
Pseudomonas aeruginosa mexR and mexEF Antibiotic Efflux Pump Variants Exhibit Increased Virulence


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
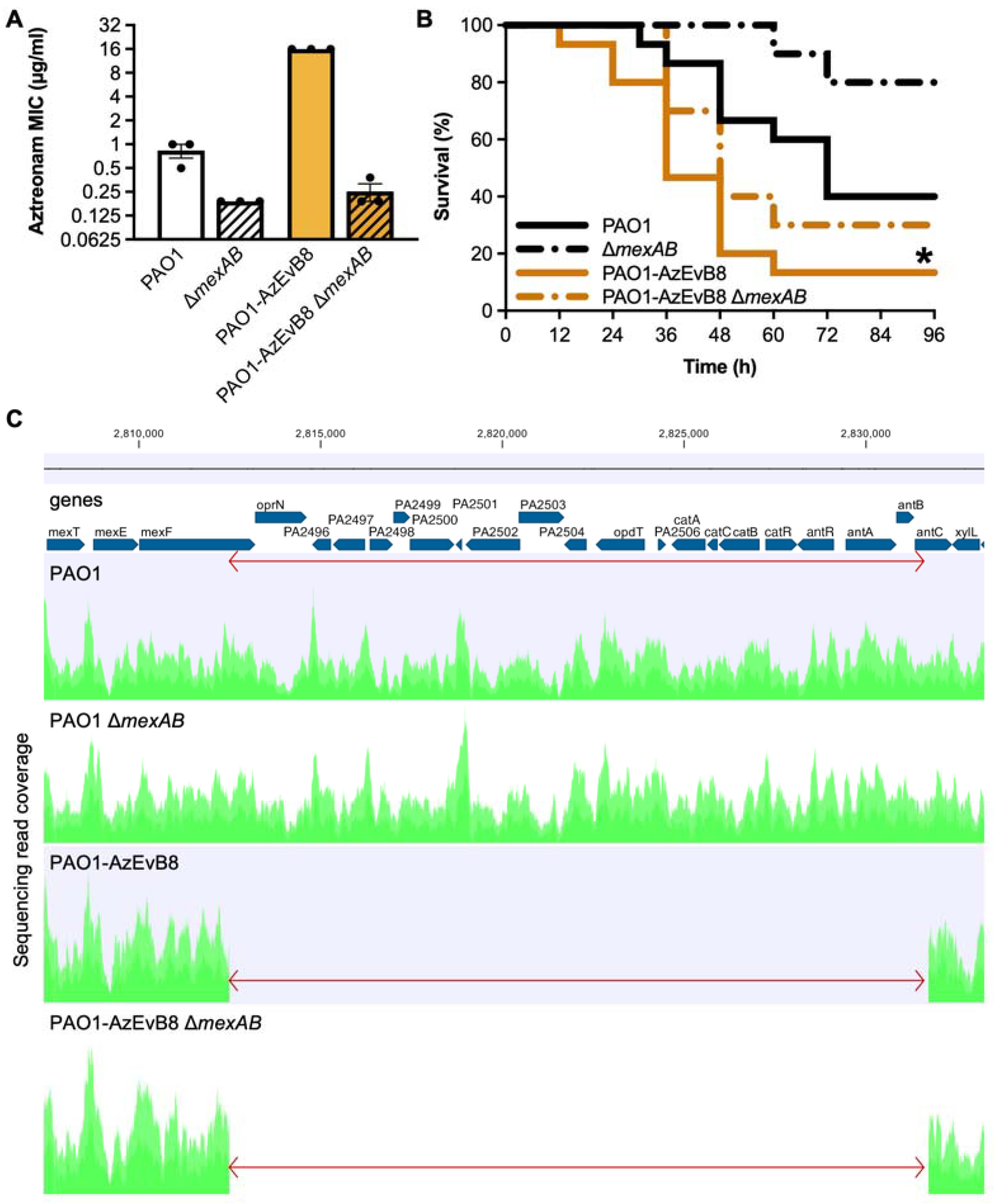
2.1. Deletion of mexAB Restores Aztreonam Susceptibility of Evolved P. aeruginosa
2.2. Deletion of mexAB Does Not Affect Virulence of Evolved P. aeruginosa
2.3. Genome Sequencing Reveals a Previously Undetected 19 kb Deletion in PAO1-AzEvB8
2.4. Mutation of mexEF Increases Swarming While mexR Mutation Does not
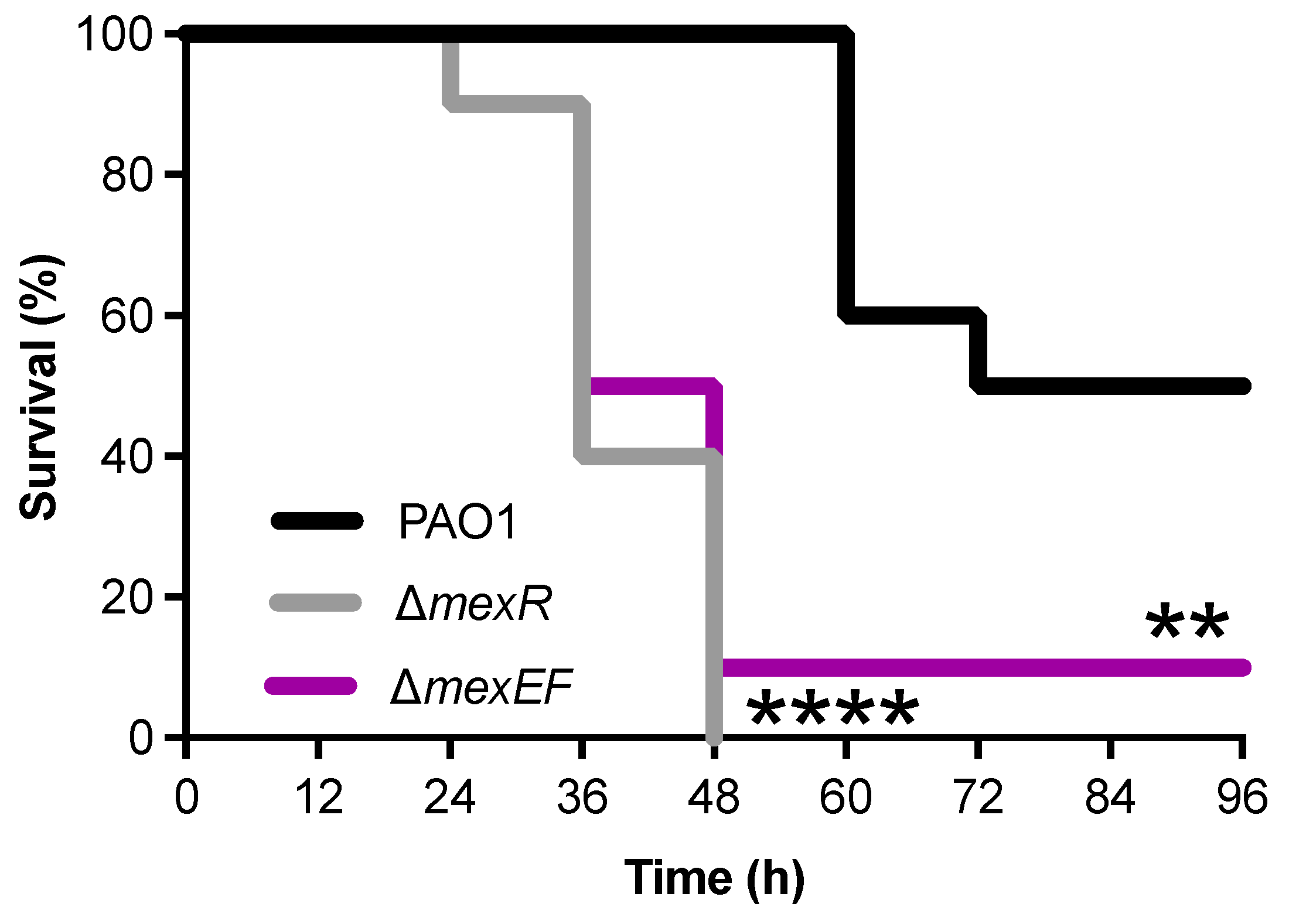
2.5. Deletion of either mexR or mexEF Increases P. aeruginosa Virulence
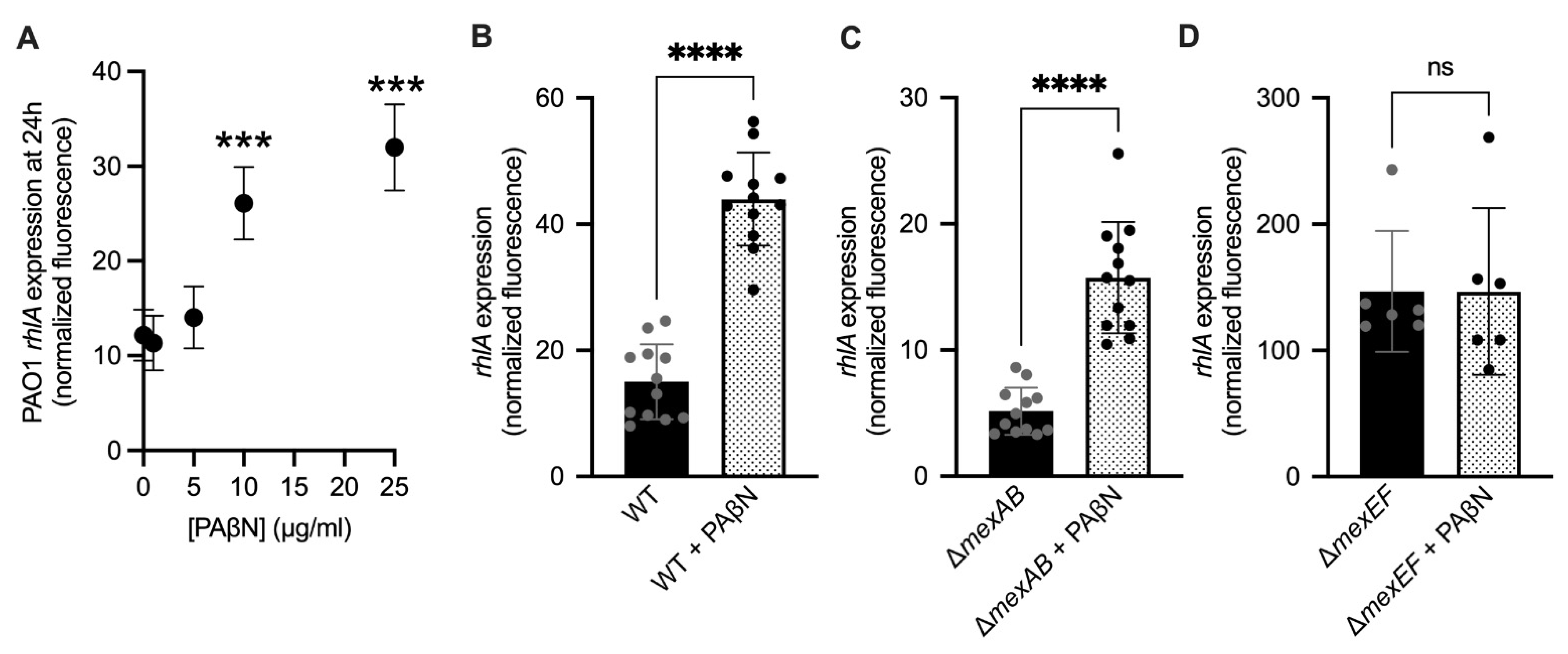
2.6. Efflux Pump Inhibition Increases Rhamnolipid Virulence Factor Expression
3. Discussion
3.1. Deletion of mexEF and Overexpression of mexAB through mexR Mutation Can Each Increase Virulence of P. aeruginosa
3.2. Understanding Why rhlA Was Not Expressed in the PAO1-AzEvB8 Strain That Exhibited Greater Swarming Than WT
3.3. Relationship of These Findings to Previous Research
3.4. Differences among Infection Models Help Explain Why Previous Studies Concluded Different Effects of Efflux Pump Inhibition on Virulence
3.5. Relevance of mexEF Mutations in P. aeruginosa Clinical Isolates
3.6. Implications for CF and the Use of Efflux Pump Inhibitor as Potential Therapies
3.7. Limitations of the Present Work
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Deletion Plasmid Construction
4.3. Transformation of P. aeruginosa Deletion Mutants
4.4. PAO1 Transformation with rhlA Reporter Plasmid
4.5. DNA Extraction, Purification, and PCR
4.6. Swarming Assay
4.7. Gradient Diffusion Antibiotic Susceptibility Testing
4.8. rhlA GFP Reporter Assay
4.9. Murine Lung Infection
4.10. Genome Sequencing and Analysis
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [Green Version]
- Moradali, M.F.; Ghods, S.; Rehm, B.H. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.J., Jr.; Wenzel, R.P. Epidemiology of infections due to Pseudomonas aeruginosa. Rev. Infect. Dis. 1984, 6 (Suppl. S3), S627–S642. [Google Scholar] [CrossRef] [PubMed]
- Parkins, M.D.; Somayaji, R.; Waters, V.J. Epidemiology, Biology, and Impact of Clonal Pseudomonas aeruginosa Infections in Cystic Fibrosis. Clin. Microbiol. Rev. 2018, 31, e00019-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Ramirez, D.; Seshadri, R.; Cullina, J.F.; Powers, C.A.; Schulert, G.S.; Bar-Meir, M.; Sullivan, C.L.; McColley, S.A.; Hauser, A.R. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. J. Clin. Microbiol. 2004, 42, 5229–5237. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.E.; Buckley, D.G.; Wu, Z.; Saenphimmachak, C.; Hoffman, L.R.; D’Argenio, D.A.; Miller, S.I.; Ramsey, B.W.; Speert, D.P.; Moskowitz, S.M.; et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2006, 103, 8487–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, H.K.; Kwon, T.; Zlosnik, J.E.; Speert, D.P.; Marcotte, E.M.; Whiteley, M. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 2010, 1, e00199-10. [Google Scholar] [CrossRef] [Green Version]
- Jorth, P.; Staudinger, B.J.; Wu, X.; Hisert, K.B.; Hayden, H.; Garudathri, J.; Harding, C.L.; Radey, M.C.; Rezayat, A.; Bautista, G.; et al. Regional Isolation Drives Bacterial Diversification within Cystic Fibrosis Lungs. Cell Host Microbe 2015, 18, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Winstanley, C.; O’Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Darch, S.E.; McNally, A.; Harrison, F.; Corander, J.; Barr, H.L.; Paszkiewicz, K.; Holden, S.; Fogarty, A.; Crusz, S.A.; Diggle, S.P. Recombination is a key driver of genomic and phenotypic diversity in a Pseudomonas aeruginosa population during cystic fibrosis infection. Sci. Rep. 2015, 5, 7649. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Williams, D.; Fothergill, J.L.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. High virulence sub-populations in Pseudomonas aeruginosa long-term cystic fibrosis airway infections. BMC Microbiol. 2017, 17, 30. [Google Scholar] [CrossRef] [Green Version]
- Faure, E.; Kwong, K.; Nguyen, D. Pseudomonas aeruginosa in Chronic Lung Infections: How to Adapt within the Host? Front. Immunol. 2018, 9, 2416. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.O.; Bjarnsholt, T.; Phipps, R.; Rasmussen, T.B.; Calum, H.; Christoffersen, L.; Moser, C.; Williams, P.; Pressler, T.; Givskov, M.; et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 2007, 153, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Zulianello, L.; Canard, C.; Kohler, T.; Caille, D.; Lacroix, J.S.; Meda, P. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 3134–3147. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, E.G.; Wangdi, T.; Kazmierczak, B.I. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect. 2011, 13, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Brady, C.; Guill, M.; Lahiri, T.; Lubsch, L.; Matsui, J.; Oermann, C.M.; Ratjen, F.; et al. Cystic Fibrosis Foundation pulmonary guideline. pharmacologic approaches to prevention and eradication of initial Pseudomonas aeruginosa infection. Ann. Am. Thorac Soc. 2014, 11, 1640–1650. [Google Scholar] [CrossRef]
- Sobel, M.L.; Hocquet, D.; Cao, L.; Plesiat, P.; Poole, K. Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 1782–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, K.; Tetro, K.; Zhao, Q.; Neshat, S.; Heinrichs, D.E.; Bianco, N. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother. 1996, 40, 2021–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Yoneyama, H.; Nakae, T. nalB-type mutations causing the overexpression of the MexAB-OprM efflux pump are located in the mexR gene of the Pseudomonas aeruginosa chromosome. FEMS Microbiol. Lett. 1999, 179, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorth, P.; McLean, K.; Ratjen, A.; Secor, P.R.; Bautista, G.E.; Ravishankar, S.; Rezayat, A.; Garudathri, J.; Harrison, J.J.; Harwood, R.A.; et al. Evolved Aztreonam Resistance Is Multifactorial and Can Produce Hypervirulence in Pseudomonas aeruginosa. mBio 2017, 8, e00517-17. [Google Scholar] [CrossRef] [Green Version]
- Suresh, M.; Nithya, N.; Jayasree, P.R.; Vimal, K.P.; Manish Kumar, P.R. Mutational analyses of regulatory genes, mexR, nalC, nalD and mexZ of mexAB-oprM and mexXY operons, in efflux pump hyperexpressing multidrug-resistant clinical isolates of Pseudomonas aeruginosa. World J. Microbiol. Biotechnol. 2018, 34, 83. [Google Scholar] [CrossRef]
- Horna, G.; Lopez, M.; Guerra, H.; Saenz, Y.; Ruiz, J. Interplay between MexAB-OprM and MexEF-OprN in clinical isolates of Pseudomonas aeruginosa. Sci. Rep. 2018, 8, 16463. [Google Scholar] [CrossRef]
- Llanes, C.; Hocquet, D.; Vogne, C.; Benali-Baitich, D.; Neuwirth, C.; Plesiat, P. Clinical strains of Pseudomonas aeruginosa overproducing MexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob. Agents Chemother. 2004, 48, 1797–1802. [Google Scholar] [CrossRef] [Green Version]
- McLean, K.; Lee, D.; Holmes, E.A.; Penewit, K.; Waalkes, A.; Ren, M.; Lee, S.A.; Gasper, J.; Manoil, C.; Salipante, S.J. Genomic Analysis Identifies Novel Pseudomonas aeruginosa Resistance Genes under Selection during Inhaled Aztreonam Therapy In Vivo. Antimicrob. Agents Chemother. 2019, 63, e00866-19. [Google Scholar] [CrossRef] [Green Version]
- Oshri, R.D.; Zrihen, K.S.; Shner, I.; Omer Bendori, S.; Eldar, A. Selection for increased quorum-sensing cooperation in Pseudomonas aeruginosa through the shut-down of a drug resistance pump. ISME J. 2018, 12, 2458–2469. [Google Scholar] [CrossRef] [Green Version]
- Kohler, T.; van Delden, C.; Curty, L.K.; Hamzehpour, M.M.; Pechere, J.C. Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 5213–5222. [Google Scholar] [CrossRef] [Green Version]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosson, P.; Zulianello, L.; Join-Lambert, O.; Faurisson, F.; Gebbie, L.; Benghezal, M.; van Delden, C.; Curty, L.K.; Kohler, T. Pseudomonas aeruginosa virulence analyzed in a Dictyostelium discoideum host system. J. Bacteriol. 2002, 184, 3027–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakata, Y.; Kondo, A.; Hoshino, K.; Yano, H.; Arai, K.; Hirotani, A.; Kunishima, H.; Yamamoto, N.; Hatta, M.; Kitagawa, M.; et al. Efflux pump inhibitors reduce the invasiveness of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2009, 34, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Rampioni, G.; Pillai, C.R.; Longo, F.; Bondi, R.; Baldelli, V.; Messina, M.; Imperi, F.; Visca, P.; Leoni, L. Effect of efflux pump inhibition on Pseudomonas aeruginosa transcriptome and virulence. Sci. Rep. 2017, 7, 11392. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Casey, M.; Frank, D.W.; Ausubel, F.M.; Drenkard, E. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect. Immun. 2003, 71, 2404–2413. [Google Scholar] [CrossRef] [Green Version]
- Kropinski, A.M.; Chadwick, J.S. The pathogenicity of rough strains of Pseudomonas aeruginosa for Galleria mellonella. Can. J. Microbiol. 1975, 21, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Marvig, R.L.; Sommer, L.M.; Molin, S.; Johansen, H.K. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 2015, 47, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Hosaka, M.; Hirai, K.; Iyobe, S. New norfloxacin resistance gene in Pseudomonas aeruginosa PAO. Antimicrob. Agents Chemother. 1990, 34, 1757–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, T.; Michea-Hamzehpour, M.; Henze, U.; Gotoh, N.; Curty, L.K.; Pechere, J.C. Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 1997, 23, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Quale, J.; Bratu, S.; Gupta, J.; Landman, D. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2006, 50, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Tomas, M.; Doumith, M.; Warner, M.; Turton, J.F.; Beceiro, A.; Bou, G.; Livermore, D.M.; Woodford, N. Efflux pumps, OprD porin, AmpC beta-lactamase, and multiresistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob. Agents Chemother. 2010, 54, 2219–2224. [Google Scholar] [CrossRef] [Green Version]
- Keating, C.L.; Zuckerman, J.B.; Singh, P.K.; McKevitt, M.; Gurtovaya, O.; Bresnik, M.; Marshall, B.C.; Saiman, L. Pseudomonas aeruginosa Susceptibility Patterns and Associated Clinical Outcomes in People with Cystic Fibrosis following Approval of Aztreonam Lysine for Inhalation. Antimicrob. Agents Chemother. 2021, 65, e02327-20. [Google Scholar] [CrossRef]
- Yoshida, K.; Nakayama, K.; Ohtsuka, M.; Kuru, N.; Yokomizo, Y.; Sakamoto, A.; Takemura, M.; Hoshino, K.; Kanda, H.; Nitanai, H.; et al. MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: Highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 2007, 15, 7087–7097. [Google Scholar] [CrossRef]
- Roux, D.; Danilchanka, O.; Guillard, T.; Cattoir, V.; Aschard, H.; Fu, Y.; Angoulvant, F.; Messika, J.; Ricard, J.D.; Mekalanos, J.J.; et al. Fitness cost of antibiotic susceptibility during bacterial infection. Sci. Transl. Med. 2015, 7, 297ra114. [Google Scholar] [CrossRef] [Green Version]
- Cowley, E.S.; Kopf, S.H.; LaRiviere, A.; Ziebis, W.; Newman, D.K. Pediatric Cystic Fibrosis Sputum Can Be Chemically Dynamic, Anoxic, and Extremely Reduced Due to Hydrogen Sulfide Formation. mBio 2015, 6, e00767-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, K.L.; Brown, S.A.; Whiteley, M. Membrane-bound nitrate reductase is required for anaerobic growth in cystic fibrosis sputum. J. Bacteriol. 2007, 189, 4449–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, K.L.; Aye, L.M.; Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J. Bacteriol. 2007, 189, 8079–8087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, K.H.; Wessel, A.K.; Palmer, G.C.; Murray, J.L.; Whiteley, M. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 2015, 112, 4110–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, C.S.; Stoltz, D.A.; Meyerholz, D.K.; Ostedgaard, L.S.; Rokhlina, T.; Taft, P.J.; Rogan, M.P.; Pezzulo, A.A.; Karp, P.H.; Itani, O.A.; et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008, 321, 1837–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, J.R.; Edwards, M.S.; Langston, C.; Baker, C.J. A mouse model of chronic pulmonary infection with Pseudomonas aeruginosa and Pseudomonas cepacia. Pediatr. Res. 1987, 22, 698–702. [Google Scholar] [CrossRef] [Green Version]
- Van Heeckeren, A.M.; Schluchter, M.D. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab. Anim. 2002, 36, 291–312. [Google Scholar] [CrossRef]
- Van Heeckeren, A.M.; Schluchter, M.D.; Drumm, M.L.; Davis, P.B. Role of Cftr genotype in the response to chronic Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L944–L952. [Google Scholar] [CrossRef] [Green Version]
- Held, K.; Ramage, E.; Jacobs, M.; Gallagher, L.; Manoil, C. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J. Bacteriol. 2012, 194, 6387–6389. [Google Scholar] [CrossRef] [Green Version]
- Hmelo, L.R.; Borlee, B.R.; Almblad, H.; Love, M.E.; Randall, T.E.; Tseng, B.S.; Lin, C.; Irie, Y.; Storek, K.M.; Yang, J.J.; et al. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat. Protoc. 2015, 10, 1820–1841. [Google Scholar] [CrossRef]
- Hoang, T.T.; Karkhoff-Schweizer, R.R.; Kutchma, A.J.; Schweizer, H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 1998, 212, 77–86. [Google Scholar] [CrossRef]
- Taylor, R.G.; Walker, D.C.; McInnes, R.R.E. coli host strains significantly affect the quality of small scale plasmid DNA preparations used for sequencing. Nucleic Acids Res. 1993, 21, 1677–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.; Priefer, U.; Pühler, A. A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Nat. Biotechnol. 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Chung, C.T.; Niemela, S.L.; Miller, R.H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. USA 1989, 86, 2172–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lequette, Y.; Greenberg, E.P. Timing and localization of rhamnolipid synthesis gene expression in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2005, 187, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Deatherage, D.E.; Barrick, J.E. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 2014, 1151, 165–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaillancourt, M.; Limsuwannarot, S.P.; Bresee, C.; Poopalarajah, R.; Jorth, P. Pseudomonas aeruginosa mexR and mexEF Antibiotic Efflux Pump Variants Exhibit Increased Virulence. Antibiotics 2021, 10, 1164. https://doi.org/10.3390/antibiotics10101164
Vaillancourt M, Limsuwannarot SP, Bresee C, Poopalarajah R, Jorth P. Pseudomonas aeruginosa mexR and mexEF Antibiotic Efflux Pump Variants Exhibit Increased Virulence. Antibiotics. 2021; 10(10):1164. https://doi.org/10.3390/antibiotics10101164
Chicago/Turabian StyleVaillancourt, Mylene, Sam P. Limsuwannarot, Catherine Bresee, Rahgavi Poopalarajah, and Peter Jorth. 2021. "Pseudomonas aeruginosa mexR and mexEF Antibiotic Efflux Pump Variants Exhibit Increased Virulence" Antibiotics 10, no. 10: 1164. https://doi.org/10.3390/antibiotics10101164