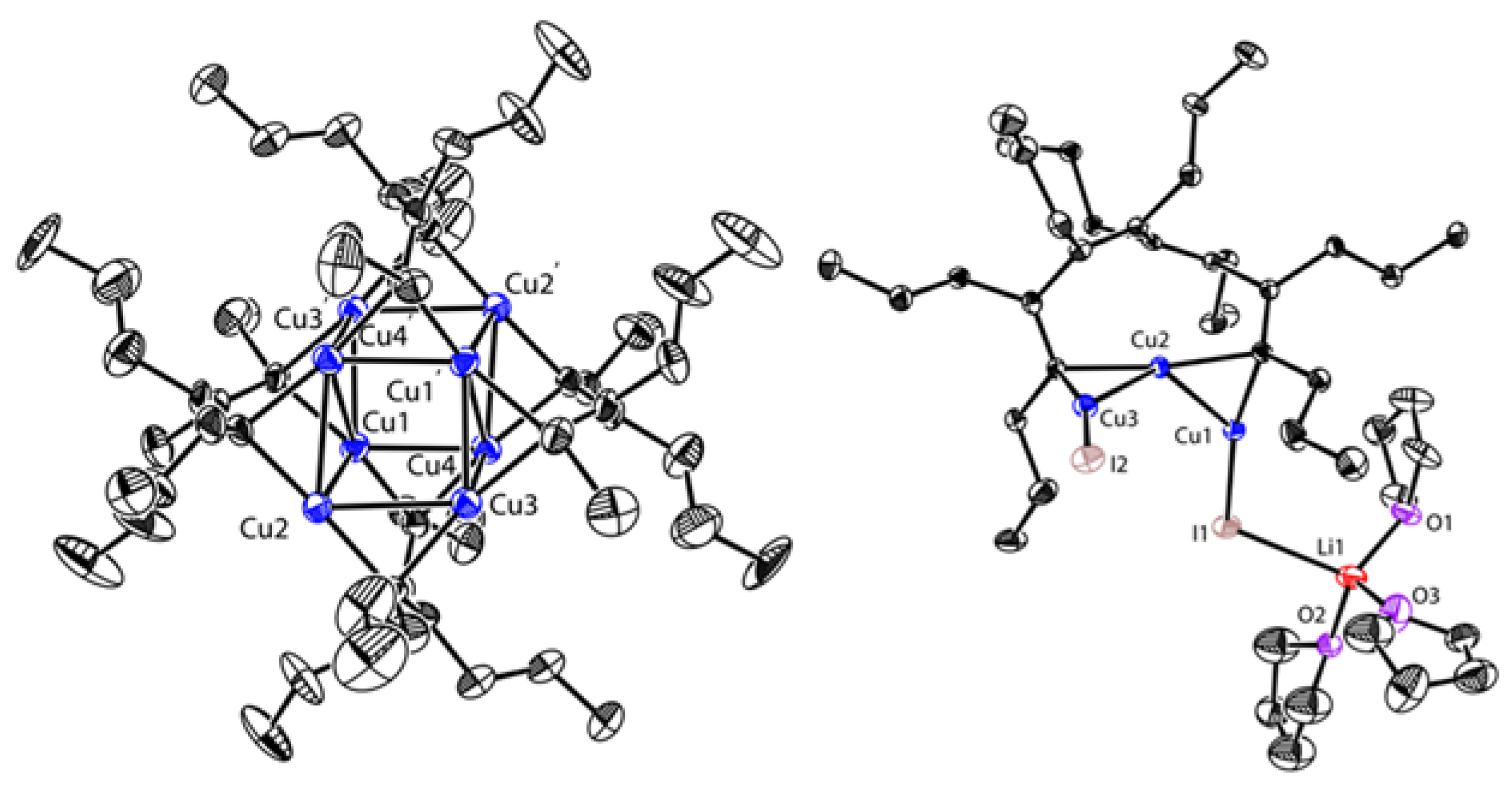
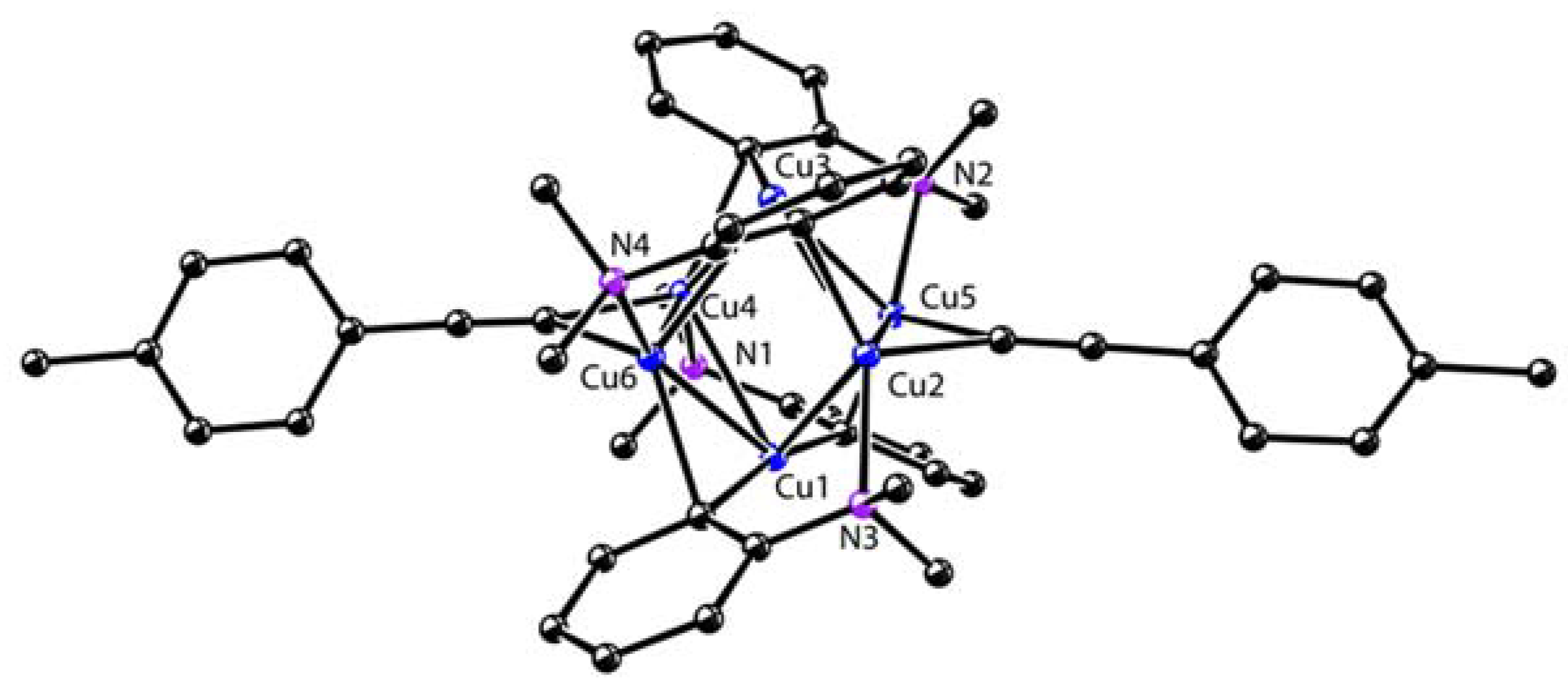
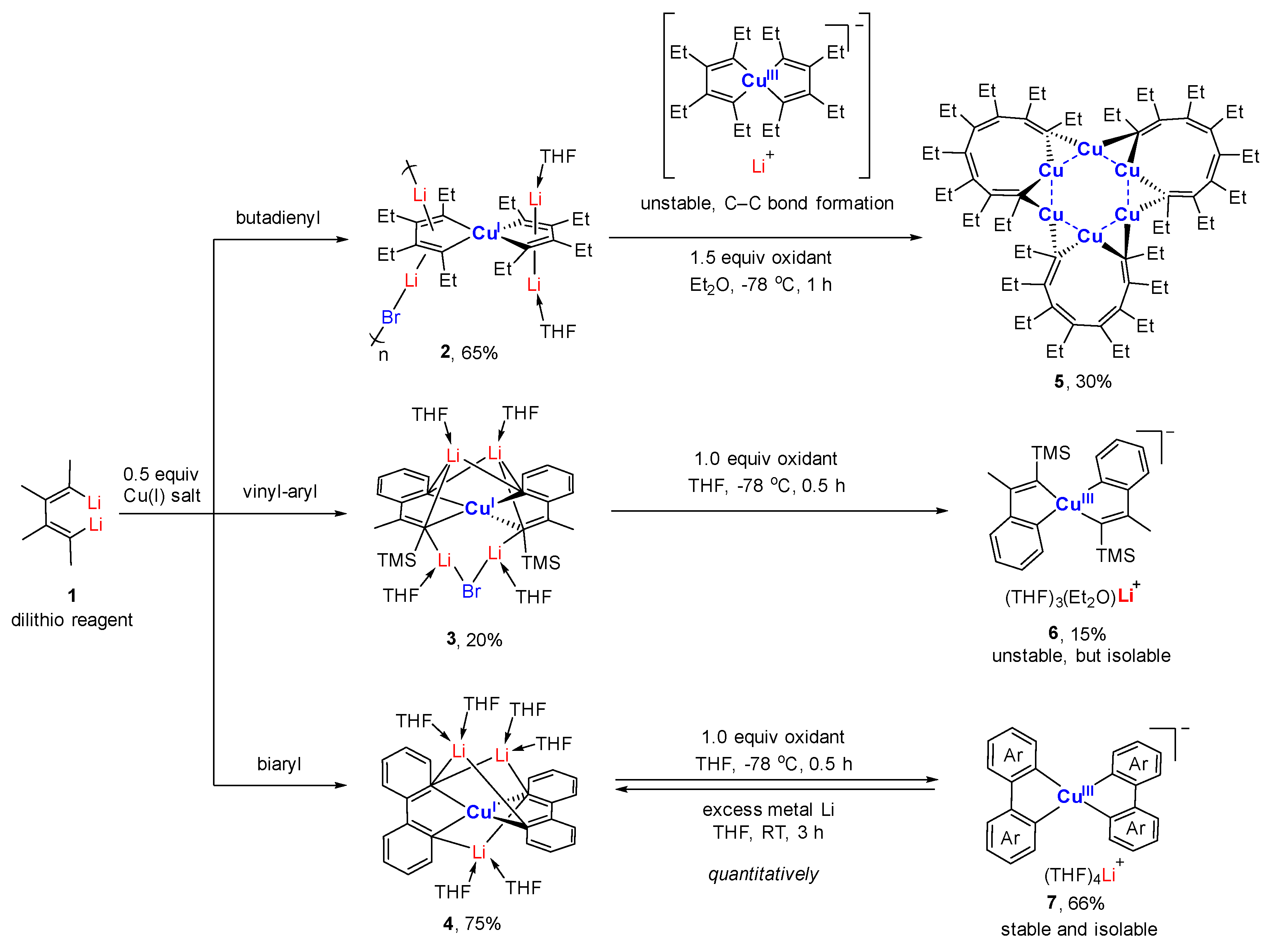
Starting from the 1,4-dilithio 1,3-butadienes 1 (dilithio reagent for short) and 0.5 equiv. of copper(I) salts, Xi’s group isolated the first series of spiro organocopper(I) compounds 2–4 (Scheme 2) [59,60]. It seems that the terminal alkenyl carbon atom is favorable for LiBr coordination, probably because it is more electron-rich than the phenyl ones. The butadienyl spiro copper complex 2 forms one-dimensional polymers with repeating units linked by Li—Br. When one vinyl moiety is replaced by a phenyl ring, LiBr forms a Li2Br fragment between the two vinyl carbon atoms of 3. No LiBr salt can be found in the biphenyl spiro copper complex 4 (Figure 1). The copper atoms are all coordinated with two dianionic ligands, forming a distorted tetrahedral geometry (dihedral angels between two five-membered chelate rings is about 63–84°). The averaged Cu(I)-C bond lengths of 2–4 are from 2.01 to 2.06 Å. Upon oxidation, either the dianionic ligand or the metal center can be oxidized depending on the electron-richness of the ligands. Compound 1 is transformed to a hexanuclear tetraenyl copper cluster 5 with a Cu6 hexagon. In this process, the oxidative dimerization of the original butadienyl ligand occurs. It might undergo a spiro Cu(III) intermediate, which is too unstable to be isolated. When 3 is treated with oxidizing agents, a spiro Cu(III) complex 6 is afforded and isolated successfully. Compound 6 is thermally unstable and is difficult to handle at room temperature. With an extended conjugated backbone, 4 is oxidized to form a very stable spiro Cu(III) complex 7. In addition, 7 can be reduced by metal lithium to regenerate 4 quantitatively. Both 6 and 7 have copper atoms with a distorted square planar geometry. The averaged Cu(III)-C bonds of 6 and 7 (1.96–1.97 Å) are noticeably shorter than the Cu(I)-C bonds of 2–4. It is noteworthy that 6 and 7 represent a novel type of unprecedented organocopper(III) complex. By virtue of Cu L2,3-edge X-ray absorption spectroscopy and experimentally calibrated electronic structure calculations, Lancaster and co-workers found that Cu(III) complex 7 has a LUMO (lowest unoccupied molecular orbital) with only ∼25% Cu 3d character, which suggests that the formal Cu(III) is not a physical d8 configuration [61].
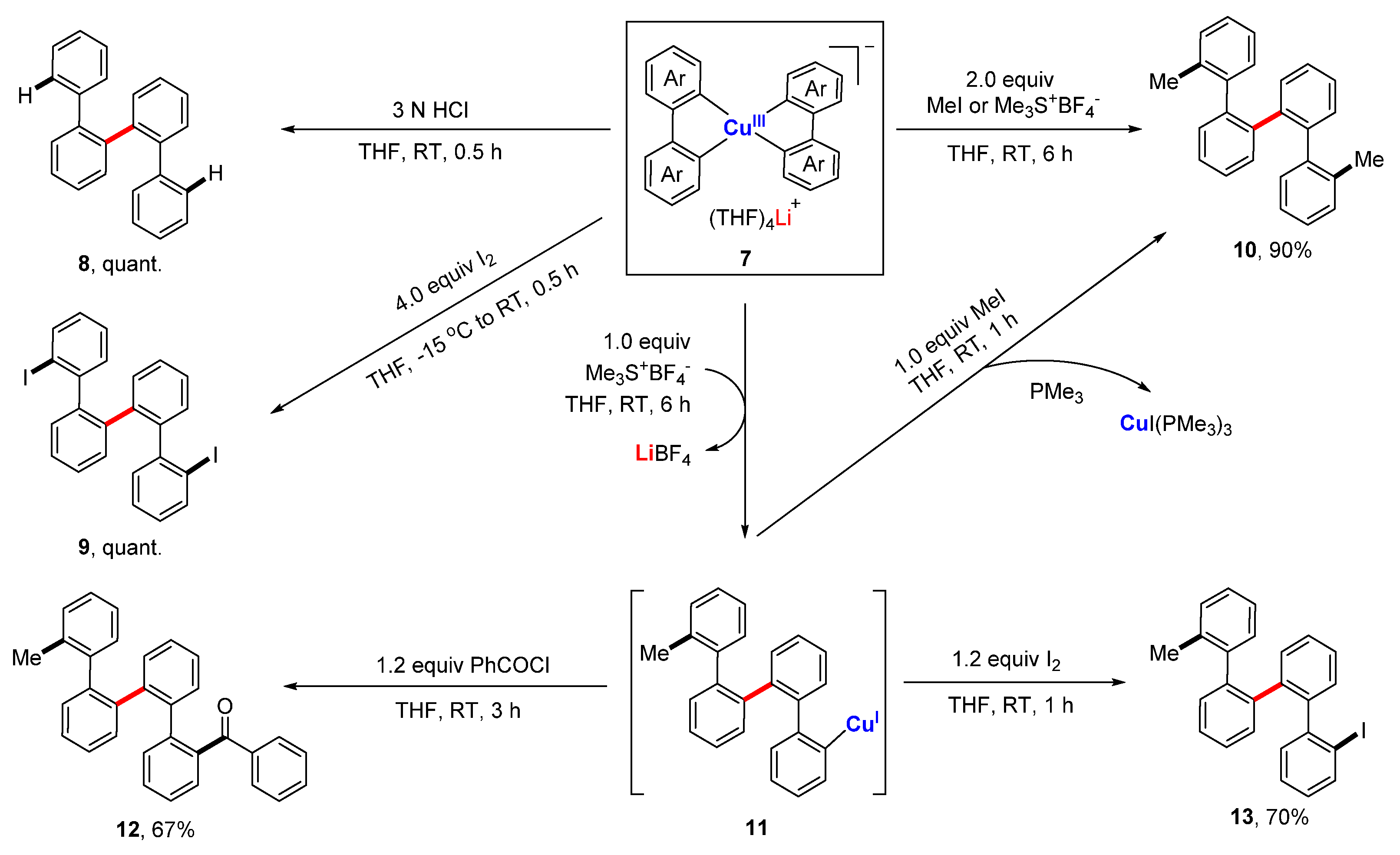
As revealed by the crystal structure of 7, all four aryl rings are at cis-positions, which, in principle, provides a favorable arrangement for reductive elimination. However, no C-C bond formation occurred when the anionic compound 7 itself was refluxed overnight. However, when treated with electrophiles, e.g., aqueous HCl solution, iodine, and alkylation reagents, 7 afforded the symmetrical quaterphenyl derivatives 8–10 facilely in almost all quantitative yields via an electrophilic attack followed by a reduction elimination (Scheme 3). A stepwise experiment showed that 7 reacted with 1.0 equiv. of methylation agent, producing a quaterphenylcopper(I) species 11, which can couple with other electrophiles to form the di-substituted quaterphenyl compounds 10, 12, and 13 in high yields. The reductive elimination of high-valent copper compounds has often been proposed as the final step to release the product, but there has been nearly no concrete experimental evidence of that until this work.
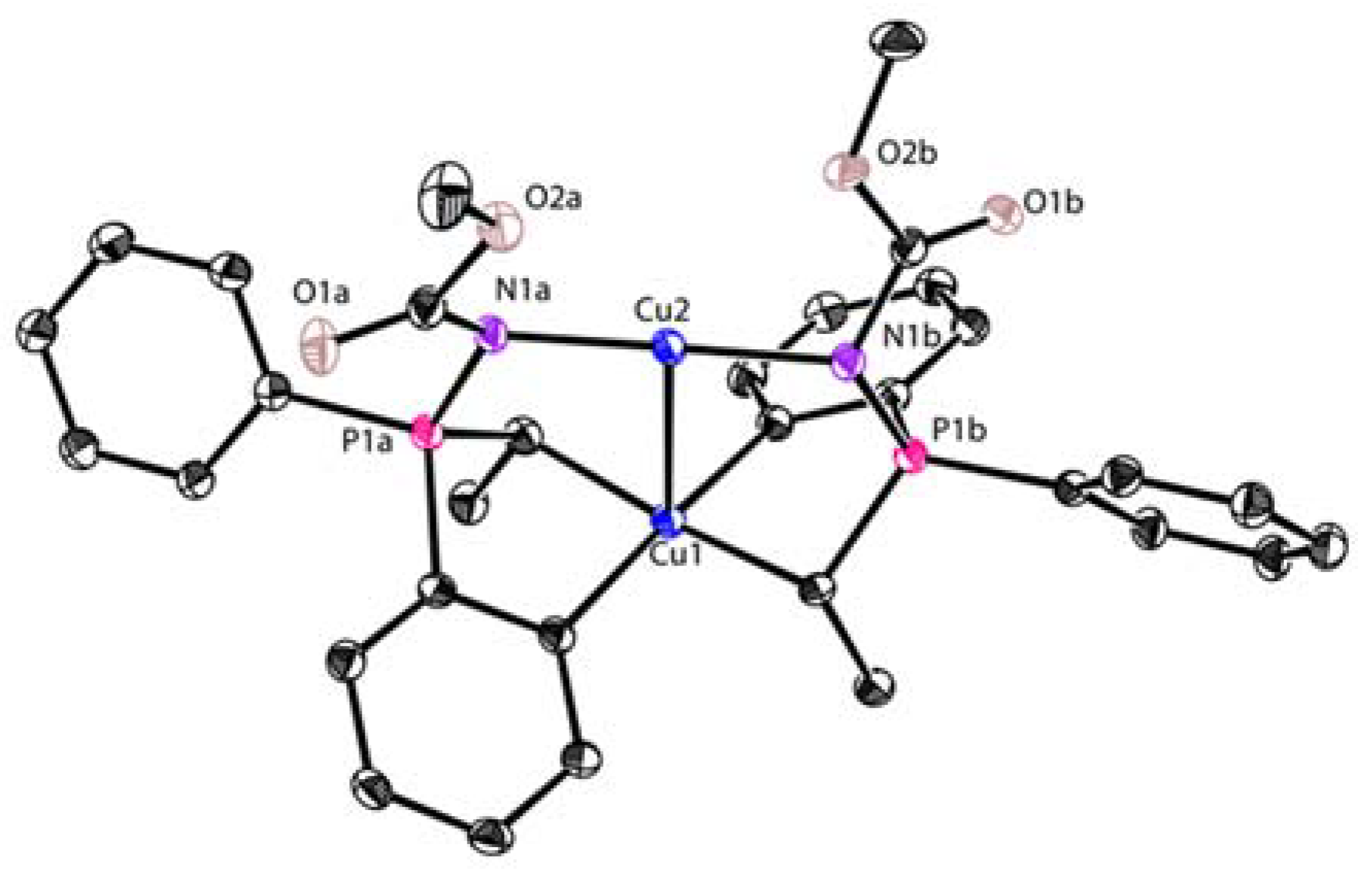
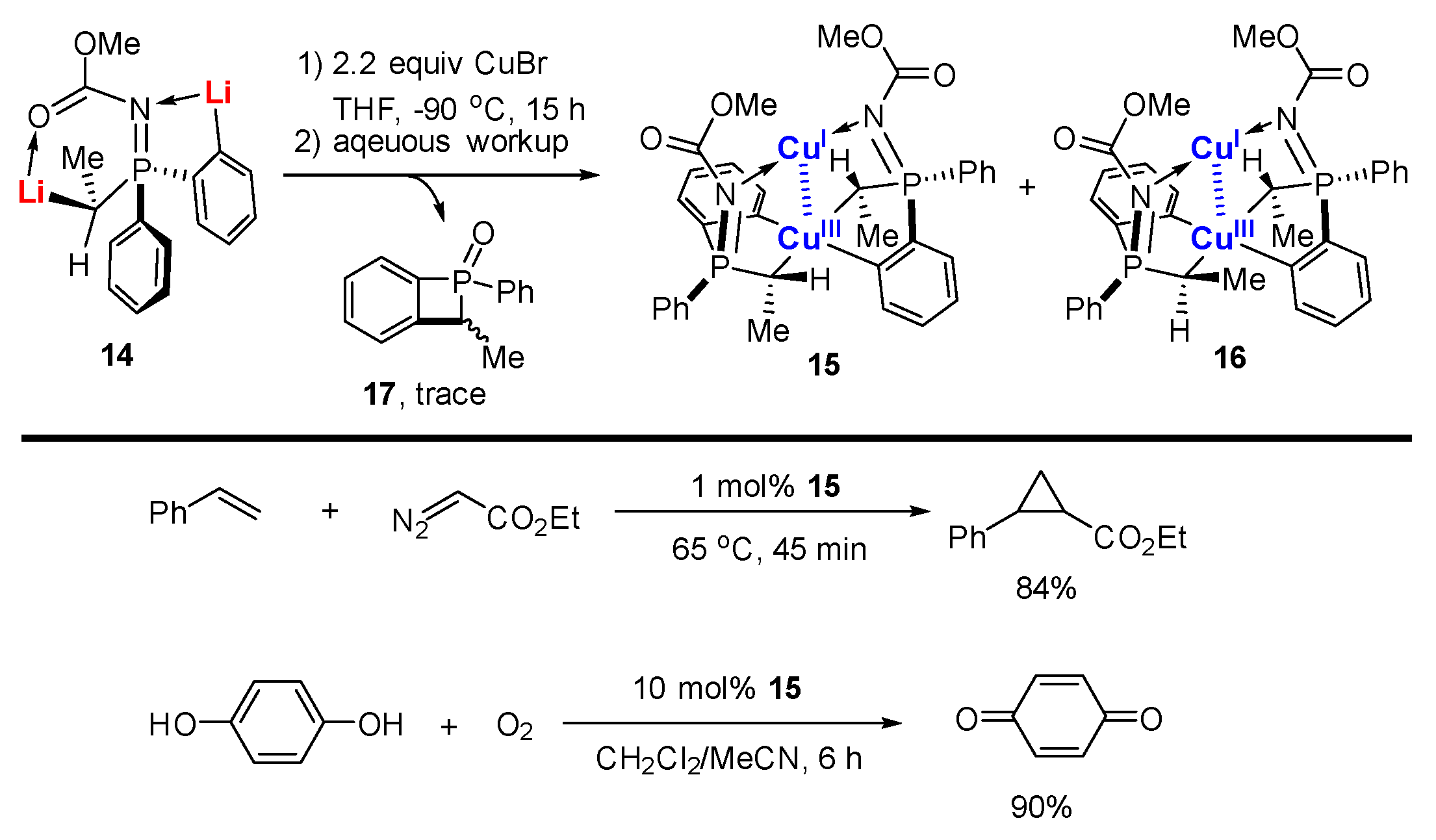
In a similar vein, López-Ortiz and co-workers reported the reaction between dilithiated phosphazene 14 with 2.2 equiv. of CuBr followed by an aqueous workup to afford two diastereotopic isomers of mixed-valent Cu(I)/Cu(III) complexes 15 and 16 in 53% combined yield, in which the Cu(III) center was chelated with two C,C-bidentate aryl-alkyl ligands (Scheme 4) [62]. A byproduct with a four-membered ring 17 was isolated as well. Both 15 and 16 can be purified by column chromatography. Accordingly, they are stable in air and moisture. Cu(III) and Cu(I) adopt a distorted square planar and linear geometry, respectively (Figure 2). The averaged Cu(III)-C bond (1.98 Å) in 15 and 16 is similar to that in 6 and 7. The oxidation states of the Cu ions in 15 were further confirmed by extended X-ray absorption fine structure (EXAFS). Furthermore, all of the pieces of evidence, including the short Cu-Cu distance (2.74 Å) in the single-crystal structure, a critical bond point in the topological analysis of the X-ray experimental Fourier map as well as a diagnostic absorption band at 343 nm, strongly supported a weak metal–metal d8-d10 bonding between two Cu ions. Compound 15 can be used as a catalyst in catalytic cyclopropanation and aerobic oxidations. The characteristic signal of 15 has been observed and suggests that 15 remains unaffected during these transformations, probably due to its rigid structural configuration and extremely high stability.
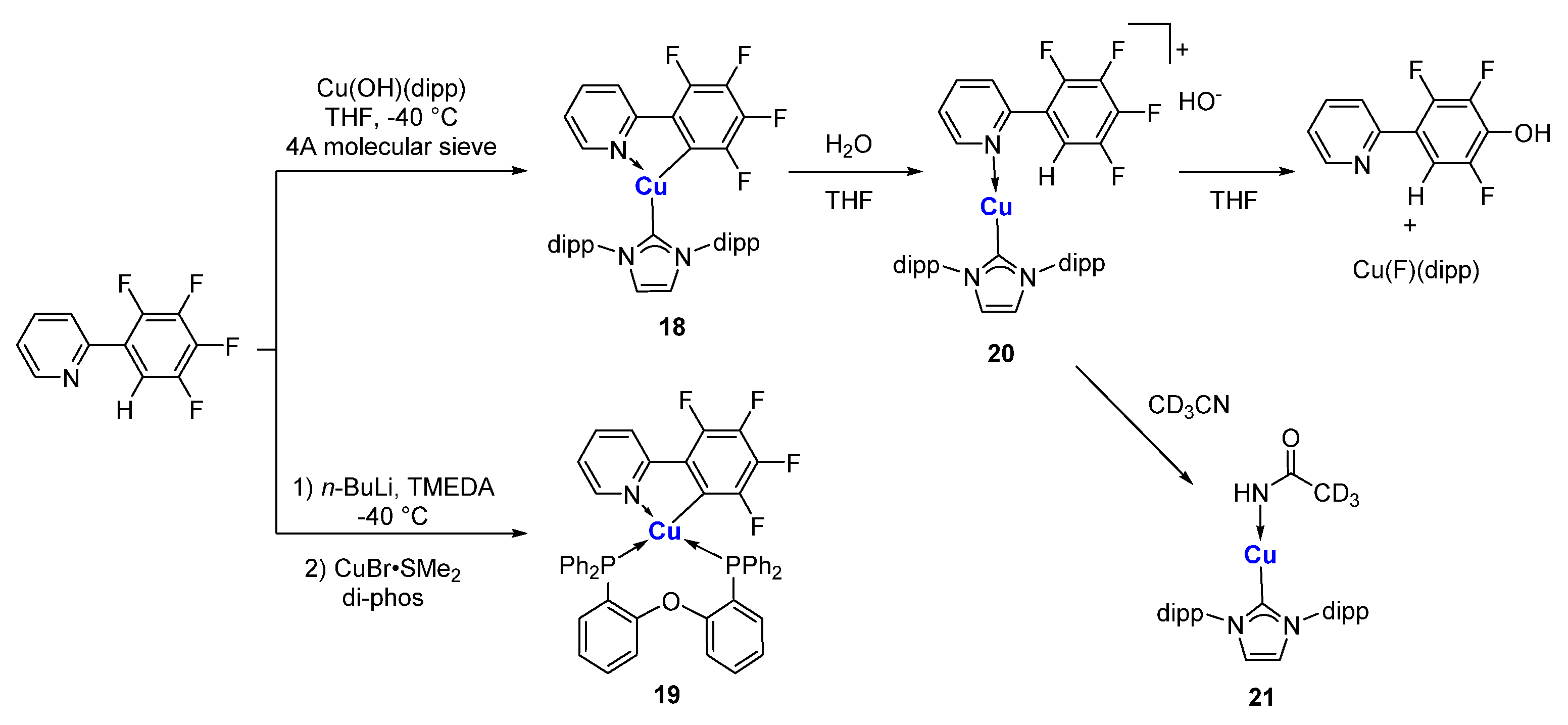
The compound 2-phenylpyridine and its derivatives are among the most popular scaffolds for photoactive transition metal complexes. Partial fluorination of the aryl ring makes the C-H bond at the 2-position very acidic. Additionally, at the same time, it increases the stability of the resulting Cu-C bond. Steffen and co-workers reported that the C,N-chelated organocopper compound 18 was obtained from a direct metalation from Cu(OH)(NHC), in which 4A molecular sieves play an essential role in removing water (Scheme 5) [63]. In the presence of water, compound 18 will inevitably undergo a hydrolysis process, regenerating the C-H bond and forming an ionic species 20. The counteranion of hydroxide in 20 will replace a fluoride at the 4-position via a nucleophilic aromatic substitution in THF to 4-hydroxyl-2-arylpyridine and copper fluoride species. When MeCN is used as the solvent, hydroxide will attack MeCN, forming a copper acetamide species 21. Similarly, a spiro complex 19 can also be prepared from lithiation and subsequent transmetalation with CuBr·SMe2 in the presence of diphosphine ligands. It has been shown in single crystal structures that 18 and 19 adopt distorted trigonal and tetrahedral coordination geometries, respectively. In solution, 18 is weakly emissive. In the solid state, the C,N-chelated organocopper compounds exhibit intense orange-red luminescence (λmax = 610 (18), 607 (19) nm) at room temperature and have phosphorescence lifetimes of τ = 8.6 (18) and 9.5 (19) μs.
3. Organocuprates with Contact Ion Pair Structures
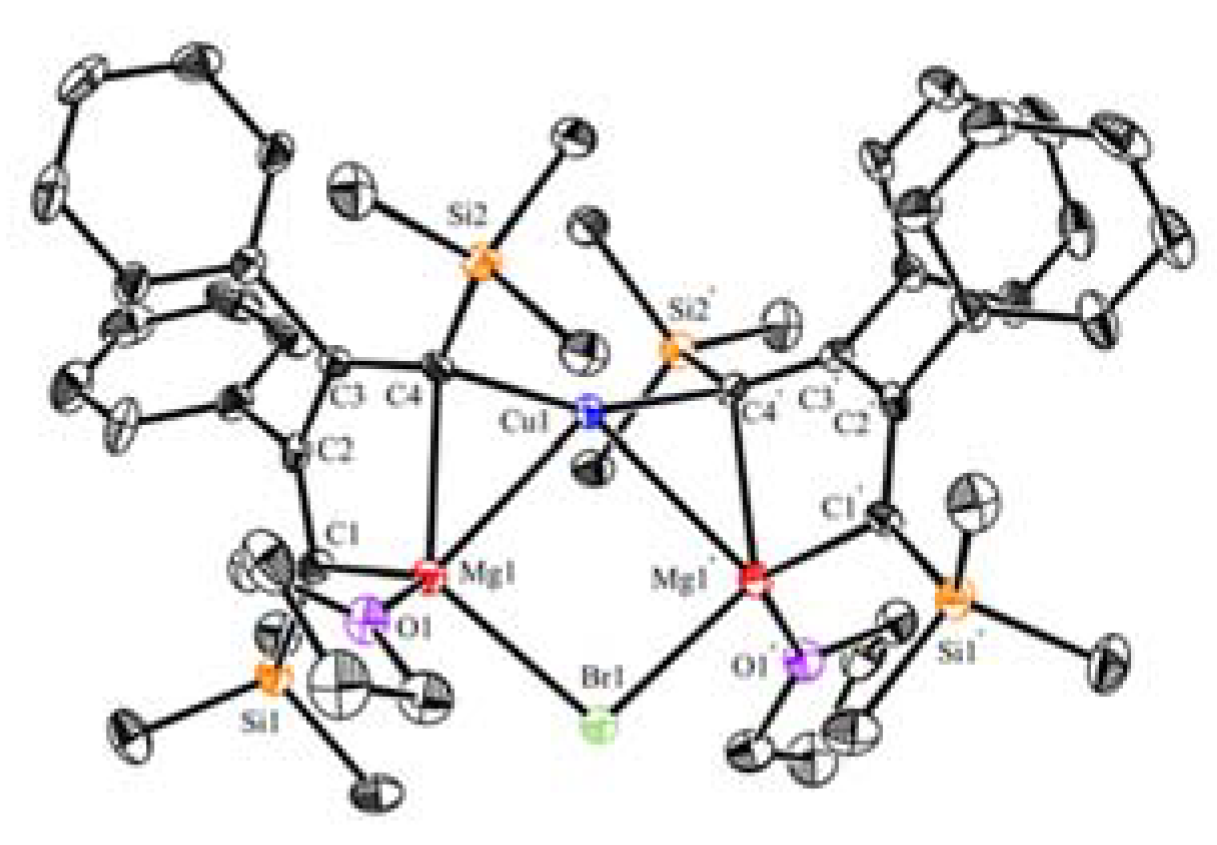
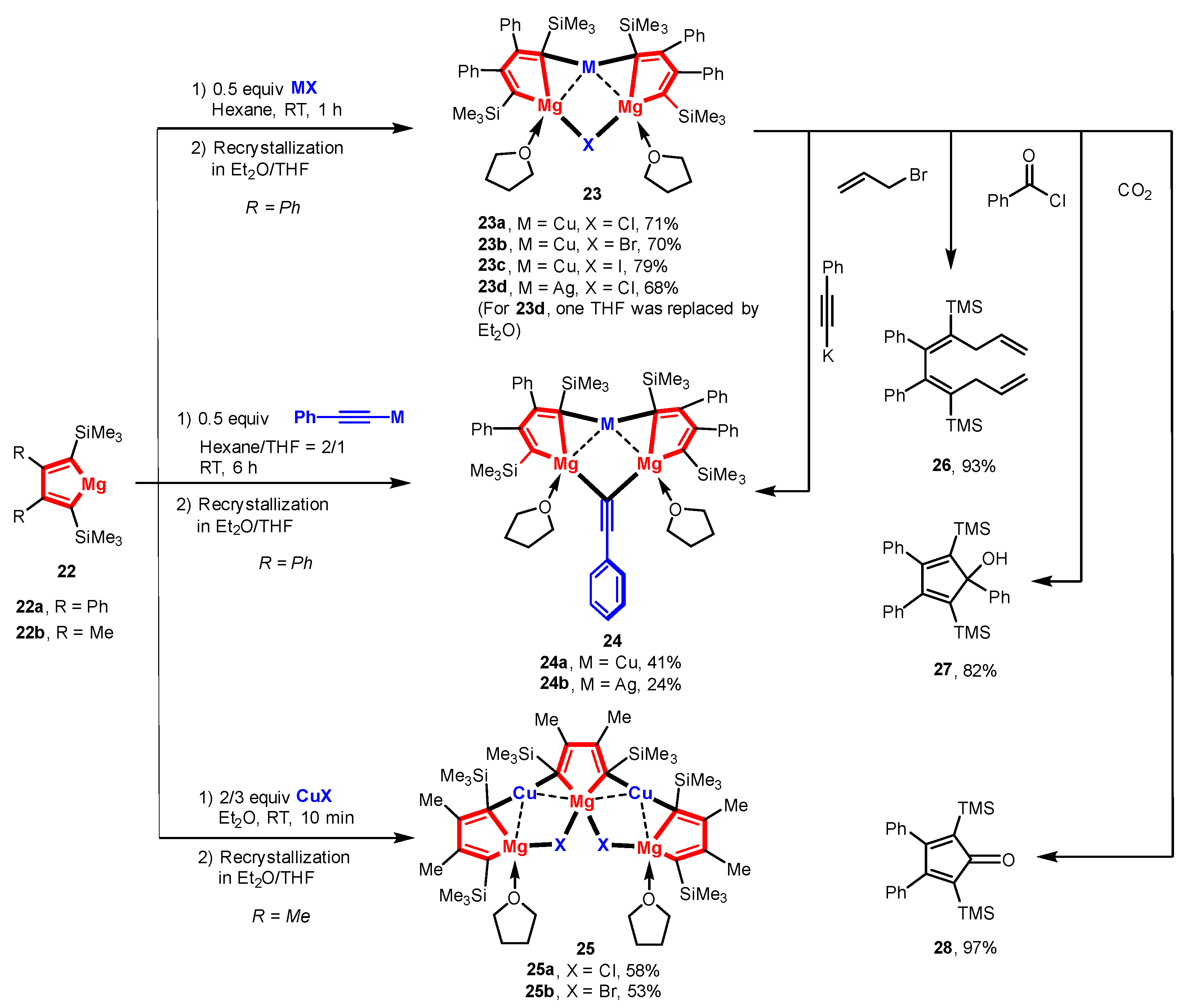
As mentioned in the Introduction, organocuprates have been extensively used in organic synthesis. Although numerous lithium organocuprates have been structurally characterized, the equally important magnesium organocuprates remain less unexplored, especially in terms of their well-defined structures. Only a few examples of aryl magnesium organocuprates have been previously reported [64]. As reported in the previous work of Xi and co-workers [35], magnesiacyclopentadienes 22, the first alkaline-earth metallocyclopentadienes, have a small C-Mg-C bite angle and a coplanar 5-membered chelate ring. The bridging character of anionic carbon atoms (Lewis base) and the coordinative unsaturation of the Mg atom (Lewis acid) make 22 available to bind other metal salts. Two equiv. of 22 reacted with 1.0 equiv. of copper halides or (phenylethynyl)copper to create magnesium organocuprates 23a–c and 24a with a Cu:Mg ratio of 1:2 (Scheme 6) [58]. In addition, inert silver halides as well as alkynyl silver compounds can apply to the above transformations, producing magnesium organoargenates 23d and 24b. The structures of forming organocuprates are dependent on the substituents at the 2,3-positions for some reason. When the substituents are changed from Ph to Me groups, 25, with a Cu:Mg ratio of 2:3, was obtained as the major product. Structural analysis and DFT calculation (AIM) revealed that as the Cu-C bonds form, one Mg-C bond becomes very weak (2.34 Å), while the other one remains unchanged (2.11 Å) in 23a. It seems that two Mg atoms provide a pre-organized position to anchor the halides. The C-Cu-C bond angle (157°) as well as the Cu-C bond lengths (1.95 Å) indicates a Cu-C-Mg 3c-2e bonding character (Figure 3). As supported by the molecular structure and DFT calculations, no bonding interaction was found between Cu-Br in 23a. On the whole, the binding of the anionic C (sp2) carbon atoms to the Cu atom and the bridging of the Mg atoms by the halides leads to a formal heterolytic cleavage of the M-X bond (M = Cu, Ag; X = halide, alkynyl) in a cooperative way along with the transfer of anionic X units from M centers to Mg centers. Accordingly, 23–25 can be considered as a resting-state intermediate of the transmetalation reaction between organomagnesium reagents and coinage-metal salts. The bridging ligand can be transformed quantitatively, e.g., 23c → 23a or 24a. The preliminary reactivity of these rigid magnesium organocuprates was provided by the reactions of 23c with various electrophiles, producing small organic molecules 26–28.
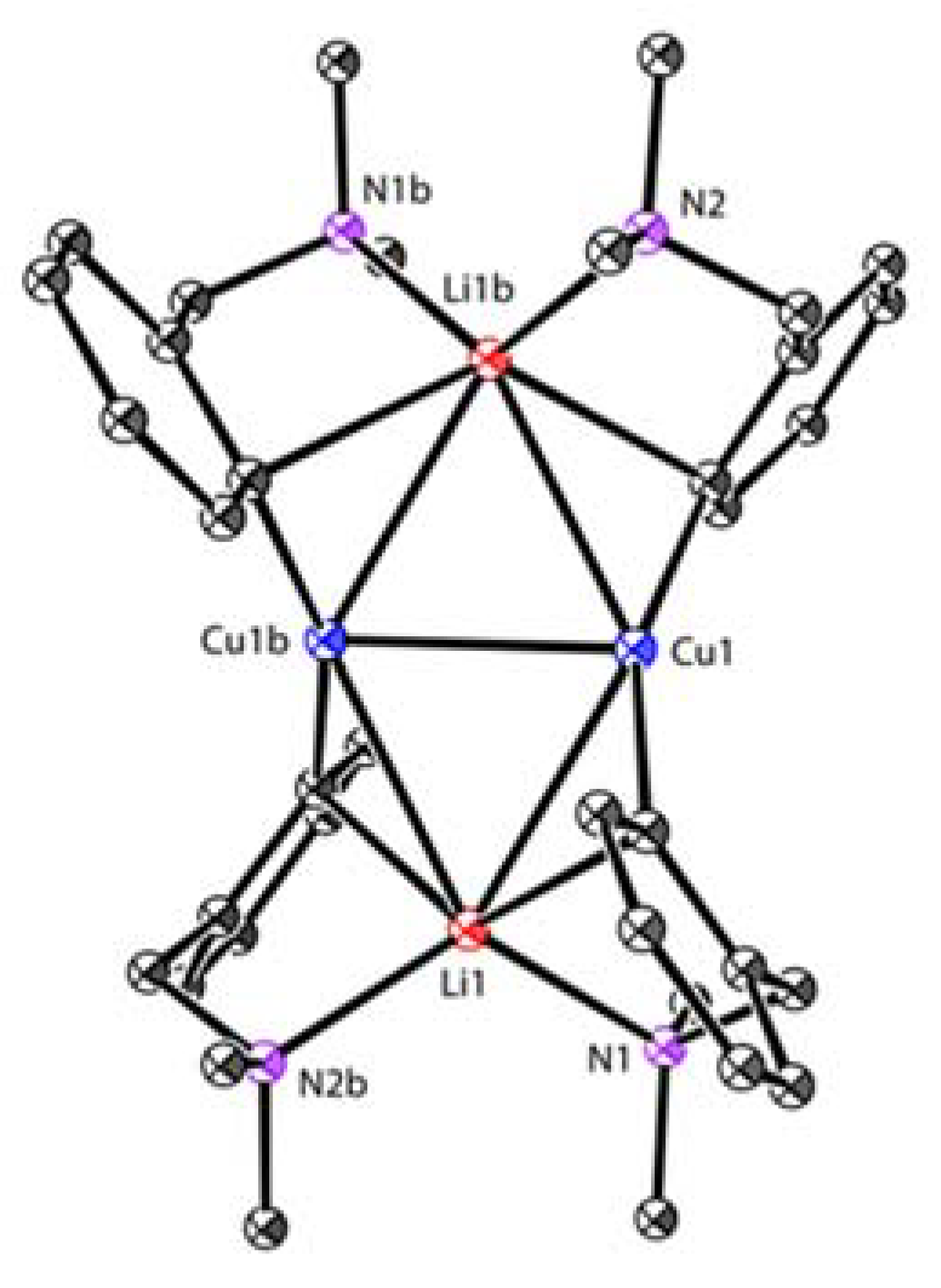
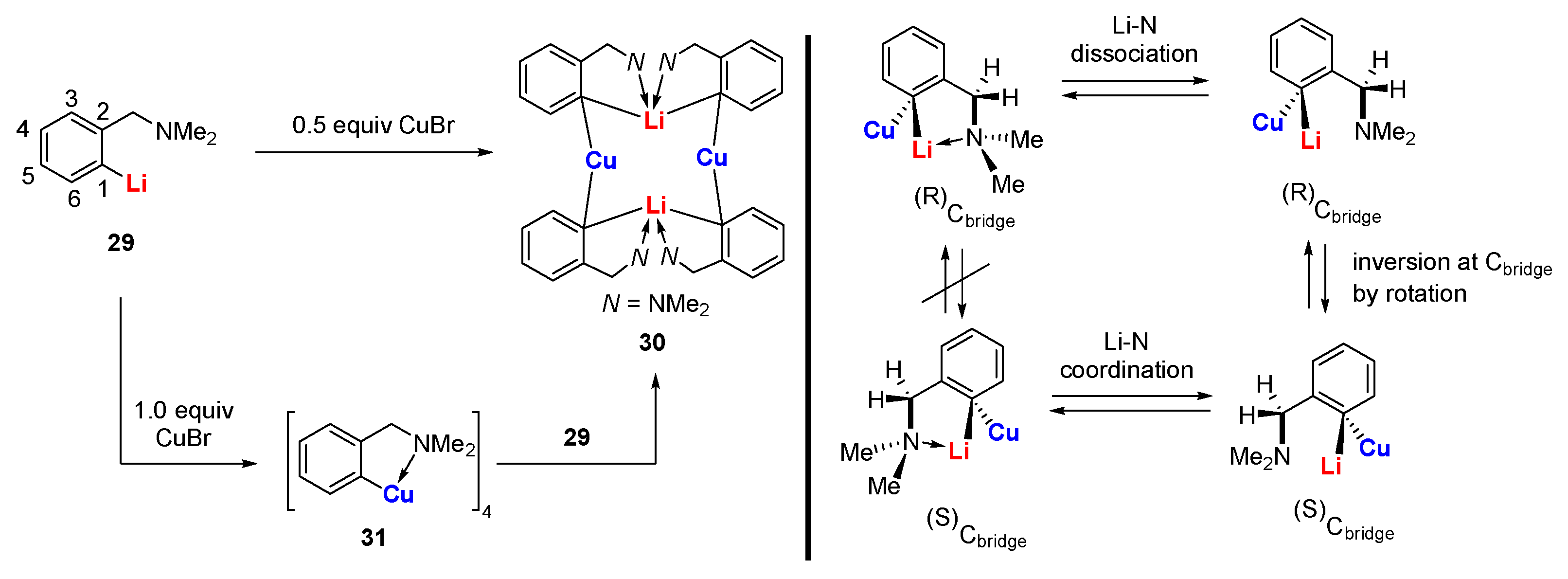
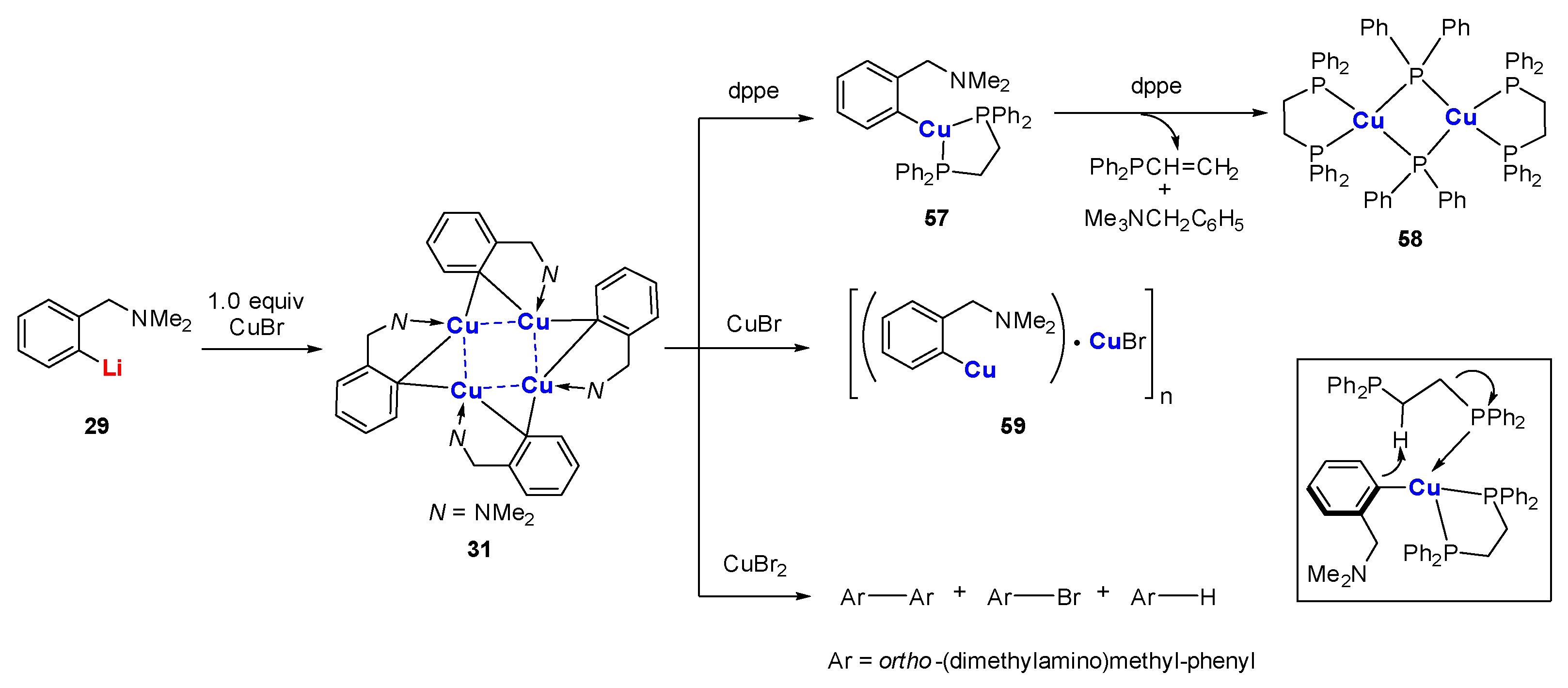
When a built-in coordinating site is added to the monoanionic phenyl skeleton at the ortho-position, it will generate a C,N-bidentate ligand, as shown in Scheme 7. With the (dimethylamino)methylphenyl (DMMP) lithium reagent 29 in hand, van Koten’s group developed two methods to synthesize the dicopper-dilithium organocuprate 30: (1) a direct transmetalation of 29 with 0.5 equiv. of CuBr and (2) an interaggregate exchange reaction between DMMP copper cluster 31 (vide infra) and 29 [65,66]. The short averaged Cu-C bonds (1.94 Å) as well as the weak averaged Li-C bonds (2.39 Å) in 30 (Figure 4) suggest that the 2c-2e bond of the Cu-C bonds has a higher character [67]. However, the split coupling of 13C-7Li (7 Hz) was still observed in solution, which indicates that the s electron density is present between the Li and C (bridge) nuclei [68]. Meanwhile, J (13C-7Li) did not change during variable-temperature NMR studies, allowing it to exclude the intraaggregate exchange processes. By monitoring the characteristic signals of methylene protons (AB pattern) and NMe2 protons (two singlets) using dynamic 1H and 13C NMR spectroscopies, van Koten and co-workers found the rotation of the 3c-2e bonded aryl groups around the C(1)-C(4) axis for the first time [69]. The C4Cu2Li2 core fragment appears to be a persistent structural feature in neutral aryl lithium organocuprate (contact ion structure [64]).
4. Binuclear Macrocyclic Copper Complexes
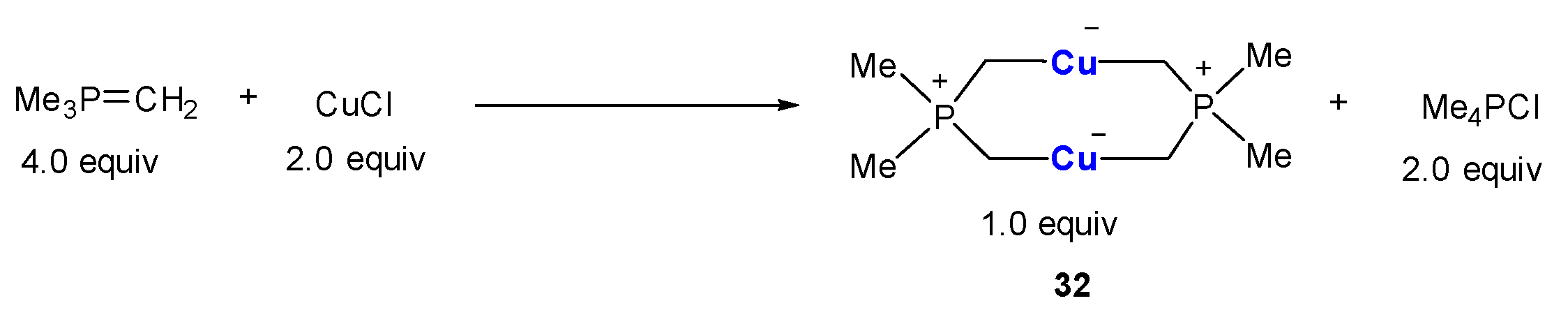
It seems that trimethylmethylenephosphorane (Me3P=CH2) shows a better stabilization effect on the Cu-C bonds than its isoelectronic analogue Me3Si-CH2. Buchner and Zangrando found that the treatment of Me3P=CH2 with CuCl afforded a binuclear cuprate complex 32 in which negative charges are formally located at the Cu atoms while positive charges are located at the phosphine atoms (Scheme 8) [70,71]. In this process, the hybridization of CH2 was changed from sp2 to sp3. The core structure of P2(CH2)4Cu2 forms a coplanar 8-membered macrocycle. The averaged Cu-C bond (1.96 Å) and C-Cu-C bond angel (176°) are in the typical ranges for organocuprates.
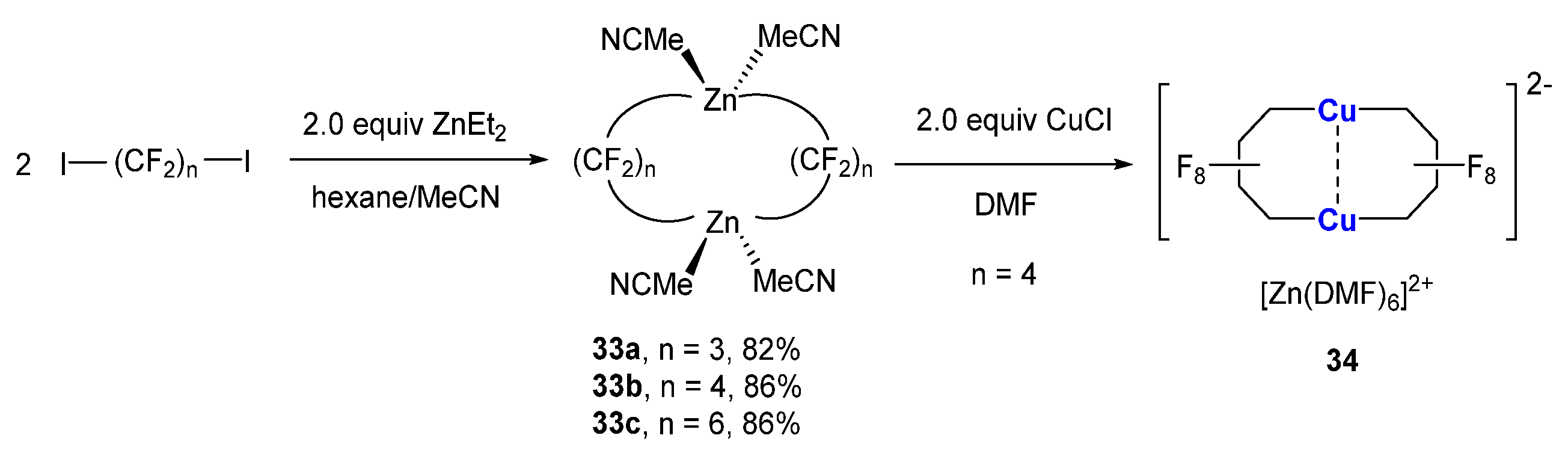
Recently, there has an increasing interest in (CF2)n-containing materials in industry. However, the methods of incorporating difluoromethylene into molecules are lagging behind other perfluroalkyl counterparts. Vicic and co-workers reported a new route to access perfluoroalkyl-based metallacycles with varied ring sizes (Scheme 9) [72]. Tetra-perfluoroalkyl-dizinc compounds 33 were obtained from the reaction between diiodoperfluoroalkanes and 2.0 equiv. of ZnEt2. The treatment of 33b with 2.0 equiv. of CuCl in DMF afforded a 10-membered dicopper compound 34. The averaged Cu-C bond (1.93 Å) is relatively shorter than that of other non-fluorinated alkyl cuprates. Comound 34 was inactive towards electrophiles, which is consistent with its strong Cu-C bond and high stability.
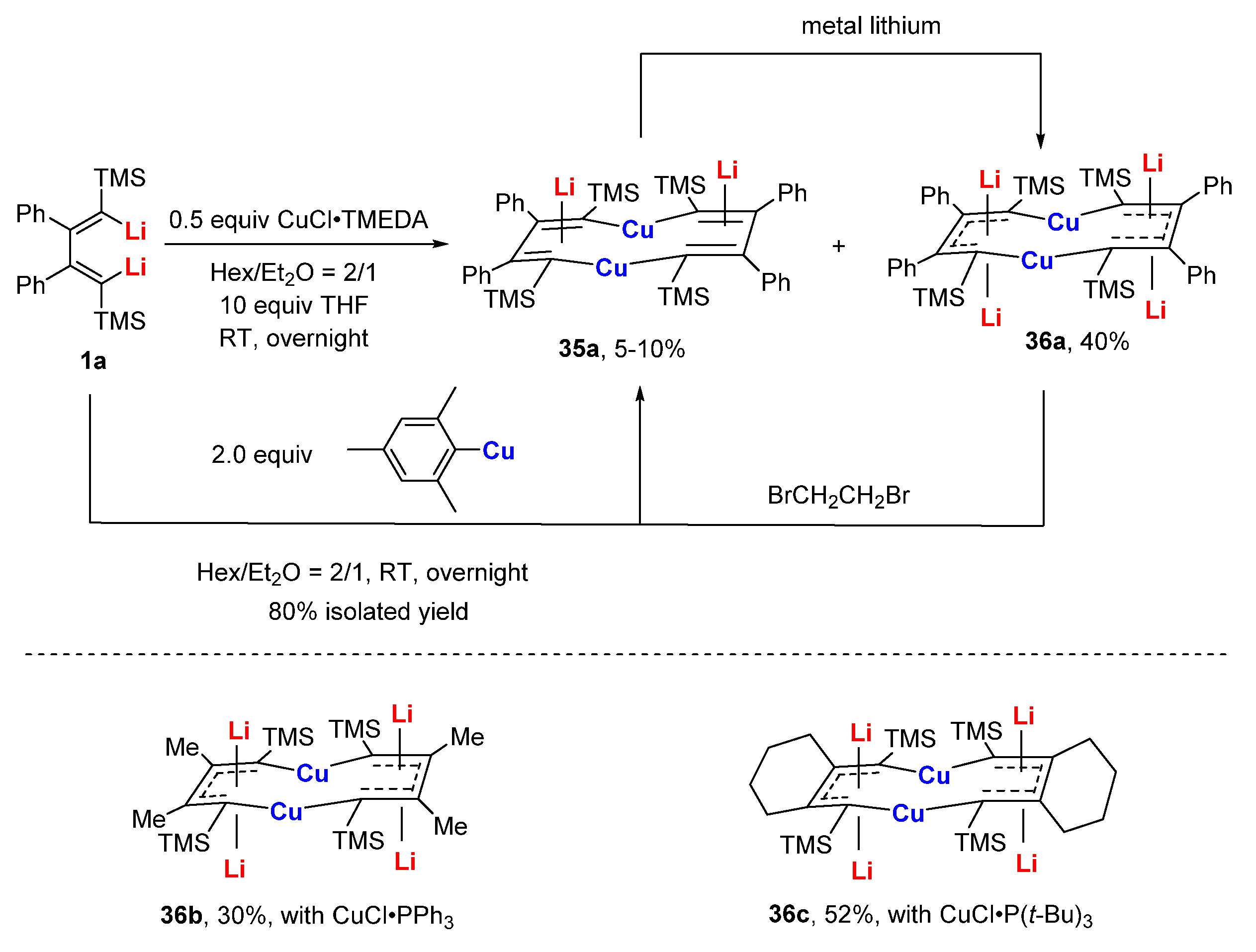
Metal-containing aromatic molecules have attracted much attention recently. Compared to six-membered metallobezenes and metallabenzynes as well five-membered metallocylcopentadienes, macrocyclic metalla-aromatics are less explored. Trans,cis,trans,cis,cis-[10]annulene is a conjugate molecule with 10-π electrons. However, it is non-aromatic due to the steric repulsion between the two internal hydrogen atoms. It was previously envisioned that the replacement of two internal CH with a transition metal would not only release the steric hindrance but would also provide electrons to the delocalized π-system. Aromatic dicupra[10]annulenes 36 with four lithium atoms were first synthesized and isolated from the reaction of dilithio reagents 1 and a CuCl complex by Xi’s group (Scheme 10) [32]. In the above reactions, 35 was isolated with two lithium atoms as a small amount of byproduct that is also able to be efficiently synthesized from dilithio reagent and mesitylcopper. More importantly, reversible two-electron transformations between 35 and 36 were observed [47]. Compared to the alternating bonds of the butadienyl skeleton in 35a (1.52, 1.37 Å), the C-C bonds of annulene moiety in 36a are remarkably averaged (1.47, 1.42 Å), suggesting a considerable π-delocalization (Figure 5). The Cu-C bonds of 36a (1.92 Å) were notably shorter than those of 35a (1.96 Å), which might be attributed to the extra two electrons posed to 36a. The Cu 2p3/2 binding energy (932.9 eV) and Cu LMM kinetic energy (915.8 eV) of 36b, as displayed by XPS data, show that the Cu atoms in 36 are more likely to be Cu(I). With the assembled experimental results (the above crystallographic data; low-frequency resonance signals of 7Li NMR: −5.1 to −6.2 ppm) and DFT calculations (negative values of ISE: −11.7 and −21.5; large negative NICS values: −9.0 to −12.8), it is safe to conclude that dicupra[10]annulenes are aromatic. AdNDP analysis has previously suggested dicupra[10]annulenes could be regarded as a 10-π aromatic system. Based on independent theoretical calculations, Grande-Aztatzi and co-workers consider dicupra[10]annulenes to be a metalla-naphthalene [73]. Zhu and co-workers suggest that dicupra[10]annulenes have 16e Craig-type Möbius aromaticity [74]. Additionally, both Zhu’s and Sundholm’s calculations reveal that lithium counterions play an important role in these organocopper complexes [74,75].
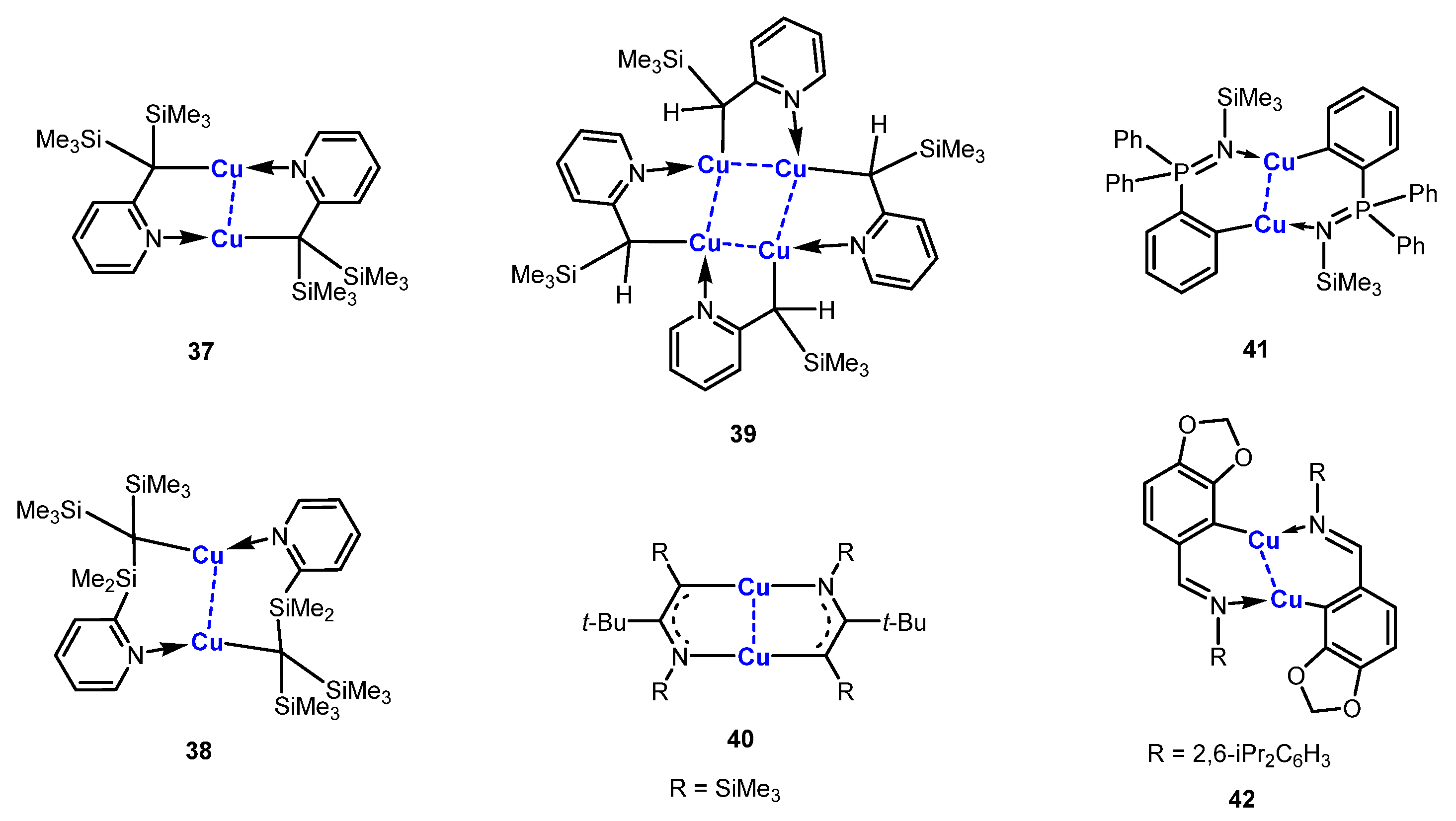
As shown in Scheme 11, the C,N-chelated dicopper compounds 37,38 and 40–42 documented in the literature are generally macrocyclic neutral species since the two negative ligand charges are exactly balanced with two Cu(I) atoms [76,77,78,79,80,81,82]. An early example of structurally well-defined alkylcopper compounds was [Me3SiCH2Cu]4, in which trimethylsilyl groups not only pose steric hindrance to increase the stability but also improve the solubility by virtue of the lipophilicity of the silicon groups [83,84]. White and others found that incorporating an ortho-bulky bis-trimethylsilyl-alkyl ligand at a pyridine can stabilize t dicopper compound 37 to a great extent [76,80]. Compound 37 can be sublimated at 160 °C without decomposition. Compund 37 features an 8-membered core structure (C4N2Cu2). When increasing the distance between the alkyl moiety and the pyridine, a dicopper compound 38 with a 10-membered ring can also be generated, as reported by Eaborn and Smith [81]. However, with a smaller steric effect, the alkyl-pyridine ligands will bind four copper atoms, generating a tetracopper complex 39 [77]. Similarly, three dicopper compounds 40–42 based on 1-azaallyl, aryl-phosphanimine, and aryl-imine ligands were synthesized from the corresponding lithiated starting materials and copper salts by Lappert [78], Stalke [79], and Schmidt [82], respectively. The Cu-C bonds of 37–40 are in the range of 1.93–1.95 Å, which suggests a mostly 2c-2e Cu-C bond, while the Cu-C bonds (1.91 Å) in 41 and 42 fall in the short end of the range observed for Cu-C 2c-2e bonds. Cyclic voltammetry and bulk electrolysis of 37 show very promising evidence of the existence of formal copper(II) alkyl species with relatively high stability.
5. Polynuclear Organocopper Clusters
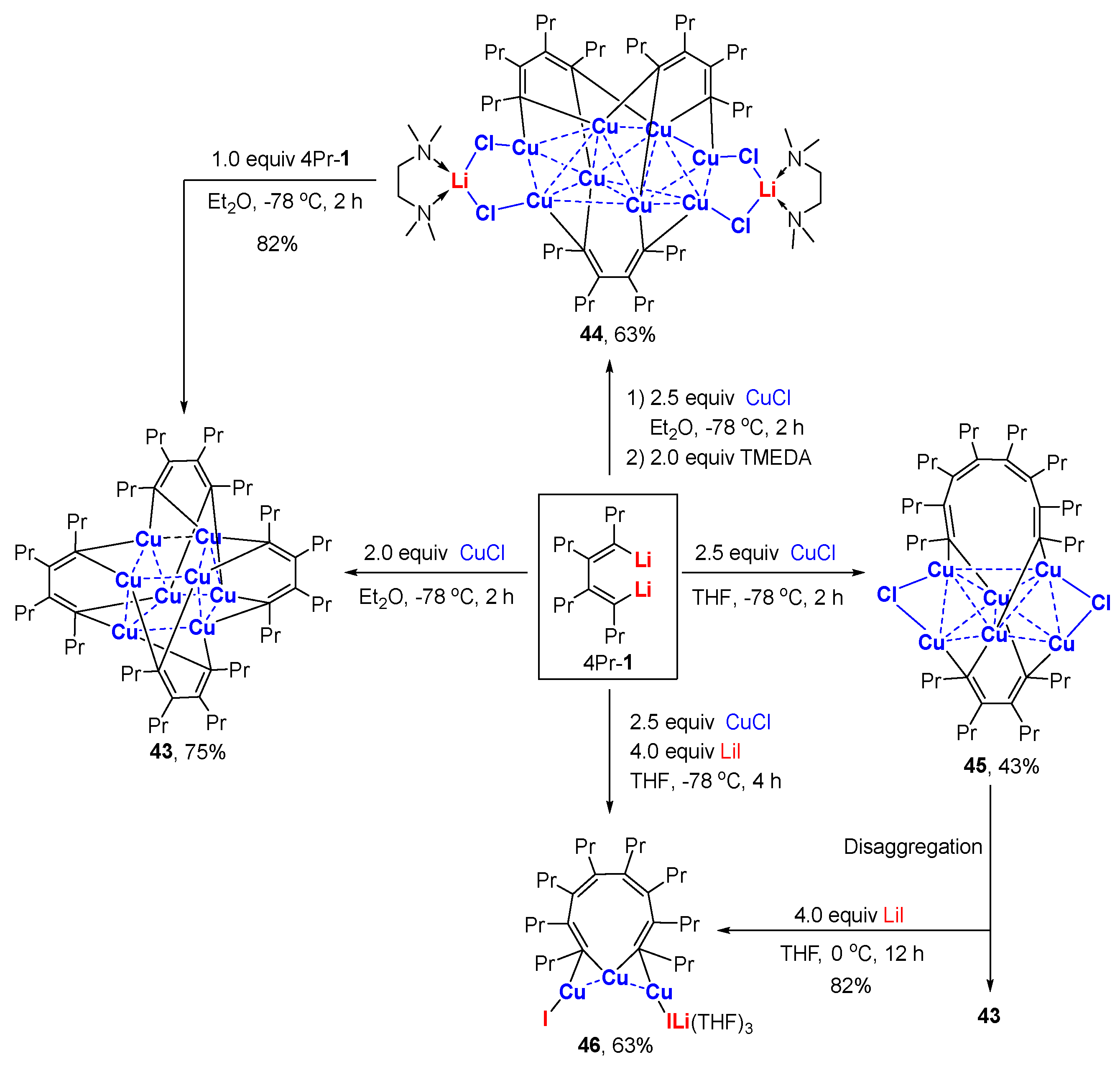
Fifteen years ago, Xi and co-workers reported that the cis-1,3-butadienyl 1,4-dicopper species, obtained from the reaction beteen dilithio reagent and 2.0 equiv. of CuCl, efficiently produced multi-substituted semibullvalenes after thermolysis at 50 ℃ in non-coordinating toluene solvent [85]. Later on, it was found that the same copper species underwent lithium iodide-assisted linear dimerization in Et2O at 0 °C to afford an all-cis octatetraenyl dicopper intermediate that could be subject to subsequent Pd-catalyzed cross-coupling reactions with halides [86]. In order to unveil the nature of butadienyl copper species, Xi and co-workers carefully conducted transmetalation reactions at a low temperature (−78 ℃) in order tto avoid uncontrollable thermal decompositions. The reaction of the dilithio reagents 4Pr-1 with 2.0 equiv. of CuCl in Et2O afforded 1,4-dicopper tetramer 43 at a 75% yield (Scheme 12) [56]. When 2.5 equiv. of CuCl was used in the presence of 2.0 equiv. of TMEDA, a different copper aggregate 44 was obtained at a 63% yield. Compound 44 could be transformed to 43 through treatment with 1.0 equiv. of dilithio reagent. Additionally, the products were also dependent on the solvents and additives, e.g., lithium salts. When THF was used as a solvent, the reaction between dilithio reagent and 2.5 equiv. of CuCl produced 45, which was linked by a 1,3-butadienyl and 1,3,5,7-octatetraenyl units at a 43% yield. In the presence of extra LiI, the above reaction produced the 1,3,5,7-octatetraenyl-1,8-dicopper compound 46 at a 63% yield. The unit of 1,3,5,7-octatetraenyl was formed from the dimerization of butadienyl copper complexes, as observed in previous work [86]. Compounds 46 and 43 can also be generated from the reaction between 45 and LiI. Structural analysis of 43–46 reveals that the Cu-C bonds and C-Cu-C bond angles are in the range of 1.96–2.08 Å and 73–84°, respectively. The close Cu-Cu distances (2.37–3.01 Å) suggests weak d10-d10 interactions in all of the copper aggregates above (Figure 6).
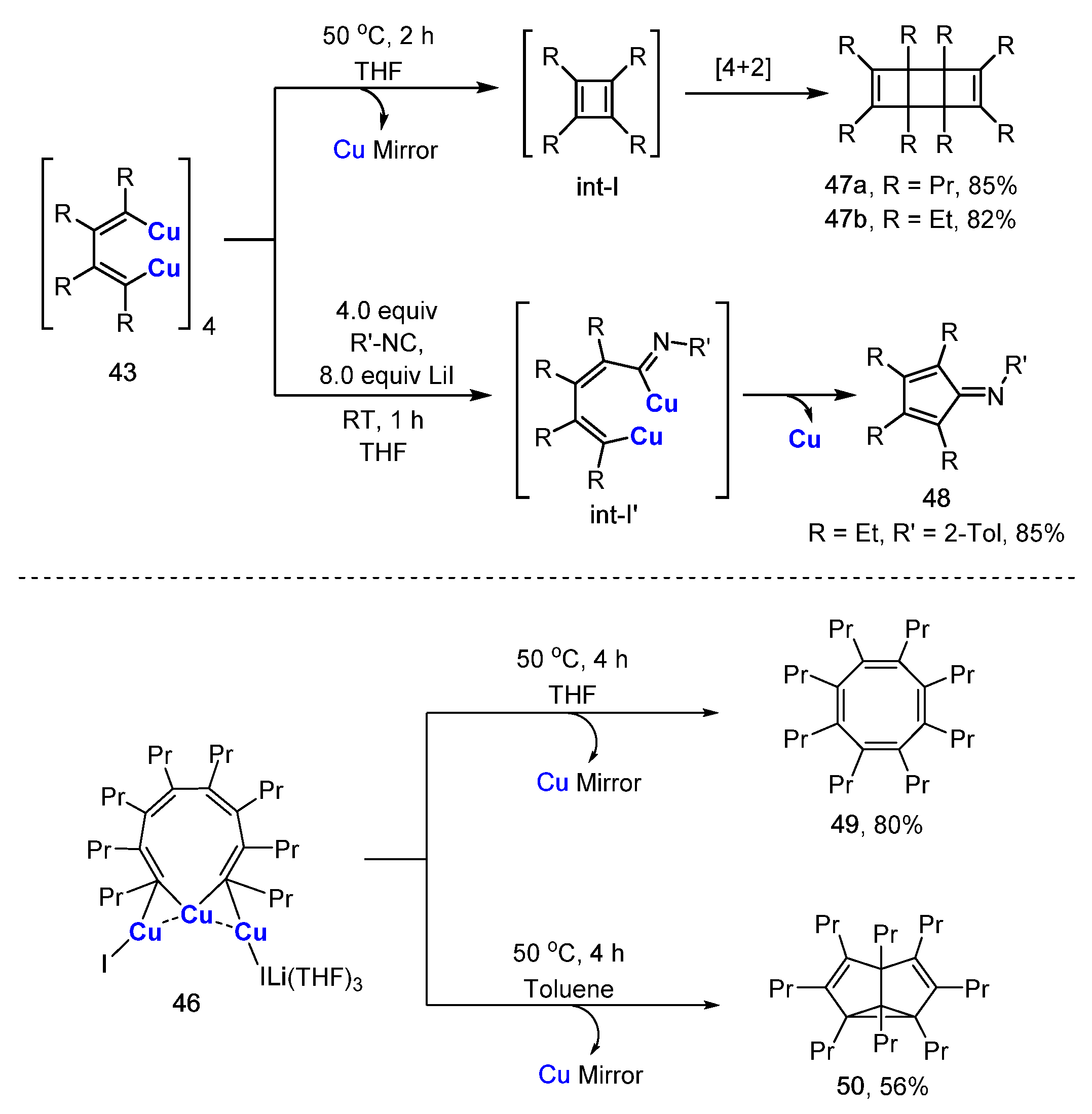
The reactivity of the above bis-enyl copper compounds was displayed by the reactions of 43 and 46 (Scheme 13). Compound 43 produced tricycle-octa-diene 47 and cyclopenta-2,4-dien-1-imine 48 via thermolysis and the insertion of isocyanide, respectively. When heated in THF, 46 afforded multi-substituted cyclooctatetraene 49. In toluene, the thermolysis of 46 produced semibullvalene 50 as the major product, which was consistent with earlier work [85].
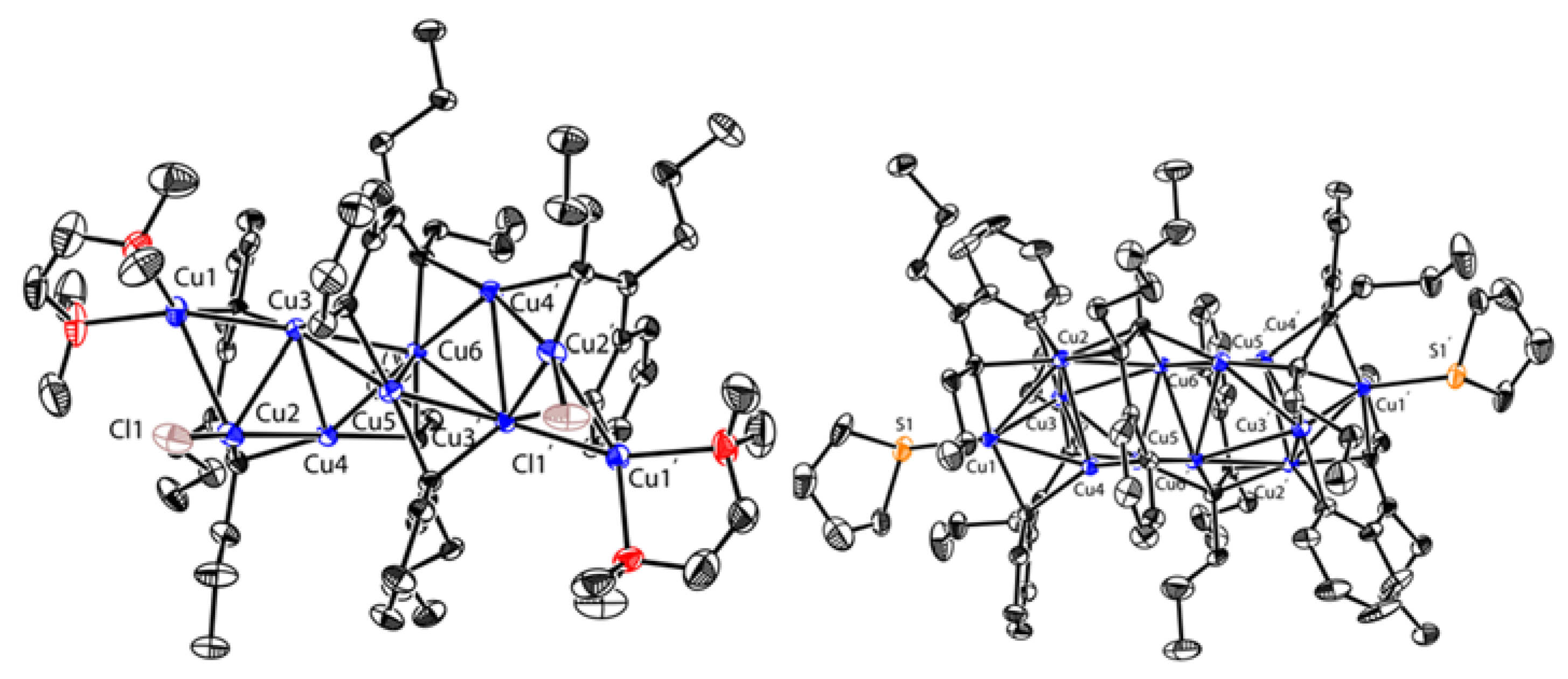
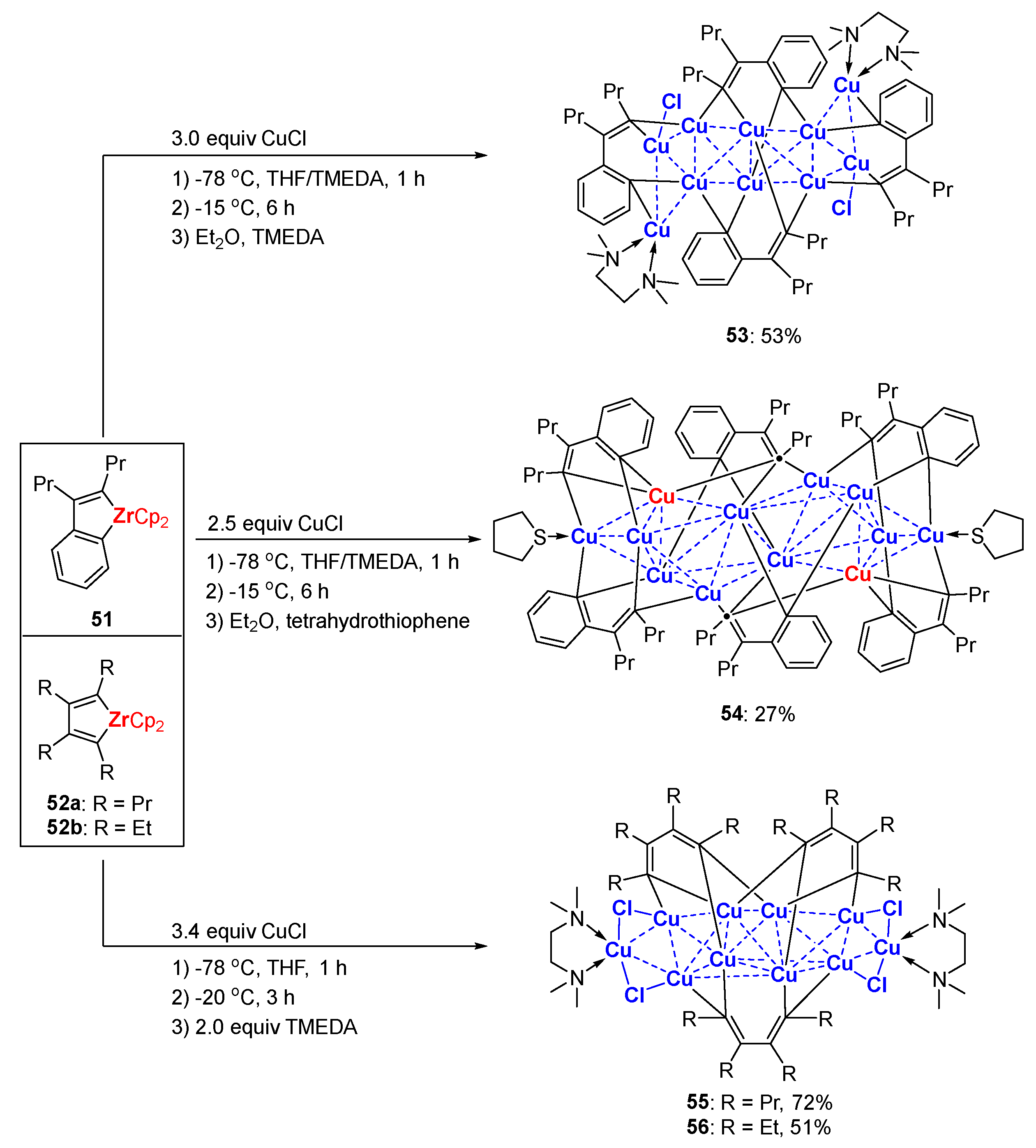
Copper-mediated reactions of zirconacyclopentadienes (or zirconaindenes) and electrophiles developed by Takahashi and others are extensively used to construct a variety of functional organic molecules [87,88]. However, organocopper intermediates remain elusive though the methodologies were reported about twenty years ago. Inspired by this work, Xi’s group revisited the transmetalation reaction between zirconium reagents and copper salts. Salt-free zirconaindenes 51 and zirconacyclopentadienes 52 were readily prepared according to the reported procedure [87]. A 1:3 reaction of 51 and CuCl in THF/ TMEDA afforded a tetrameric styrenyl copper aggregate 53 at a 53% isolated yield (Scheme 14) [57]. When 51 was treated with 2.5 equiv. of CuCl, a hexameric styrenyl copper aggregate 54 was obtained at a 27% isolated yield after crystallization in Et2O and tetrahydrothiophene (THT). Zirconacyclopentadienes 52a,b reacted with 3.4 equiv. of CuCl in THF, affording trimeric butadienyl copper aggregates 55 and 56 at 72% and 51% yields, respectively. The additional ancillary ligand (TMEDA vs. THT) could change the aggregate state of the styrenyl clusters. In terms of molecular structures, 53 consists of ten copper atoms, while 54 is a hexanuclear cluster with twelve copper atoms. The lengths of the Cu-C(alkenyl) bonds (1.98–2.01 Å) are comparable to those of the Cu-C(aryl) bonds (1.98–2.02 Å), as shown in 53 (Figure 7). The binding mode of 54 looks more complicated and also more unique. Compound 54 comprises two unusual μ3-alkenyl carbon atoms (black dots: C17, C17′) and the corresponding two tri-coordinated copper atoms (red: Cu2, Cu2′). Compounds 55 and 56 share identical core copper structures. In addition, 55 and 56 can be regarded as variants of 44 by replacing the two lateral lithium atoms with copper atoms. The substituents of the butadienyl skeleton seem to have an influence on the stability of their copper clusters. In Et2O, 55 is relatively stable below 10 ℃, whereas 56 is would thermally decompose and release the copper mirror at 10 ℃ within 0.5 h. The reactivity of 55 was provided by its coupling reaction with diiodobenzene, producing multi-substituted naphthalene.
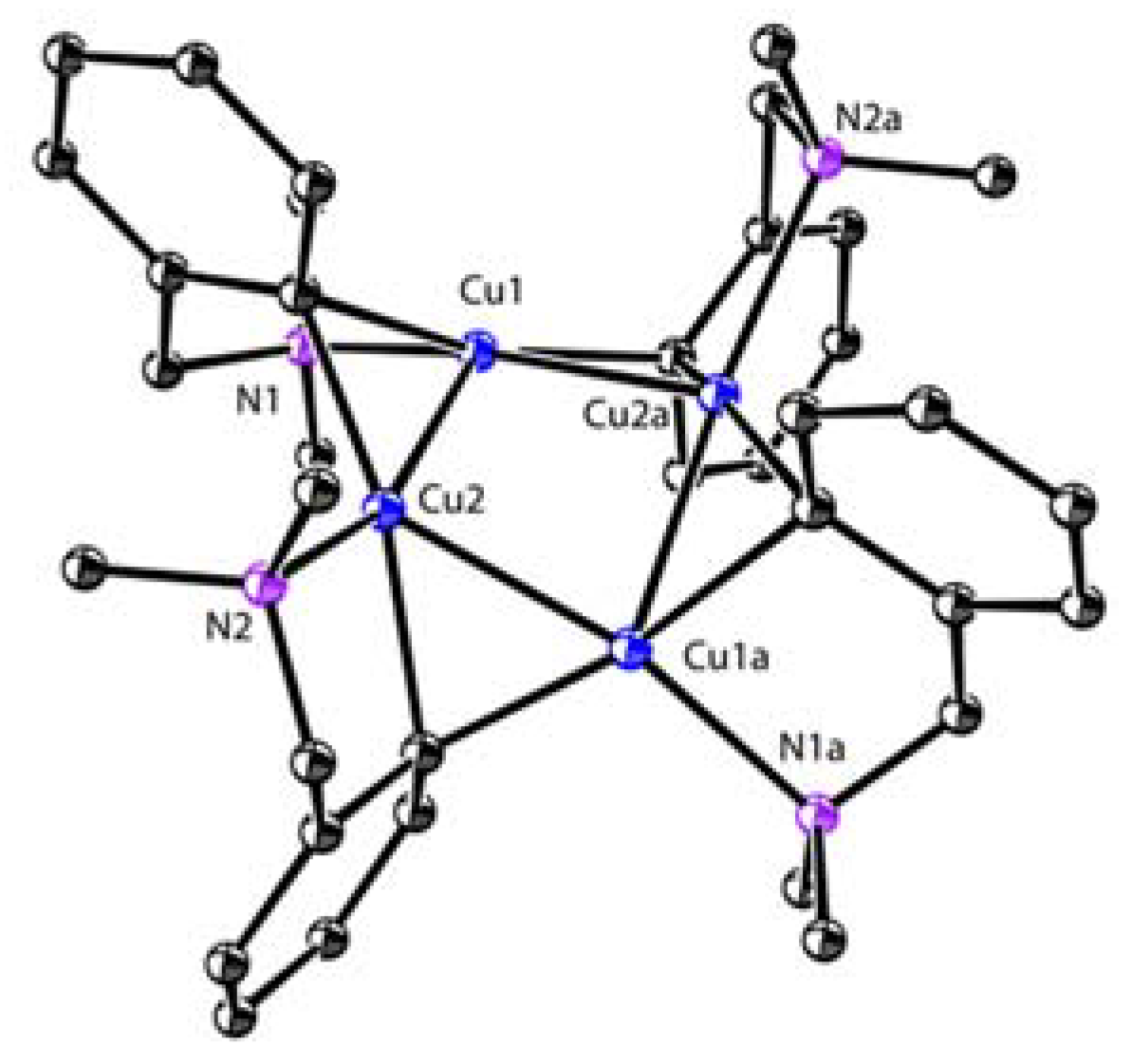
The intramolecular stabilization effect developed by van Koten was initially used to synthesize organocopper clusters. The reaction of 29 with 1.0 equiv. of CuBr afforded a LiBr-free tetranuclear copper cluster 31, which is thermally stable up to 170–185 ℃ (phenylcopper: slow decomposition at room temperature) [89,90]. The molecular structure of 31 comprises four copper atoms in a butterfly arrangement (Scheme 15) [91,92]. Each Cipso bridges two copper atoms via the 3c-2e Cu-C bonds (1.97–2.16 Å). Each copper atom has a distorted trigonal planar geometry (Figure 8). Variable temperature 1H NMR spectroscopy shows that the methylene and NMe2 signals are singlets that are independent of the temperature, which suggest a weak Me2N→Cu interaction in solution. Whereas the reaction between 31 and 1.0 equiv. of dppe afforded a 1:1 donor–acceptor copper complex 57, the addition of excess dppe resulted in an unusual C-P bond cleavage with the formation of a phosphidocopper complex 58, Ph2PCH=CH2, and Ar-H [93,94]. This process can be explained by a concerted mechanism, as shown in the box. A 1:1 reaction of 31 with CuBr affords a polymeric complex 59. When cupric halides were used instead, the oxidative coupling products Ar-Ar, Ar-X, and Ar-H were obtained [95]. In addition, van Koten and co-workers reported a serial of organocopper aggregates with the N-C-C-C-Cu connectivity supported on other C,N-bidentate ligands, such as 8-(dimethylamino)naphthyl [96], 2-oxazoline-4-methylphenyl [97,98], and (2-(dimethylamino)phenyl)-1-vinyl [99,100,101,102].
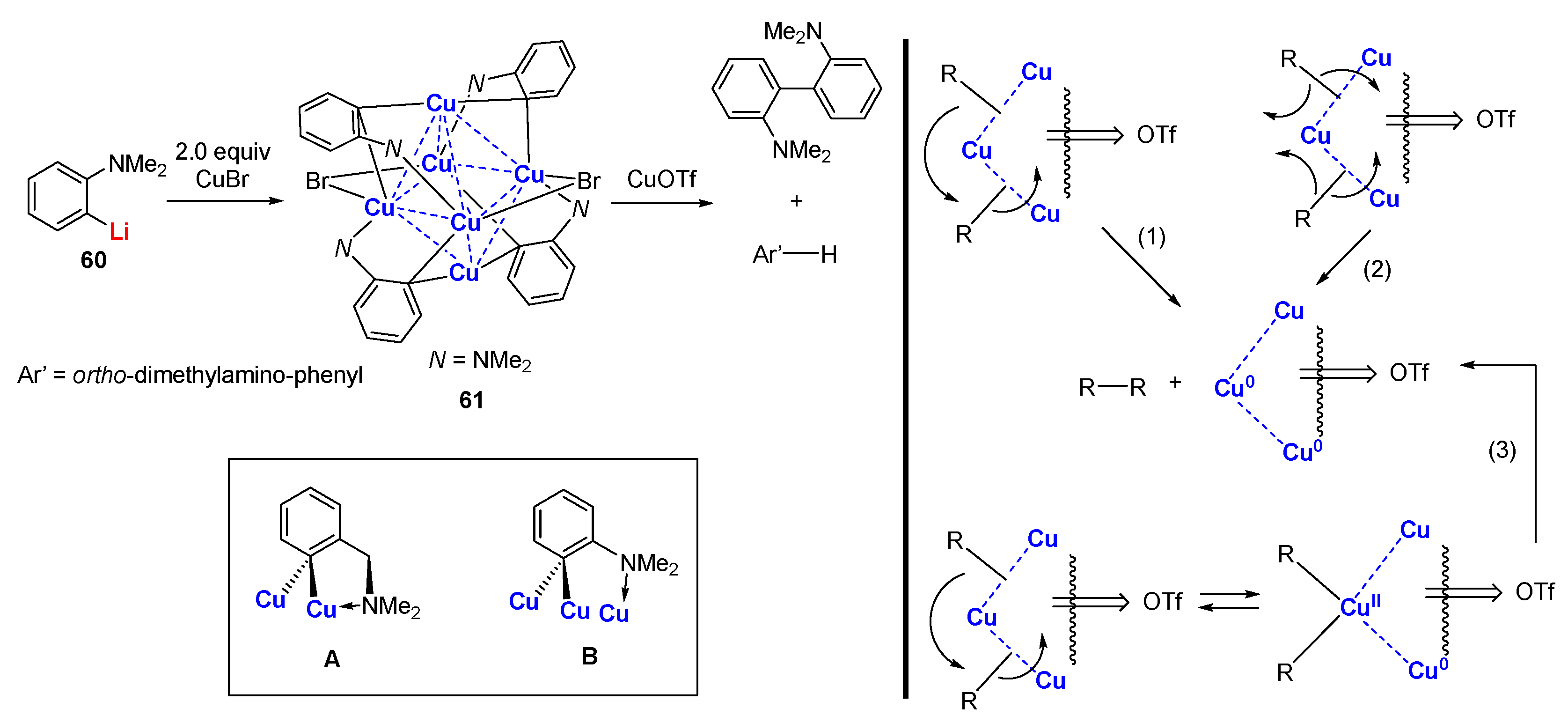
When a built-in heteroatom moiety is directly attached to the phenyl skeleton, the C,N-bidentate ligand displays different features when binding to the copper atoms (N-C-C-Cu connectivity). Van Koten reported that the treatment of 60 with copper bromide affords a hexanuclear copper cluster 61 that includes six copper atoms in a distorted octahedral arrangement (Scheme 16) [103,104,105,106]. Each bromide atom bridges the trans-equatorial edges. Each C,N-bidentate ligand bridges three copper atoms on the face of an octahedron. Molecular weight determinations and 1H NMR spectroscopy supported that 61 retained its aggregate structure in solution. Me2N was observed as two broadened singlets (1.84, 2.92 ppm) at room temperature, suggesting a relatively strong Me2N→Cu bonding interaction in contrast to the one seen in 31. When the temperature increases to 90 ℃, the two singlets coalesced to a single singlet (2.42 ppm). In 31, the Cipso atom and Me2N bind to the same copper atom, leading to a 5-membered chelate ring (A), whereas in 61, Me2N→Cu coordination takes place with a different copper atom to which the aryl group is not bonded (B), which is due to the steric effects. When 61 was treated with CuOTf, a selective, quantitative coupling of biaryls Ar’-Ar’ occurred, which was unexpected [107,108]. Only a trace amount of Ar’-H was formed, which excluded the pathway involving free radicals. The charge transfer from the core coppers to the strongly electron accepting OTf groups, which reduces the electron density of the core skeleton, accounts for the C-C bond formation. The proposed mechanism (3), including the valence disproportionation inside the metal core followed by the reductive elimination of Ar’-Ar’, rationalized the large influence of the identity of the anions.
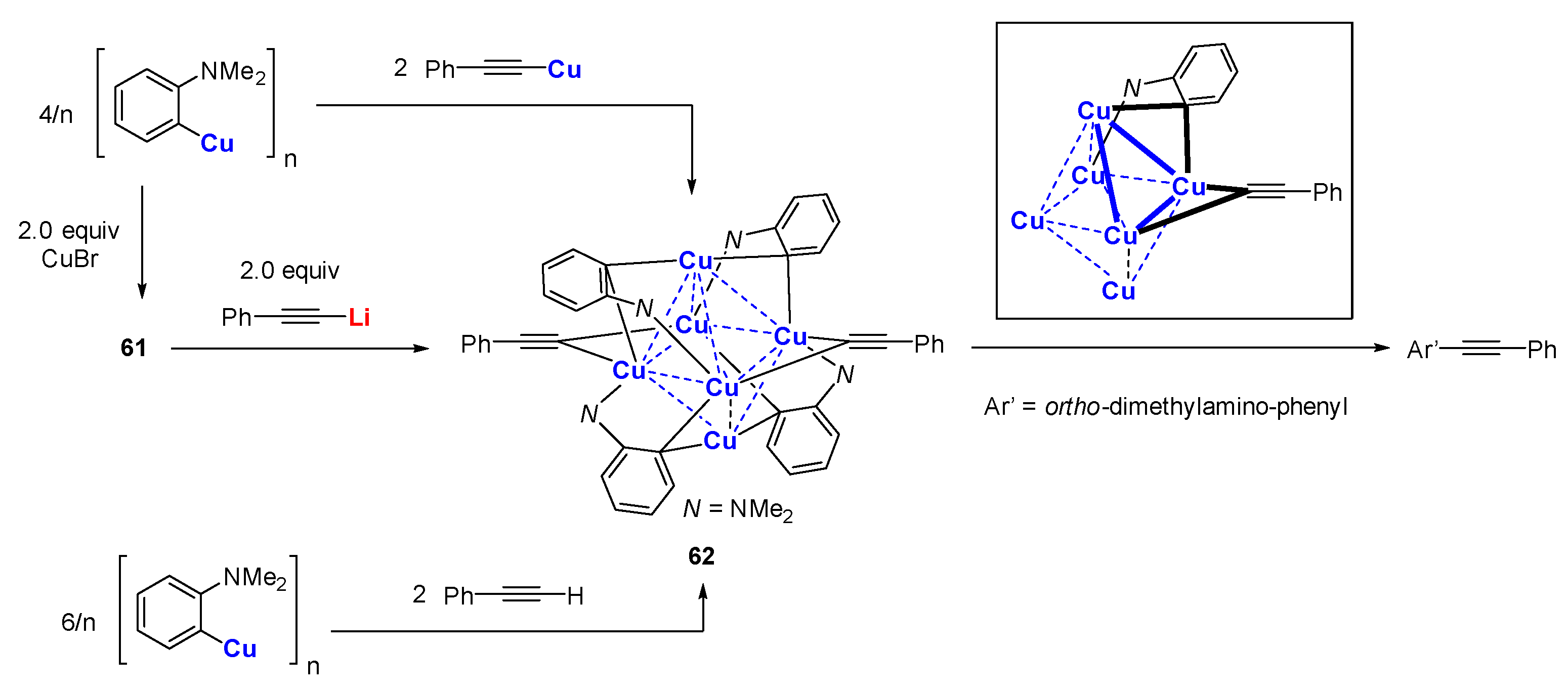
Based on the earlier work shown in Scheme 16, van Koten reported that 62 could be synthesized via three methods (Scheme 17) [109,110,111,112]. The thermolysis of Ar’4(CCPh)2Cu6 62 results in the exclusive formation of the mixed coupling product Ar’-CCPh. The absence of Ar’-H or PhCC-H from hydrogen abstraction indicates that the reaction undergoes a concerted pathway rather than proceed via free radicals. The molecular structure of 62 comprises exclusively triangular copper faces occupied by one Ar’ and one PhCC ligand, whereas Cu3Ar’2 or Cu3(CCPh)2 faces are absent, explaining the high selectivity of the asymmetric C-C bond formation (Figure 9).
6. Conclusions and Perspective
In this review, we have summarized the structurally well-defined organocopper compounds based on C,N- and C,C-bidentate ligands, from mononuclear copper spiro compounds to polynuclear copper clusters. It was shown that the intramolecular stabilization effect of the C,N-bidentate ligands and the cooperative effect of the C,C-bidentate ligands (cis-1,3-butadienyl) can both enhance the solubility and stability of the resulting organocopper compounds. In the molecular structures, copper(I) atoms adopt linear, trigonal, or tetrahedral geometries, depending on the coordination numbers. In charge-neutral copper aggregates, the electron-deficient 3c-2e Cu-C bonds are predominant, whereas in more electron-rich organocuprates, the Cu-C bonds have a much higher degree of 2c-2e bond character.
The strategy for enhancing the intramolecular coordination was further developed by adding two build-in heteroatom moieties to the aryl ring. Consequently, the original ligands turn into tridentate pincer ligands, e.g., C,N,N and N,C,N-ligands [29], which are now widely used in organic synthesis and small molecules activations. Another point of interest in organocopper chemistry is high-valent organocopper compounds [113]. It was found that the macrocyclic ligands with three intramolecular heteroatom moieties, e.g., C,N,N,N-cyclic ligands, were excellent platforms to support organocopper(III) compounds. Moreover, there has been increasing interest in more challenging organocopper(II) compounds in recent years since many Cu-catalyzed reactions may have Cu(II)-C containing species as their intermediates.
Author Contributions
Literature search and draft preparation, L.L.; discussion and reading, H.C. and Z.Y.; review and editing, J.W.; Supervision, Z.X. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (No. 21690061).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ley, S.V.; Thomas, A.W. Modern Synthetic Methods for Copper-Mediated C(aryl)–O, C(aryl)–N, and C(aryl)–S Bond Formation. Angew. Chem. Int. Ed. 2003, 42, 5400–5449. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Sperotto, E.; van Klink, G.P.M.; van Koten, G.; de Vries, J.G. The mechanism of the modified Ullmann reaction. Dalton Trans. 2010, 39, 10338–10351. [Google Scholar] [CrossRef] [Green Version]
- Beletskaya, I.P.; Cheprakov, A.V. The Complementary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 2012, 31, 7753–7808. [Google Scholar] [CrossRef]
- Gephart, R.T.; Warren, T.H. Copper-Catalyzed sp3 C–H Amination. Organometallics 2012, 31, 7728–7752. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, C.; Jiao, N. Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chem. Soc. Rev. 2012, 41, 3464–3484. [Google Scholar] [CrossRef]
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev. 2013, 113, 6234–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evano, G.; Blanchard, N. Copper-Mediated Cross-Coupling Reactions; Evano, G., Blanchard, N., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Lazreg, F.; Nahra, F.; Cazin, C.S.J. Copper–NHC complexes in catalysis. Coord. Chem. Rev. 2015, 293–294, 48–79. [Google Scholar] [CrossRef] [Green Version]
- McCann, S.D.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst. Acc. Chem. Res. 2015, 48, 1756–1766. [Google Scholar] [CrossRef] [Green Version]
- Jordan, A.J.; Lalic, G.; Sadighi, J.P. Coinage Metal Hydrides: Synthesis, Characterization, and Reactivity. Chem. Rev. 2016, 116, 8318–8372. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Sengupta, S. Organocopper Reagents: Substitution, Conjugate Addition, Carbo/Metallocupration, and Other Reactions. Org. React. 1992, 135–631. [Google Scholar]
- Surry, D.S.; Spring, D.R. The oxidation of organocuprates-an offbeat strategy for synthesis. Chem. Soc. Rev. 2006, 35, 218–225. [Google Scholar] [CrossRef]
- Breit, B.; Schmidt, Y. Directed Reactions of Organocopper Reagents. Chem. Rev. 2008, 108, 2928–2951. [Google Scholar] [CrossRef]
- Ullenius, C.; Christenson, B.L. Organocuprate addition to α,β-unsaturated compounds: Synthetic and mechanistic aspects. Pure Appl. Chem. 1988, 60, 57–64. [Google Scholar] [CrossRef]
- Woodward, S. Decoding the ‘black box’ reactivity that is organocuprate conjugate addition chemistry. Chem. Soc. Rev. 2000, 29, 393–401. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Tawney, P.O. Factors Determining the Course and Mechanisms of Grignard Reactions. II. The Effect of Metallic Compounds on the Reaction between Isophorone and Methylmagnesium Bromide. J. Am. Chem. Soc. 1941, 63, 2308–2316. [Google Scholar] [CrossRef]
- Krause, N. Modern Organocopper Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002. [Google Scholar]
- Rappoport, Z.; Marek, I. The Chemistry of Organocopper Compounds; John Wiley & Sons Ltd.: Chichester, UK, 2009. [Google Scholar]
- Piazza, C.; Knochel, P. Sterically Hindered Lithium Dialkylcuprates for the Generation of Highly Functionalized Mixed Cuprates through a Halogen–Copper Exchange. Angew. Chem. Int. Ed. 2002, 41, 3263–3265. [Google Scholar] [CrossRef]
- Zanardi, A.; Novikov, M.A.; Martin, E.; Benet-Buchholz, J.; Grushin, V.V. Direct Cupration of Fluoroform. J. Am. Chem. Soc. 2011, 133, 20901–20913. [Google Scholar] [CrossRef]
- Ebert, G.; Rieke, R.D. Direct formation of organocopper compounds by oxidative addition of zerovalent copper to organic halides. J. Org. Chem. 1984, 49, 5280–5282. [Google Scholar] [CrossRef]
- Gilman, H.; Jones, R.G.; Woods, L.A. The Preparation of Methylcopper and some Observations on the Decomposition of Organocopper Compounds. J. Org. Chem. 1952, 17, 1630–1634. [Google Scholar] [CrossRef]
- Jukes, A.E. The Organic Chemistry of Copper. In Advances in Organometallic Chemistry; Stone, F.G.A., West, R., Eds.; Academic Press: New York, NY, USA, 1974; Volume 12, pp. 215–322. [Google Scholar]
- Costa, G.; Camus, A.; Gatti, L.; Marsich, N. On phenylcopper. J. Organomet. Chem. 1966, 5, 568–572. [Google Scholar] [CrossRef]
- Stephens, R.D.; Castro, C.E. The Substitution of Aryl Iodides with Cuprous Acetylides. A Synthesis of Tolanes and Heterocyclics. J. Org. Chem. 1963, 28, 3313–3315. [Google Scholar] [CrossRef]
- van Koten, G. A view of organocopper compound and cuprates. J. Organomet. Chem. 1990, 400, 283–301. [Google Scholar] [CrossRef]
- van Koten, G. Organocopper Compounds: From Elusive to Isolable Species, from Early Supramolecular Chemistry with RCuI Building Blocks to Mononuclear R2–nCuII and R3–mCuIII Compounds. A Personal View. Organometallics 2012, 31, 7634–7646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, W.-X.; Xi, Z. Organo-di-Lithio Reagents: Cooperative Effect and Synthetic Applications. In Organo-di-Metallic Compounds (or Reagents): Synergistic Effects and Synthetic Applications; Xi, Z., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–41. [Google Scholar]
- Zhang, W.-X.; Xi, Z. Organometallic intermediate-based organic synthesis: Organo-di-lithio reagents and beyond. Org. Chem. Front. 2014, 1, 1132–1139. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, Y.; Chi, Y.; Liu, L.; Zhang, W.-X.; Xi, Z. Aromatic Dicupra[10]annulenes. J. Am. Chem. Soc. 2016, 138, 60–63. [Google Scholar] [CrossRef]
- Green, J.C.; Green, M.L.H.; Parkin, G. The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds. Chem. Commun. 2012, 48, 11481–11503. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, W.-X.; Xi, Z. 1,3-Butadienylzinc Trimer Formed via Transmetalation from 1,4-Dilithio-1,3-butadienes: Synthesis, Structural Characterization, and Application in Negishi Cross-Coupling. Organometallics 2012, 31, 5546–5550. [Google Scholar] [CrossRef]
- Wei, J.; Liu, L.; Zhan, M.; Xu, L.; Zhang, W.-X.; Xi, Z. Magnesiacyclopentadienes as Alkaline-Earth Metallacyclopentadienes: Facile Synthesis, Structural Characterization, and Synthetic Application. Angew. Chem. Int. Ed. 2014, 53, 5634–5638. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, W.-X.; Xi, Z. Dianions as Formal Oxidants: Synthesis and Characterization of Aromatic Dilithionickeloles from 1,4-Dilithio-1,3-butadienes and [Ni(cod)2]. Angew. Chem. Int. Ed. 2015, 54, 5999–6002. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, Y.; Zhang, W.-X.; Xi, Z. 1,3-Butadienyl Dianions as Non-Innocent Ligands: Synthesis and Characterization of Aromatic Dilithio Rhodacycles. Angew. Chem. Int. Ed. 2015, 54, 9986–9990. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Y.C.; Wei, J.; Wang, Y.; Wang, Z.; Zhang, W.-X.; Xi, Z. The First Lutetacyclopentadienes: Synthesis, Structure, and Diversified Insertion/C–H Activation Reactivity. Chem. Eur. J. 2015, 21, 6686–6689. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Zhang, W.-X.; Xi, Z. Lithium Aluminate Complexes and Alumoles from 1,4-Dilithio-1,3-Butadienes and AlEt2Cl. lnorg. Chem. 2015, 54, 10695–10700. [Google Scholar] [CrossRef]
- Xu, L.; Wang, Y.; Wang, Y.-C.; Wang, Z.; Zhang, W.-X.; Xi, Z. Sandwich Lutetacyclopentadiene with the Coordination of Lithium to the Diene Unit: Synthesis, Structure, and Transformation. Organometallics 2016, 35, 5–8. [Google Scholar] [CrossRef]
- Ma, W.; Yu, C.; Chi, Y.; Chen, T.; Wang, L.; Yin, J.; Wei, B.; Xu, L.; Zhang, W.-X.; Xi, Z. Formation and ligand-based reductive chemistry of bridged bis-alkylidene scandium(III) complexes. Chem. Sci. 2017, 8, 6852–6856. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.; Liu, L.; Zhang, W.-X.; Xi, Z. Synthesis and Structural Characterization of Butadienylcalcium-based Heavy Grignard Reagents and a Ca4[O] Inverse Crown Ether Complex. Angew. Chem. Int. Ed. 2017, 56, 9188–9192. [Google Scholar] [CrossRef]
- Zhang, Y.; Chi, Y.; Wei, J.; Yang, Q.; Yang, Z.; Chen, H.; Yang, R.; Zhang, W.-X.; Xi, Z. Aromatic Tetralithiodigalloles with a Ga–Ga Bond: Synthesis and Structural Characterization. Organometallics 2017, 36, 2982–2986. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Chi, Y.; Zhang, X.; Zhang, W.-X.; Xi, Z. Spiro Metalla-aromatics of Pd, Pt, and Rh: Synthesis and Characterization. J. Am. Chem. Soc. 2017, 139, 5039–5042. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, L.; Zhang, Y.; Yu, H.T.; Zhang, W.-X.; Xi, Z. Selective Transformation of Well-Defined Alkenyllithiums to Alkenylmagnesiums via Transmetalation. Chem. Eur. J. 2017, 24, 3186–3191. [Google Scholar] [CrossRef]
- Wei, B.; Zhang, W.-X.; Xi, Z. Well-defined styryl and biphenyl calcium complexes from dilithio compounds and calcium iodide: Synthesis, structure and reactivity toward nitrous oxide. Dalton Trans. 2018, 47, 12540–12545. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Y.; Zhang, W.-X.; Xi, Z. Reversible Two-Electron Redox Reactions Involving Tetralithio/Dilithio Palladole, Platinacycle, and Dicupra[10]annulene. Organometallics 2019, 38, 2807–2811. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Chen, T.; Huang, Z.; Zhang, W.-X.; Xi, Z. Dilithio Spiro Zincacyclopentadienes and Dizinca[10]cycles: Synthesis and Structural Characterization. Organometallics 2019, 38, 2174–2178. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Zhu, M.; Chi, Y.; Zhang, W.-X.; Ye, S.; Xi, Z. Tetralithio Metalla-aromatics with Two Independent Perpendicular Dilithio Aromatic Rings Spiro-fused by One Manganese Atom. Angew. Chem. Int. Ed. 2019, 58, 9625–9631. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Z.; Zhang, W.-X.; Xi, Z. Indacyclopentadienes and Aromatic Indacyclopentadienyl Dianions: Synthesis and Characterization. Chem. Eur. J. 2019, 25, 4218–4224. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Ma, W.; Zhang, W.-X.; Xi, Z. Mono- and Bis-Titanium Complexes Bridged by 2-Butene Tetraanion: Synthesis and Structural Characterization. Organometallics 2020, 39, 793–796. [Google Scholar] [CrossRef]
- Yu, C.; Zhong, M.; Zhang, Y.; Wei, J.; Ma, W.; Zhang, W.-X.; Ye, S.; Xi, Z. Butadienyl Diiron Complexes: Nonplanar Metalla-Aromatics Involving σ-Type Orbital Overlap. Angew. Chem. Int. Ed. 2020, 59, 19048–19053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, B.; Zhong, M.; Zhang, W.-X.; Xi, Z. Cyclic Bis-alkylidene Complexes of Titanium and Zirconium: Synthesis, Characterization, and Reaction. Chem. Eur. J. 2020, 26, 16472–16479. [Google Scholar] [CrossRef]
- Zheng, Y.; Cao, C.-S.; Ma, W.; Chen, T.; Wu, B.; Yu, C.; Huang, Z.; Yin, J.; Hu, H.-S.; Li, J.; et al. 2-Butene Tetraanion Bridged Dinuclear Samarium(III) Complexes via Sm(II)-Mediated Reduction of Electron-Rich Olefins. J. Am. Chem. Soc. 2020, 142, 10705–10714. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Y.; Zhang, W.-X.; Wei, J.; Ye, S.; Xi, Z. A tris-spiro metalla-aromatic system featuring Craig-Möbius aromaticity. Nat. Commun. 2021, 12, 1319. [Google Scholar] [CrossRef]
- Geng, W.; Wei, J.; Zhang, W.-X.; Xi, Z. Isolable and Well-Defined Butadienyl Organocopper(I) Aggregates: Facile Synthesis, Structural Characterization, and Reaction Chemistry. J. Am. Chem. Soc. 2014, 136, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Geng, W.; Yang, Q.; Zhang, W.-X.; Xi, Z. Well-Defined Butadienyl Organocopper(I) Aggregates from Zirconacyclopentadienes and CuCl: Synthesis and Structural Characterization. Organometallics 2015, 34, 4198–4201. [Google Scholar] [CrossRef]
- Liu, L.; Wei, J.; Chi, Y.; Zhang, W.-X.; Xi, Z. Structure and Reaction Chemistry of Magnesium Organocuprates Derived from Magnesiacyclopentadienes and Copper(I) Salts. Angew. Chem. Int. Ed. 2016, 55, 14762–14765. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, M.; Yu, H.-T.; Zhang, W.-X.; Xi, Z. Organocopper(III) Spiro Complexes: Synthesis, Structural Characterization, and Redox Transformation. J. Am. Chem. Soc. 2017, 139, 13688–13691. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, M.; Yu, H.-T.; Zhang, W.-X.; Xi, Z. Formation of a Hexanuclear Octatetraenyl Organocopper(I) Aggregate via Oxidation of Spiro Butadienyl Organocuprate. Organometallics 2018, 37, 845–847. [Google Scholar] [CrossRef]
- DiMucci, I.M.; Lukens, J.T.; Chatterjee, S.; Carsch, K.M.; Titus, C.J.; Lee, S.J.; Nordlund, D.; Betley, T.A.; MacMillan, S.N.; Lancaster, K.M. The Myth of d8 Copper(III). J. Am. Chem. Soc. 2019, 141, 18508–18520. [Google Scholar] [CrossRef] [PubMed]
- García-López, J.; Yañez-Rodríguez, V.; Roces, L.; García-Granda, S.; Martínez, A.; Guevara-García, A.; Castro, G.R.; Jiménez-Villacorta, F.; Iglesias, M.J.; López-Ortiz, F. Synthesis and Characterization of a Coupled Binuclear CuI/CuIII Complex. J. Am. Chem. Soc. 2010, 132, 10665–10667. [Google Scholar] [CrossRef]
- Molteni, R.; Edkins, K.; Haehnel, M.; Steffen, A. C–H Activation of Fluoroarenes: Synthesis, Structure, and Luminescence Properties of Copper(I) and Gold(I) Complexes Bearing 2-Phenylpyridine Ligands. Organometallics 2016, 35, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.P. The structures of lithium and magnesium organocuprates and related species. Coord. Chem. Rev. 2011, 255, 1226–1251. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. Novel aryl-bridged tetranuclear copper–lithium cluster compounds: Synthesis and characterization by 13C and 1H nuclear magnetic resonance spectroscopy of bis-{2-[(dimethylamino)methyl]phenyl}copper(I) lithium. J. Chem. Soc. Chem. Commun. 1972, 940–941. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. Group IB organometallic chemistry: XXIX. Synthetic and structural aspects of polynuclear arylcopperlithium compounds Ar4Cu2Li2 (“arylcuprates”) and interaggregate exchange phenomena in Ar4Cu4/Ar4Li4/Ar4Cu2Li2 systems. J. Organomet. Chem. 1979, 174, 367–387. [Google Scholar] [CrossRef]
- van Koten, G.; Jastrzebski, J.T.B.H.; Muller, F.; Stam, C.H. Crystal structure of the neutral diarylcuprate Cu2Li2(C6H4CH2NMe2-2)4: The asymmetric bonding configuration of organo groups bridging copper and lithium. J. Am. Chem. Soc. 1985, 107, 697–698. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. Group IB organometallic chemistry. 30. Stereochemistry of arylmetal-1B, aryllithium, and arylmetal-1B-lithium clusters Ar4M2Li2(M = copper, silver, or gold). Detection of rotation of three-center two-electron bonded aryl groups around the C(1)-C(4) axis. J. Am. Chem. Soc. 1979, 101, 6593–6599. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. Intramolecular dynamics of 3c-2e bonded aryl groups in polynuclear aryl-copper, -silver and -gold derivatives. J. Organomet. Chem. 1979, 171, C39–C43. [Google Scholar] [CrossRef] [Green Version]
- Schmidbaur, H.; Adlkofer, J.; Buchner, W. A New, Unusually Stable Type of Organo-copper and -silver Compounds. Angew. Chem. Int. Ed. Engl. 1973, 12, 415–416. [Google Scholar] [CrossRef]
- Nardin, G.; Randaccio, L.; Zangrando, E. Copper(I)-alkyl bonding in the dinuclear copper(I) compound [(CH3)2P(CH2)2]2Cu2. J. Organomet. Chem. 1974, 74, C23–C25. [Google Scholar] [CrossRef]
- Kaplan, P.T.; Xu, L.; Chen, B.; McGarry, K.R.; Yu, S.; Wang, H.; Vicic, D.A. Mild, Safe, and Versatile Reagents for (CF2)n Transfer and the Construction of Fluoroalkyl-Containing Rings. Organometallics 2013, 32, 7552–7558. [Google Scholar] [CrossRef]
- Grande-Aztatzi, R.; Mercero, J.M.; Matito, E.; Frenking, G.; Ugalde, J.M. The aromaticity of dicupra[10]annulenes. Phys. Chem. Chem. Phys. 2017, 19, 9669–9675. [Google Scholar] [CrossRef] [Green Version]
- An, K.; Shen, T.; Zhu, J. Craig-Type Möbius Aromaticity and Antiaromaticity in Dimetalla[10]annulenes: A Metal-Induced Yin-and-Yang Pair. Organometallics 2017, 36, 3199–3204. [Google Scholar] [CrossRef]
- Dimitrova, M.; Sundholm, D. The aromatic character of [10]annulenes and dicupra[10]annulenes from current density calculations. Phys. Chem. Chem. Phys. 2018, 20, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papasergio, R.I.; Raston, C.L.; White, A.H. Synthesis of pyridine functionalised, sterically hindered lithium and copper (I) alkyls; crystal structures of [{2-(Me3Si)2C(M)C5H4N}2](M = Li or Cu), dimeric compounds free of multicentre bonding. J. Chem. Soc. Chem. Commun. 1983, 1419–1420. [Google Scholar] [CrossRef]
- Papasergio, R.I.; Raston, C.L.; White, A.H. Syntheses and crystal structures of complexes [M2R2][M = Cu, Ag, or Au; R = 2-C(SiMe3)2C5H4N] and [Cu4R’4][R’ = 2-CH(SiMe3)C5H4N]; electrochemical generation of [Cu2R2]2+. J. Chem. Soc. Dalton Trans. 1987, 3085–3091. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Lappert, M.F.; Layh, M. Synthesis and molecular structures of copper(I) 1-azaallyls. J. Chem. Soc. Dalton Trans. 1998, 1619–1624. [Google Scholar] [CrossRef]
- Wingerter, S.; Gornitzka, H.; Bertrand, G.; Stalke, D. The Deprotonated Iminophosphorane o-C6H4PPh2P=NSiMe3 as a Novel Chelating Ligand in Organocopper(I) and -zinc(II) Chemistry. Eur. J. Inorg. Chem. 1999, 1999, 173–178. [Google Scholar] [CrossRef]
- van den Ancker, T.R.; Bhargava, S.K.; Mohr, F.; Papadopoulos, S.; Raston, C.L.; Skelton, B.W.; White, A.H. Syntheses and crystal structures of binuclear gold(I), silver(I) and copper(I) complexes containing bulky pyridyl functionalised alkyl ligands. J. Chem. Soc. Dalton Trans. 2001, 3069–3072. [Google Scholar] [CrossRef] [Green Version]
- Eaborn, C.; Hill, M.S.; Hitchcock, P.B.; Smith, J.D. Synthesis and structures of compounds of Groups 11 and 12 containing the ligand C(SiMe3)2(SiMe2C5H4N-2). J. Chem. Soc. Dalton Trans. 2002, 2467–2472. [Google Scholar] [CrossRef]
- Beck, J.F.; Schmidt, J.A.R. Isolation and characterization of main group and late transition metal complexes using orthometallated imine ligands. Dalton Trans. 2012, 41, 860–870. [Google Scholar] [CrossRef]
- Lappert, M.F.; Pearce, R. Trimethylsilylmethylcopper, a stable copper(I) alkyl. J. Chem. Soc. Chem. Commun. 1973, 24–25. [Google Scholar] [CrossRef]
- Jarvis, J.A.J.; Pearce, R.; Lappert, M.F. Silylmethyl and related complexes. Part 4. Preparation, properties, and crystal and molecular structure of tetrakis[(trimethylsilylmethyl)-copper(I)], an alkyl-bridged, square-planar, tetranuclear copper(I) cluster. J. Chem. Soc. Dalton Trans. 1977, 999–1003. [Google Scholar] [CrossRef]
- Wang, C.; Yuan, J.; Li, G.; Wang, Z.; Zhang, S.; Xi, Z. Metal-Mediated Efficient Synthesis, Structural Characterization, and Skeletal Rearrangement of Octasubstituted Semibullvalenes. J. Am. Chem. Soc. 2006, 128, 4564–4565. [Google Scholar] [CrossRef]
- Wei, J.; Wang, Z.; Zhang, W.-X.; Xi, Z. Construction of Octaalkyl-Substituted and Decasubstituted all-cis-Octatetraenes via Linear Dimerization of 1,4-Dicopper-1,3-butadienes and Subsequent Cross-Coupling with Halides. Org. Lett. 2013, 15, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Li, Y. Zirconacyclopentadienes in Organic Synthesis. In Titanium and Zirconium in Organic Synthesis; Marek, I., Ed.; Wiley-VCH: Weinheim, Germany, 2002; pp. 50–83. [Google Scholar]
- Yan, X.; Xi, C. Conversion of Zirconacyclopentadienes into Metalloles: Fagan–Nugent Reaction and Beyond. Acc. Chem. Res. 2015, 48, 935–946. [Google Scholar] [CrossRef] [PubMed]
- van Koten, G.; Leusink, A.J.; Noltes, J.G. Stable arylcopper compounds containing 2-(dimethylamino)methyl or 2-methoxymethyl groups at the aryl nucleus. J. Chem. Soc. D 1970, 1107–1108. [Google Scholar] [CrossRef] [Green Version]
- van Koten, G.; Leusink, A.J.; Noltes, J.G. Synthesis and properties of some 2-(dimethylamino)methyl-substituted arylcopper compounds. J. Organomet. Chem. 1975, 84, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Guss, J.M.; Mason, R.; Sotofte, I.; van Koten, G.; Noltes, J.G. A tetranuclear cluster complex of copper(I) with bridging aryl ligands: The crystal structure of (4-methyl-2-cupriobenzyl)dimethylamine. J. Chem. Soc. Chem. Commun. 1972, 446–447. [Google Scholar] [CrossRef] [Green Version]
- van Koten, G.; Noltes, J.G. Structural characterization of some 2-(dimethylamino)methyl-substituted phenylcopper compounds R4Cu4. J. Organomet. Chem. 1975, 84, 129–138. [Google Scholar] [CrossRef] [Green Version]
- van Koten, G.; Noltes, J.G. Complex-forming reactions of an arylcopper(I) cluster compound with organophosphines. An unexpected C(alkyl)–P bond cleavage in 1,2-bis(diphenylphosphino)ethane. J. Chem. Soc. Chem. Commun. 1972, 452–453. [Google Scholar] [CrossRef] [Green Version]
- van Koten, G.; Noltes, J.G.; Spek, A.L. Group IB organometallic chemistry: XXXVII. Complex forming reactions of polynuclear arylcopper compounds: Calk–P bond cleavage in 1,2-bis(diphenylphosphino)ethane (diphos) by (2-Me2NCH2C6H4)4Cu4 and crystal structure of [(C6H5)2PCu·diphos]2·2C6H6. J. Organomet. Chem. 1978, 159, 441–463. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. The reactions of 2-[(dimethylamino)methyl]phenylcopper and -lithium tetramer with cuprous and cupric halides. J. Organomet. Chem. 1975, 84, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Wehman, E.; van Koten, G.; Knotter, M.; Spelten, H.; Heijdenrijk, D.; Mak, A.N.S.; Stam, C.H. 8-(Dimethylamino)naphthylcopper(I), a novel stable organocopper compound with unusual structural features. Its synthesis, crystal structure (X-ray), and reactivity. J. Organomet. Chem. 1987, 325, 293–309. [Google Scholar] [CrossRef]
- Wehman, E.; van Koten, G.; Jastrzebski, J.T.B.H. Functionally substituted arylcopper compounds; crystal structure of the copper bromide complex of [2-(4,4-dimethyl-2-oxazoline)-4-methylphenylcopper]. J. Organomet. Chem. 1986, 302, C35–C39. [Google Scholar] [CrossRef] [Green Version]
- Wehman, E.; van Koten, G.; Jastrzebski, J.T.B.H.; Rotteveel, M.A.; Stam, C.H. Functionally substituted arylcopper compounds: Synthesis, structure, and reactivity of [2-(2-oxazolinyl)aryl]copper(I) species. Organometallics 1988, 7, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- ten Hoedt, R.W.M.; van Koten, G.; Noltes, J.G. Synthesis and characterization of the polynuclear organocopper–copper halide compound [(4-MeC6H4)MeC=C(C6H4NMe2-2)]2Cu4Br2 containing bridging vinyl groups. J. Organomet. Chem. 1978, 161, C13–C16. [Google Scholar] [CrossRef]
- ten Hoedt, R.W.M.; van Koten, G.; Noltes, J.G. Group IB organometallic chemistry: XXXI. Synthesis and characterization of tetranuclear Me2N- and Me2NCH2-substituted diarylpropenylcopper–copper anion compounds (Vi2Cu4X2) containing bridging propenyl ligands. Isolation of a thermally stable mixed diarylpropenyl/arylcopper compound (Vi2Cu4Ar2). J. Organomet. Chem. 1979, 179, 227–240. [Google Scholar] [CrossRef]
- ten Hoedt, R.W.M.; van Koten, G.; Noltes, J.G. Group IB organometallic chemistry: XXXIV. Thermal behaviour and chemical reactivity of tetranuclear Me2N-substituted diarypropenylcopper-copper anion (Vi2Cu4X2) and mixed diarylpropenyl/organocopper (Vi2Cu4R2) compounds. J. Organomet. Chem. 1980, 201, 327–342. [Google Scholar] [CrossRef]
- Noltes, J.G.; ten Hoedt, R.W.M.; van Koten, G.; Spek, A.L.; Schoone, J.C. Group IB organometallic chemistry: XXXV. Crystal and molecular structure of [Cu4(4-MeC6H4)-MeC=C(C6H4NMe2-2)2(C6H4NMe2-2)2], a tetranuclear organocopper compound containing bridging alkenyl and aryl groups. J. Organomet. Chem. 1982, 225, 365–376. [Google Scholar] [CrossRef]
- van Koten, G.; Leusink, A.J.; Noltes, J.G. Stable arylcopper compounds containing methoxy, dimethylamino, diphenylphosphino or dimethylsulfamoyl groups at the aryl nucleus. Inorg. Nucl. Chem. Lett. 1971, 7, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Guss, J.M.; Mason, R.; Thomas, K.M.; van Koten, G.; Noltes, J.G. Copper(I)-aryl bonds in cluster complexes: The structure of [Cu(2-Me2NC6H4)]4(CuBr)2·1.5C6H6. J. Organomet. Chem. 1972, 40, C79–C80. [Google Scholar] [CrossRef]
- van Koten, G.; Leusink, A.J.; Noltes, J.G. Synthesis and characterization of arylcopper compounds containing the methoxy or dimethylamino group as a built-in ligand. J. Organomet. Chem. 1975, 85, 105–114. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. Group IB organometallic chemistry: XVI. Complex formation between 2-(dimethylamino)-phenylcopper with copper(I) or silver halides. Synthesis and structural characterization of hexanuclear (2-Me2NC6H4)4Cu6-nMnX2 (M = Cu or Ag) cluster compounds. J. Organomet. Chem. 1975, 102, 551–563. [Google Scholar] [CrossRef]
- van Koten, G.; Jastrzebski, J.T.B.H.; Noltes, J.G. Group IB organometallic chemistry. 21. Selective formation of biaryls via interaction of polynuclear arylcopper compounds with copper(I) trifluoromethanesulfonate [copper(I) triflate]. J. Org. Chem. 1977, 42, 2047–2053. [Google Scholar] [CrossRef]
- van Koten, G.; Jastrzebski, J.T.B.H.; Noltes, J.G. Interaction of arylmetal compounds of group 1B with copper(I) and silver(I) trifluoromethanesulphonate; a novel biaryl synthesis and a route to hexanuclear aryl-CuAg, -AgAu, and -CuAu clusters. J. Chem. Soc. Chem. Commun. 1977, 203–204. [Google Scholar] [CrossRef]
- van Koten, G.; Noltes, J.G. Synthesis and specific intramolecular C–C coupling reactions of novel hexanuclear copper cluster complexes Ar4R2Cu6. J. Chem. Soc. Chem. Commun. 1974, 575–576. [Google Scholar] [CrossRef] [Green Version]
- ten Hoedt, R.W.M.; van Koten, G.; Noltes, J.G. Group IB organometallic chemistry: XIX. Synthesis and characterization of mixed-organocopper cluster compounds R4R’2Cu6 containing aryl and acetylide ligands. J. Organomet. Chem. 1977, 133, 113–121. [Google Scholar] [CrossRef]
- van Koten, G.; ten Hoedt, R.W.M.; Noltes, J.G. Group IB organometallic chemistry. 20. The role of mixed organocopper cluster compounds RnR’mCun+m in selective carbon-carbon coupling reactions of 2- and 4-(dimethylamino)phenylcopper with copper arylacetylides. J. Org. Chem. 1977, 42, 2705–2711. [Google Scholar] [CrossRef]
- ten Hoedt, R.W.M.; Noltes, J.G.; van Koten, G.; Spek, A.L. Group 1B organometallic chemistry. Part 25. Crystal and molecular structure of 1,2,3;1,4,5;2,3,6;4,5,6-tetrakis-μ3-2-dimethylaminophenyl-2,5;3,4-bis-μ2-4-tolylethynyl-octahedro-hexacopper(I). J. Chem. Soc. Dalton Trans. 1978, 1800–1806. [Google Scholar] [CrossRef]
- Liu, L.; Xi, Z. Organocopper(III) Compounds with Well-defined Structures Undergo Reductive Elimination to Form C-C or C-Heteroatom Bonds. Chin. J. Chem. 2018, 36, 1213–1221. [Google Scholar] [CrossRef]
Scheme 1.
Fragments in the molecular structures with characteristic connectivity. The lines do not necessarily represent 2c-2e or 3c-2e bonds.
Scheme 1.
Fragments in the molecular structures with characteristic connectivity. The lines do not necessarily represent 2c-2e or 3c-2e bonds.


Figure 1.
Molecular structures of 4, 5, and 7. Reprinted with permission from [59,60]. Copyright 2017 and 2018 American Chemical Society.

Scheme 3.
Reductive elimination of organometallic spiro Cu(III) complexes.

Scheme 4.
Mixed-valent bimetallic Cu(I)/Cu(III) compounds [62].
Scheme 4.
Mixed-valent bimetallic Cu(I)/Cu(III) compounds [62].

Figure 2.
Molecular structure of 15; drawn based on [62].
Figure 2.
Molecular structure of 15; drawn based on [62].

Scheme 5.
C,N-chelated organocopper compounds [63].
Scheme 5.
C,N-chelated organocopper compounds [63].

Scheme 6.
Rigid magnesium organocuprates [58].
Scheme 6.
Rigid magnesium organocuprates [58].

Figure 3.
Molecular structure of 23b. Modified with permission from [58]. Copyright 2016 John Wiley and Sons.
Figure 3.
Molecular structure of 23b. Modified with permission from [58]. Copyright 2016 John Wiley and Sons.


Figure 4.
Molecular structure of 30; drawn based on [67].
Figure 4.
Molecular structure of 30; drawn based on [67].


Scheme 9.
Perfluoro-dicupra-cyclodecanes [72].
Scheme 9.
Perfluoro-dicupra-cyclodecanes [72].


Figure 5.
Molecular structures of 35a and 36a. Modified with permission from [32]. Copyright 2016 American Chemical Society.
Figure 5.
Molecular structures of 35a and 36a. Modified with permission from [32]. Copyright 2016 American Chemical Society.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 12.
Conjugate C,C-bidentate organocopper aggregates [56].
Scheme 12.
Conjugate C,C-bidentate organocopper aggregates [56].

Figure 6.
Molecular structures of 35a and 36a; drawn based on [56].
Figure 6.
Molecular structures of 35a and 36a; drawn based on [56].

Scheme 13.
Reactivity of bis-enyl organocopper aggregates.

Scheme 14.
Styrenyl and budadienyl organocopper aggregates [57].
Scheme 14.
Styrenyl and budadienyl organocopper aggregates [57].

Figure 7.
Molecular structures of 53 and 54; drawn based on [57].
Figure 7.
Molecular structures of 53 and 54; drawn based on [57].

Scheme 15.
Synthesis and reactivity of C,N-chelated tetranuclear organocopper cluster [89,90,91,92,93,94,95].

Figure 8.
Molecular structure of 31; drawn based on [92].
Figure 8.
Molecular structure of 31; drawn based on [92].

Scheme 16.
C,N-chelated hexanuclear organocopper cluster and reactivities [103,104,105,106,107,108].

Scheme 17.
Selective coupling from the thermolysis of hexanuclear organocopper cluster [109,110,111,112].

Figure 9.
Molecule structure of 62; drawn based on [109].
Figure 9.
Molecule structure of 62; drawn based on [109].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, L.; Chen, H.; Yang, Z.; Wei, J.; Xi, Z. C,C- and C,N-Chelated Organocopper Compounds. Molecules 2021, 26, 5806. https://doi.org/10.3390/molecules26195806
AMA Style
Liu L, Chen H, Yang Z, Wei J, Xi Z. C,C- and C,N-Chelated Organocopper Compounds. Molecules. 2021; 26(19):5806. https://doi.org/10.3390/molecules26195806
Chicago/Turabian StyleLiu, Liang, Hui Chen, Zhenqiang Yang, Junnian Wei, and Zhenfeng Xi. 2021. "C,C- and C,N-Chelated Organocopper Compounds" Molecules 26, no. 19: 5806. https://doi.org/10.3390/molecules26195806