
The Genetics of Sleep Disorders in Children: A Narrative Review
by
 ,
,
Greta Mainieri
1 ,
,
Angelica Montini
1, 
Antonio Nicotera
2,
Gabriella Di Rosa
2,
Federica Provini
1,3,* and
Giuseppe Loddo
4 1
Department of Biomedical and Neuromotor Sciences, University of Bologna, 40138 Bologna, Italy
2
Unit of Child Neurology and Psychiatry, Department of Human Pathology of the Adult and Developmental Age, “Gaetano Barresi” University of Messina, 98124 Messina, Italy
3
IRCCS Istituto Delle Scienze Neurologiche di Bologna, 40139 Bologna, Italy
4
Azienda USL di Bologna, 40124 Bologna, Italy
*
Author to whom correspondence should be addressed.
Brain Sci. 2021, 11(10), 1259; https://doi.org/10.3390/brainsci11101259
Submission received: 31 July 2021
/
Revised: 17 September 2021
/
Accepted: 20 September 2021
/
Published: 23 September 2021
(This article belongs to the Special Issue Genetics of Neurological Disorders in Children)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Sleep is a universal, highly preserved process, essential for human and animal life, whose complete functions are yet to be unravelled. Familial recurrence is acknowledged for some sleep disorders, but definite data are lacking for many of them. Genetic studies on sleep disorders have progressed from twin and family studies to candidate gene approaches to culminate in genome-wide association studies (GWAS). Several works disclosed that sleep-wake characteristics, in addition to electroencephalographic (EEG) sleep patterns, have a certain degree of heritability. Notwithstanding, it is rare for sleep disorders to be attributed to single gene defects because of the complexity of the brain network/pathways involved. Besides, the advancing insights in epigenetic gene-environment interactions add further complexity to understanding the genetic control of sleep and its disorders. This narrative review explores the current genetic knowledge in sleep disorders in children, following the International Classification of Sleep Disorders—Third Edition (ICSD-3) categorisation.
1. Introduction
Sleep is a universal, essential behaviour, persistent throughout evolution and across different species [1]. In humans, the regular development of a mature sleep system from childhood to adulthood is an important milestone, with potential consequences for both neurological and health concerns [2]. Consequently, the prompt identification of sleep disorders in children is of the utmost importance, in order to address potential sequelae, treat comorbid conditions and relieve the burden on other family members [2]. Sleep is a dynamic process and could be influenced by external factors (environment, medical comorbidities, drugs), whose effect may possibly lead to some sleep disorders [3,4]. On the other hand, several sleep disorders recognise a genetic predisposition and the current expansion of advanced molecular techniques might serve for a better identification and consequent treatment of the different disorders [5,6,7,8,9]. Over the last decades, a bidirectional relationship between genetic factors and sleep has been recognised; specific genes may influence circadian regulation, neurotransmission and signalling pathways involved in sleep processes, whereas sleep may affect, in turn, gene expression [10,11,12]. Moreover, a differentiated gene expression during sleep and wakefulness is recognised, mostly depending on the different biological functions of the two states. In fact, genes encoding proteins involved in energy metabolism and oxidative cellular response, upregulated during wakefulness, leave the place to genes regulating protein biosynthesis, synaptic downscaling and cell membrane processes during sleep [13]. Sleep has proved to play a fundamental role in synaptic plasticity, with implications in the achievement and upholding of recently learned motor and cognitive skills, especially relevant for paediatric populations [2,14]. Moreover, the inter-relationships between sleep disorders and other neurological and psychiatric conditions, on the one hand, and metabolic/cardiovascular disturbances, on the other, are becoming increasingly clear, with implications both in children and adults [6,15,16,17,18,19,20]. Genetic studies are showing that a common genetic background might play a pivotal role, constituting a linking point among these different conditions.
In this narrative review, we specifically focused on the principal sleep disorders affecting paediatric populations, following the six main categories of the International Classification of Sleep Disorders—Third Edition (ICSD-3): Insomnia, Sleep-related Breathing Disorders, Central Disorders of Hypersomnolence, Circadian Rhythm Sleep-Wake Disorders, Parasomnias and Sleep-Related Movement Disorders [21]. Among the different sleep disorders, genetic components assume weights of variable importance. The breakthrough of genome-wide association studies (GWAS) has consistently expanded the genetic literature, also as regards sleep disorders. The aim of our paper is to summarise the current evidence of the genetics of sleep disorders, including the most recent GWA studies. To our knowledge, this is the first paper attempting to provide the present state of the art of the genetic aspects of all sleep disorders in paediatric populations. We emphasise the possible connections between genetics and the different sleep conditions, together with the relationships with neuropsychiatric or metabolic disorders.
2. Methods
A narrative literature research has been performed on Pubmed and Scopus research databases, with no restriction on articles’ publication date. Published papers up to July 2021 were included. We also included case series or case reports, when relevant genetic findings were reported. We used the following keywords: “insomnia” AND (“genetics” or “genes”), “sleep-related breathing disorders” AND (“genetics” or “genes”), “central disorders of hypersomnolence” AND (“genetics” or “genes”), “circadian rhythm sleep-wake disorders” AND (“genetics” or “genes”), “parasomnias” AND (“genetics” or “genes”), “sleep-related movement disorders” AND (“genetics” or “genes”). We only considered works in English language. Additional papers were inserted using the references of the selected works, when relevant.
3. Insomnia
Insomnia is defined as the difficulty in initiating and/or maintaining sleep, early awakening or poor sleep quality in the presence of adequate opportunity and circumstance for sleep. Daytime consequences include reduced scholar performance and social dysfunction due to mental (impaired attention, concentration or memory), mood (decreased mood or irritability) and behavioural disturbance (hyperactivity, impulsivity or aggression) [21]. Chronic insomnia affects 20% to 30% of children [21,22], reaching a prevalence of up to 86% in those with neurodevelopmental disabilities [23]. Among the different subtypes included in chronic insomnia (psychophysiological, idiopathic, paradoxical insomnia, inadequate sleep hygiene, behavioural insomnia of childhood), two, in particular, are typical of infancy. “Idiopathic insomnia” occurs by definition in infancy or early childhood [21]. Even if no consistent genetic markers have been identified, idiopathic insomnia is presumed to have a genetically determined base or congenital alterations in the sleep-inducing or arousal system [21]. The “behavioural insomnia of childhood” is divided into “sleep onset association” and “limit setting” subtypes. The first one derives from altered learned behaviours (such as the need to be rocked or to eat) or the child’s dependency on a specific environment (car, light, etc.) or objects, whereas the “limit-setting” type is characterised by bedtime restriction due to the lack of appropriate parental rules [21,24]. The diagnosis of insomnia disorder is clinical. Video-polysomnography (VPSG) may be warranted if a sleep breathing disorder or parasomnia/sleep-related complex motor behaviours are suspected [21].
The relationship between insomnia and genetic factors is intricate. The insomniac phenotype is thought of as a consequence of inheriting many alleles that exert small but cumulative effects on sleep regulatory circuits [25], associated with the influence of epigenetic mechanisms [26]. Data from twin and family studies indicate a degree of heritability ranging from 14% to 71% in children [27,28,29]. Several gene polymorphisms involved in the circadian clock, immune regulation and neurotransmitter pathways have been identified. Polymorphisms in circadian genes (CLOCK gene, PER3, peroxisome proliferator-activated receptor-c coactivator-1a) [30,31,32,33] and neuromodulators involved in the sleep-wake regulation have been associated with insomnia, sometimes in association with depressive symptoms, especially when serotoninergic pathways were involved [34,35,36,37]. From GWAS, several other polymorphisms have been linked to insomnia or prolonged sleep latencies, such as those in genes involved in bipolar disorder and schizophrenia (ROR1 and PLCB1) [38], a subunit of voltage-dependent calcium channels [39], circadian genes (ULF1) [40] and GABA and monoamine signalling [41]. In addition, a strong association with the MEIS 1 locus, strongly related with restless leg syndrome (RLS) [42] was identified, suggesting a pleiotropic effect of this gene and implications in phenotypic overlap (RLS comorbid with insomnia) [15,16]. To date, the largest GWAS included 1,331,010 participants and identified 202 genomic risk loci for insomnia, pointing out a causal effect of insomnia on depression, diabetes and cardiovascular disease. Interestingly, this study evaluated the different gene expression in different types of brain cells and revealed an enrichment of risk genes for insomnia in cortical areas as well as the striatum, supporting the involvement of the striato-cortical network in insomnia pathophysiology [17].
In summary, risk genes for insomnia have been associated with circadian clock genes, pathways regulating sleep-wake mechanisms and psychiatric and cardio-metabolic conditions. The identification of insomnia risk genes would be helpful to clarify the neurobiological mechanisms of insomnia. Furthermore, it could widen the opportunity for insomnia prevention or early and specific treatment, screening genes that influence the individual response to certain drugs.
4. Sleep-Related Breathing Disorders
Sleep-related breathing disorders are characterised by irregularities of respiration during sleep and are grouped into obstructive sleep apnoea and central sleep apnoea disorders, sleep-related hypoventilation disorders and sleep-related hypoxemia disorders [21].
Obstructive Sleep Apnea Syndrome (OSAS) is characterised by repetitive episodes of complete (apnea) or partial (hypopnea) upper airway obstruction occurring during sleep. These events often result in a reduction in blood oxygen saturation and are usually concluded by brief arousals from sleep [21]. OSAS prevalence in children ranges from 1 to 5.7% [21,43] and could be associated with excessive daytime sleepiness (especially in older children), developmental, behavioural and learning issues including attention disturbances, hyperactivity, moodiness, irritability and impaired performance [21]. Diagnostic criteria include the evidence of one or more obstructive/mixed apnoeas, or hypopnea, per hour of sleep on polysomnography or a pattern of obstructive hypoventilation, defined as at least 25% of total sleep time with hypercapnia (PaCO2 > 50 mm Hg) in association with one or more of the following: snoring; paradoxical or obstructed breathing during sleep; sleepiness, hyperactivity, behavioural or learning problems [21]. Polysomnography is the standard diagnostic test for the diagnosis of OSA in children [44].
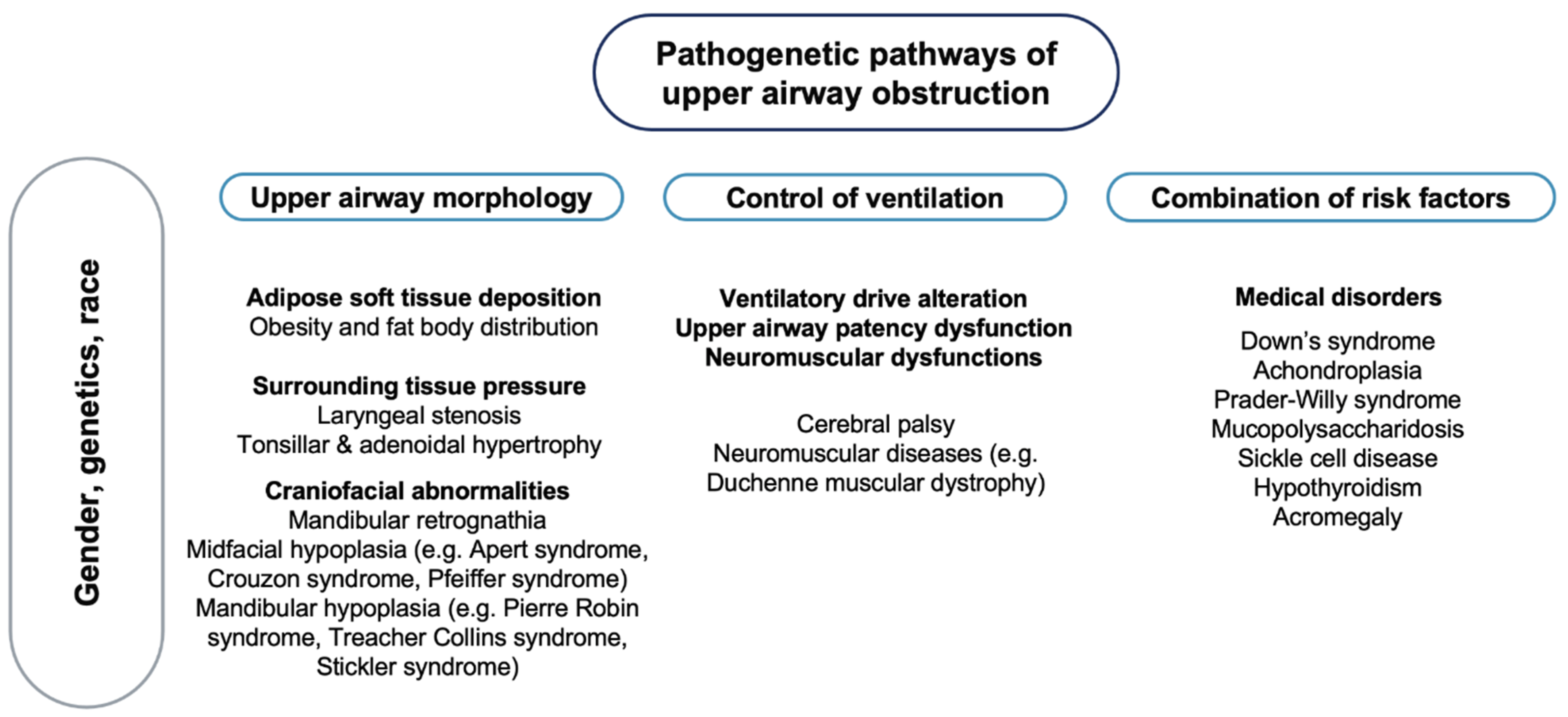
OSA is characterised by a complex phenotype, involving different pathogenetic factors [45] (Figure 1), with no recognised monogenic predisposition [46].
Most cases of OSA do not exhibit classical Mendelian patterns of inheritance, suggesting multifactorial pathogenesis, where many common variants with small or moderate genetic effects determine disease heritability [47] as well as different phenotypic expressions of the disorder. In a study on 445 first-degree relatives of 115 children with OSA, 26.6% of the adult and 12.2% of the paediatric relatives had symptoms suggestive of OSA, supporting the hypothesis of a possible role of a genetic mechanism in the aetiology of this syndrome [48]. It has been speculated that the craniofacial structure, body habitus and ventilatory control mechanism play a role in the pathogenesis of OSA [45]. The involvement of genetic factors in the craniofacial phenotypic dysmorphology associated with breathing dysfunctions has been reported in a study on 50 children with Class III malocclusion, disclosing silent mutations in the PHOX2B genotype in 32% of patients and none of the controls [49]. The PHOX2B gene is involved in human development, especially in the neural crest, a group of cells in the early embryo from which many tissues in the face and skull originate [50]. Furthermore, several neural crest cells migrate to form parts of the autonomic nervous system, which controls many functions and, above all, breathing [51]. Genetic factors involved in metabolic pathways and cognitive functions also seem to play a relevant role in the pathogenesis of OSA in children. In a study on 229 children with and without apnoea and 412 relatives, being overweight had a significant modification effect on the familial aggregation and the heritability [52]. The link between OSA in children and being overweight can be explained by different mechanisms. Children with OSA have been reported to have an increased expression of the Liver X receptors (LXRs), nuclear receptors that play a central role in the transcriptional control of lipid metabolism [53], as well as higher plasma fatty-acid binding protein 4, a cytosolic protein abundantly expressed in adipocytes and macrophages [54]. Contrasting data have been reported on the association between OSA and Apolipoprotein E (APOE) gene [47]. The APOE gene is involved in lipid metabolism, but it also has three common alleles with effects on the nervous system [55]. Of these, the APOE ε4 allele has been more frequently reported in children with OSA and particularly in those who develop neurocognitive deficits [56]. In addition, polymorphisms within the NADPH oxidase (NOX) gene or its functional subunits may account for important components of the variance in cognitive deficits associated with OSA in children [57]. NOX has been shown to mediate neural cell loss in the context of intermittent hypoxia during sleep [58]. A genome-wide gene expression study in non-obese children with OSA showed an altered gene expression in circulating leukocytes regulating and modulating the inflammatory response [59]. Other studies have underlined the association between paediatric OSA and genes involved in endothelial dysfunction and inflammatory responses (Forkhead box P3, interferon regulatory factor 1, macrophage migration inhibitory factor, endothelial nitric oxide synthase and interleukin-10) [60,61,62,63,64].
In conclusion, both in children and adults, genetic factors may influence anatomical structures or metabolic pathways associated with OSAS. However, the pathogenesis of OSAS is multifactorial, and further studies are needed to better characterise the genetic role. Central sleep apnoea (CSA) is defined as the absence of chest and abdominal movements associated with a cessation of airflow for more than 20 s, or lasting more than two baseline respiratory cycles if associated with an arousal, awakening or oxygen desaturation of at least 3% [65]. The ICSD-3 distinguishes CSA with Cheyne–Stokes breathing (which will not be discussed in this review because it occurs in individuals over 60), CSA due to a medical disorder without Cheyne–Stokes respiration, CSA due to high-altitude periodic breathing, CSA due to a medication or substance, treatment-emergent CSA, primary or idiopathic CSA (usually occurring in middle-aged/elderly individuals), primary sleep apnoea of infancy and prematurity [21].
In newborns and infants, CSAs are physiological, especially in the context of a sigh, movement and/or REM sleep [66]. In otherwise healthy 1–18-year-old individuals, the prevalence of central apnoeas, lasting 10–18 s, reached 30% [67]. Primary CSA of infancy is characterised by apnoea or cyanosis in infants of at least 37 weeks with recurrent, prolonged central apnoeas lasting ≥20 s with periodic breathing for ≥5% of total sleep time on polysomnographic recordings [21]. Similarly, primary CSA of prematurity affects infants of less than 37 weeks. Both disorders can be associated with hypoxemia and bradycardia [21], with a risk of neurodevelopmental disturbances [68]. A genetic basis on twins with primary CSA of prematurity has been described [69], but conclusive data are lacking for most CSA subtypes. Genes involved in hypoxic response such as the Endothelial PAS domain-containing protein 1 (EPAS1) or the Hypoxia inducible factors (HIF1A and HIF2A) have been linked to CSA due to high-altitude periodic breathing, because of a possible role in the genetic adaptation of high-altitude hypoxia [70,71]. In the paediatric population, CSA occurs more commonly in association with an underlying medical disorder, especially the Arnold–Chiari malformation, brainstem disease or syndromic conditions [72]. Indeed, a genetic contribution in CSA is mostly related to these secondary forms. Specifically, genetic factors linked to secondary CSA are those involved in neuromuscular disorders [66], Trisomy 21, Joubert syndrome [72], Prader–Willi syndrome [73], achondroplasia [74] and other rare genetic diseases, namely spondyloepiphyseal dysplasia congenita, Pierre Robin syndrome with Cornelia de Lange syndrome and Potocki–Lupski syndrome [73,75].
Sleep-related hypoventilation disorders are characterised by insufficient sleep-related ventilation resulting in an abnormally elevated arterial partial pressure of CO2 during sleep [21]. The congenital central alveolar hypoventilation syndrome (CCHS) is a rare genetic condition characterised by alveolar hypoventilation due to a deficient autonomic central control of ventilation and a diffuse autonomic dysfunction [76]. Cyanosis, feeding difficulties, hypotonia or central apnoea are common features of the disorder. Different mutations of the PHOX2B gene (mostly heterozygous) have been reported in CCHS. This gene encodes a transcription factor expressed in the developing hindbrain and peripheral nervous system as well as in all noradrenergic and visceral motor and branchiomotor neurons of the cranial nerves. Its expression has also been detected in neuronal groups involved in the medullary control reflexes of autonomic functions [77]. The most frequent mutation is an in-frame tandem duplication of tracts of different lengths of the polyalanine stretch in the exon 3 [51,77,78,79]. The number of duplications is negatively correlated to anthropometric measures such as mandible breadth, nasolabial angle, lateral lip height and mandible-face width index [80]. Sleep-related hypoventilation and autonomic dysregulation in the absence of PHOX2B gene mutation is encountered in Late-Onset Central Hypoventilation with hypothalamic dysfunction (ROHHAD) [21]. This disorder affects patients aged 2–3 years old and is characterised by at least two of the following: obesity, endocrine abnormalities of hypothalamic origin, severe emotional or behavioural disturbances, tumours of neural origin [21]. The mortality rate is estimated at 50–60% due to hypoventilation, cardiopulmonary failure and cardiopulmonary arrest [81]. The similarities between ROHHAD and CCHS suggest a possible genetic involvement in the aetiology of the ROHHAD syndrome, although some studies ruled out the presence of mutations in several candidate genes [82,83]. Epigenetic mechanisms have been hypothesised in some reports on the discordant presentation of ROHHAD syndrome in monozygotic twins [84].
In conclusion, CCHS is one of the rare monogenetic disorders of central respiratory control associated with mutations of the PHOX2B gene. The discovery of the association between CCHS and the PHOX2B gene helped clinicians in the diagnostic work-up of the disorder. The screening for the PHOX2B mutations has been proposed as an integral part of genetic counselling and prenatal screening as well as a potential target for gene therapy [85].
Sleep-related hypoxemia disorder is characterised by arterial oxygen saturation during sleep ≤90% in children for ≥5 min, as documented by polysomnography, out-of-centre sleep testing or nocturnal oximetry without hypoventilation. Obstructive sleep apnoea or CSA may be present, but these are not believed to be the principal cause of hypoxemia. The underlying pathologies are airway or pulmonary parenchymal disease, chest wall disorder, pulmonary hypertension or neuromuscular disorders. Consequences include pulmonary artery hypertension, cor pulmonale or neurocognitive dysfunction. The genetic patterns related to sleep-related hypoxemia reflect those of the underlying inherited conditions, such as muscular dystrophies or cystic fibrosis [21].
5. Central Disorders of Hypersomnolence
This group of disorders includes conditions whose primary complaint is excessive daytime sleepiness [21].
Narcolepsy is characterised by daily periods of irrepressible need to sleep or daytime lapses into sleep. Other associated features are hypnagogic or hypnopompic hallucinations, sleep paralysis and the disruption of nocturnal sleep. The ICSD-3 indicate a Narcolepsy type 1 (NT1) diagnosis if one or both of the following are present: (1) cataplexy and a mean sleep latency ≤8 min and two or more sleep-onset REM periods on a Multiple Sleep Latency Test; (2) the cerebrospinal fluid (CSF) hypocretin-1 concentration is either ≤110 pg/or <1/3 of the mean value obtained in normal individuals with the same standardised assay. Narcolepsy type 2 (NT2) is characterised by the absence of cataplexy with a CSF hypocretin-1 concentration >110 pg/or >1/3 of the mean value obtained in normal individuals with the same standardised assay [21]. It should be considered that, in children with severe depression, excessive daytime sleepiness can be reported. In these cases, CSF hypocretin can be decreased without sleep-onset REM periods on a Multiple Sleep Latency Test. Therefore, diagnosing narcolepsy involves an accurate clinical history (including a good psychiatric history) next to a number of tests performed under carefully controlled conditions. Cases with incongruous results benefit from review and re-testing after an interval of several months or longer [86].
Narcolepsy incidence is 0.83 per 100,000 person–years in children, pre-adolescents and adolescents aged 5 to 19 years [87]. Contrary to what happens with adults, in children the naps tend to be longer (30 to 90 min) and not consistently followed by a refreshed feeling [88]. Cataplexy may often present with facial hypotonia characterised by droopy eyelids, mouth opening, tongue protrusion or gait unsteadiness [89]. Depression, aggressive behaviours, attentional difficulties with school-related learning problems, impaired quality of life, obesity and precocious puberty are associated with the disorder [90,91,92,93]. The majority of cases of narcolepsy are sporadic, but cases of familial narcolepsy have been reported [94]. In some familial cases, mutations in genes modulating the immune response were found, such as the genes encoding orexin (HCRT), myelin oligodendrocyte glycoprotein (MOG) and P2Y purinoceptor 11 (P2RY11) [95,96,97]. Family and twin studies indicate that narcolepsy arises from an interaction between the environment and a predisposed genetic substrate [98]. This substrate is represented by the human leukocyte antigen (HLA) class II region, which encodes molecules that present antigenic peptides to CD4+ T cells. HLA DQB1*0602 and DQA1*0102 are the primary susceptibility alleles for narcolepsy predisposition, although other HLA class II alleles can be encountered in patients with narcolepsy [99,100,101,102,103]. Considering that HLA-DQB1*06:02 is expressed in 86–98% of patients with NT1, in 40–50% of patients with NT2 [97], but also in 5–38% of the general population [101,102], the presence of a histocompatibility antigen is, per se, insufficient to precipitate narcolepsy. Human narcolepsy is therefore best explained by a two-threshold hypothesis, with an interplay between genetic susceptibility (in which the immune system is involved) and environmental factors such as major life events, systemic illness or injury [104]. GWAS provided further evidence of polymorphisms in genes involved in the immune response, such as the gene-encoding T-cell receptor α-constant domain (TRAC) [105] or the gene-encoding tumour necrosis factor ligand superfamily member 4 (TNFSF4) [106].
The Kleine–Levin Syndrome (KLS) is characterised by recurrent episodes of excessive sleepiness and sleep duration persisting from two days to five weeks, in association with at least one of these symptoms: cognitive dysfunction, altered perception, eating disorder or disinhibited behaviour. The episodes usually recur more than once a year and at least every 18 months. The patient has a normal alertness, cognitive function, behaviour and mood between the episodes [21]. The prevalence of the disorder is estimated at around 1 to 2 cases per million. Adolescence (second decade) is the usual age of onset [107,108]. Diagnosis is mainly clinical [109]; it is necessary to exclude other disorders that can mimic its symptoms, such as structural insults of the central nervous system, encephalitis and toxic, metabolic or psychiatric disorders [21]. Social and occupational complications, long-term memory deficits and a higher risk of psychiatric disorders are encountered in patients affected [21]. Most cases of KLS are sporadic; familial occurrence (3 to 8% of relatives) has been reported in some studies [110,111], with some reports of concordant twin pairs suggesting genetic or shared environmental effects [111,112]. An autoimmune mechanism may confer susceptibility to the development of the disorder, as reported in a study on 30 patients with KLS in which an HLA-DQB1*0201 frequency was 28.3% [109]. A worldwide case-control GWAS in 673 KLS cases and ethnically matched 15,341 controls found a strong significant association within the 3′region of the Tetratricopeptide Repeat And Ankyrin Repeat Containing 1 (TRANK1) gene locus, previously associated with bipolar disorder and schizophrenia. Pathway analysis also revealed the involvement of circadian regulation genes. This study also showed that variants in the TRANK1 gene region may predispose to KLS in participants with a difficult birth, suggesting that the TRANK1 gene region modulates newborns’ response to brain injury, resulting in mental and neurological health consequences [113].
In summary, Central Disorders of Hypersomnolence are HLA-associated, multigenic and environmentally influenced conditions. In narcolepsy, the utility of HLA genotyping as a diagnostic tool is controversial and the clinical history next to the Multiple Sleep Latency Test and the hypocretin measurement remain the gold standard for the diagnosis.
6. Circadian Rhythm Sleep-Wake Disorders
The circadian system is a complex, endogenous and essential organisation of biological rhythms, whose oscillations are entrained to the 24-h light-dark cycle. In the mammalian circadian system, this internal clock is genetically determined, self-perpetuating and located in the suprachiasmatic nucleus of the hypothalamus. The master clock must dynamically coordinate and adapt to both dark-light regulation and the body’s internal signals, in order to produce a functional network responding to environmental demand [19,21,114,115,116]. The disruption of the internal clock organisation or a mismatch between the individual’s circadian sleep-wake propensity and the 24-h environmental influences might induce a Circadian Rhythm Sleep-Wake Disorder (CRSWD). For the diagnosis of a CRSWD, the individual must also complain of insomnia, daytime sleepiness or both and be in significant distress due to an impairment in mental, physical and social areas of normal functioning [21].
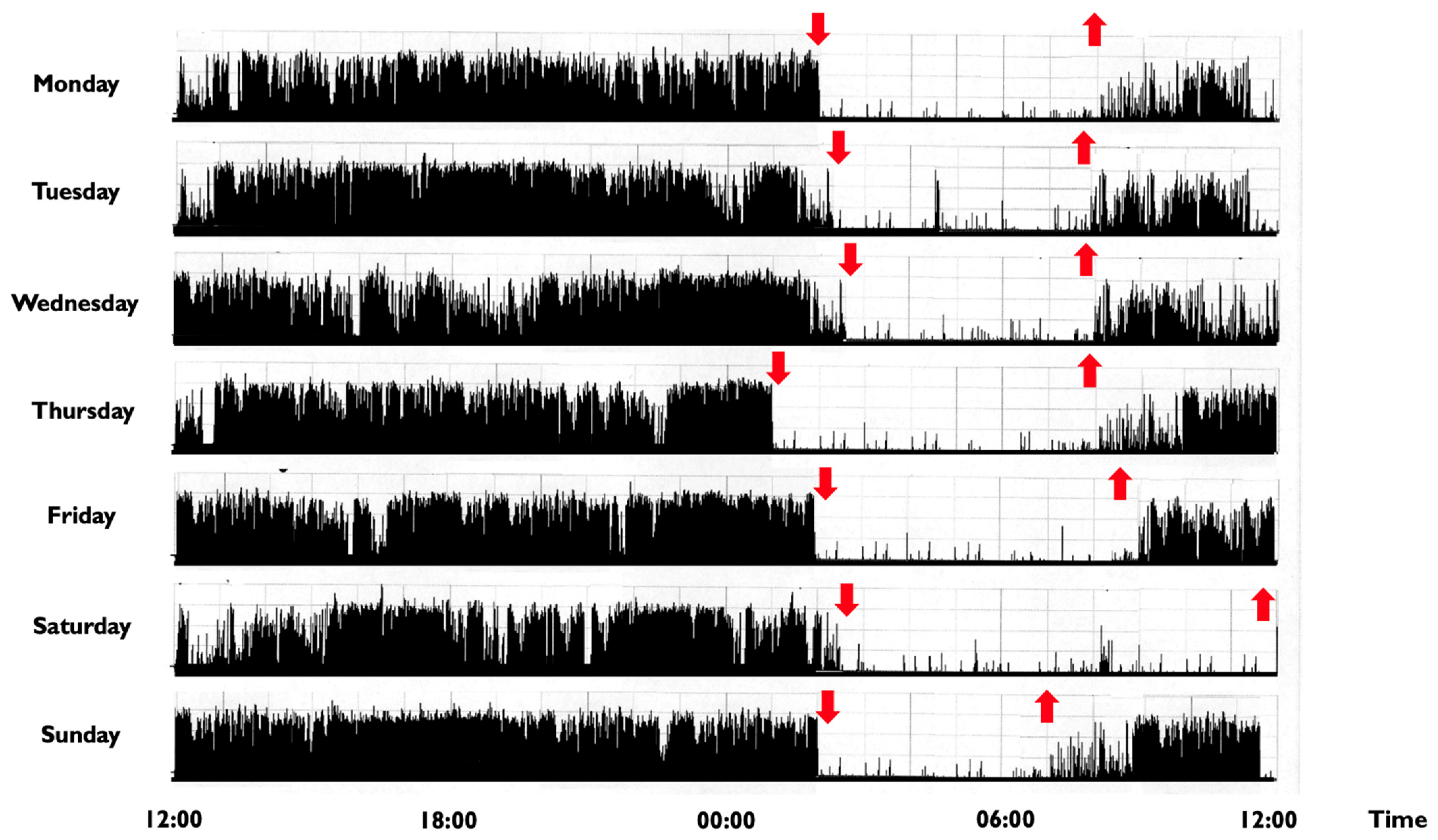
In children and adolescents, a CRSWD might occur in about 10–18% of individuals [2], with a prevalence for the Delayed Sleep-Wake Phase Disorder (DSPD) [117]. The DSPD involves difficulty falling asleep at the desired bedtime, producing a significant delay in the major sleep episode, as well as difficulty awakening at the required clock time (Figure 2) [21].
In addition to clinical history, the diagnosis may benefit from a sleep diary and actigraphic recordings, while a VPSG is not required, unless a comorbid sleep disorder is suspected. In DSPD, a positive family history is reported in about 40% of individuals and polymorphisms in some genes, such as PER3, CLOCK and arylalkylamine n-acetyltransferase have been suggested to play a role in the disorder [18,21,118]. The PER3 gene is of particular interest because it causes opposite phenotypes depending on the length of a common variable number tandem repeat (VNTR) polymorphism in the coding region [13]. Human carriers of the long allele (five repetitions of the VNTR) were found to be associated with an extreme morning preference, while the shorter allele was related to extreme evening preference and DSPD [118,119,120]. Moreover, carriers of the long allele displayed increased slow-wave sleep, theta and frontal delta activity, as well as a more pronounced detrimental effect of sleep deprivation, indicating the important role of this variant also in sleep structure and homeostasis [5,13,18,32]. CLOCK mutations/polymorphisms might be associated with the evening preference, as suggested by animal models in which a CLOCK gene mutation led to phase delays of the rhythm for body temperature, locomotor activity and wake duration [120]. In 2017, a case of familial DSPD linked to a mutation in cryptochrome circadian clock 1 (CRY1) has been reported [121]. CRY1 is one of the main transcriptional inhibitors in the negative feedback loop of the molecular circadian clock, representing a critical regulator of circadian period length.
Contrary to DSPD, the Advanced Sleep Phase Disorder (ASPD) is characterised by the earlier occurrence of the major sleep period due to the difficulty in staying awake until the desired bedtime along with early morning awakening [21]. The ASPD is less frequent than DSPD in paediatric populations [2]; a correlation with preterm birth has been demonstrated [122,123]. Children or adolescents with ASPD fall asleep immediately after or even before dinner, during public events or schoolwork [2]. Familial cases have been described, and the Familial Advanced Sleep Phase Disorder (FASPD) is recognised as a heritable phenotype of the disorder, with few genetic mutations described so far [18,121,124,125]. In the first FASPD family, a mutation of the PER2 gene was identified [124]. This mutation produced an alteration in the PER2 phosphorylation state, responsible for an acceleration of the circadian period [119,120]. Similarly, the second causative mutation for FASPD was encountered in human Casein Kinase I delta (CKIδ), an essential core component of the circadian clock involved in the phosphorylation of PER proteins. By altering its function of phosphorylating PER proteins, the mutation caused an acceleration of the 24h period and advanced sleep phases [119,120,125]. In another family, a missense mutation in the human Cryptochrome 2 (CRY2) gene has been described. This mutation alters the conformation of CRY2, leading to an easier degradation by a ubiquitin complex, causing a shortened circadian period with phase-advanced behavioural rhythms [126]. Interestingly, two rare variants in the gene PER3 were identified in a family with FASPD, whose individuals also displayed high depressive and seasonality scores. The authors suggested that PER3 might constitute a link between sleep and mood disorders, especially in the refinement of these two processes to adapt to periodic seasonal changes [127]. Finally, a study researching genes for the “extreme early bird” phenotype disclosed a mutation in the human DEC2 gene (hDEC2) [128]. DEC2 is a transcription factor regulating the circadian clock in mammals, although its definite role in sleep regulation has not been completely elucidated [129]. The described individuals carrying the mutation exhibited a “Natural Short Sleeper” phenotype with a lifelong daily sleep time need of about 6 h [120,128]. Another variant of this gene mutation showed, in humans, a short sleep phenotype together with resistance to sleep deprivation, suggesting that, beyond the circadian clock system, this gene might be implied in sleep homeostatic processes [130].
In addition, given the complexity of the signalling pathways involved in the circadian clock gene system, an increasingly expanding ground for genetic studies has been flourishing over recent years, with special attention directed at chronotype [18,119,120,131]. So far, slightly more than 300 chronotype-associated loci have been identified from GWAS [18,132,133]. Together with more predictable variants of genes of the circadian clock core, other variants affecting different physiologic pathways were linked to the individual chronotype and included: variants related to the development and functioning of retinal ganglion cells; variants in genes regulating appetite or insulin secretion; variants involved in non-essential habits such as nicotine and caffeine metabolism [133].
In summary, an alteration of circadian system genes appears to be associated with a wide variety of conditions, going from multiple sleep disorders (insomnia, sleep breathing disorders etc.) to the alteration of sleep homeostasis and various medical conditions, such as cardio-metabolic disturbances or mood disorders. As a consequence, a body of evidence is growing up relating the evening chronotype or a DSPD to neurodevelopmental disorders and mood disturbances, including major depression, bipolar disorder and schizophrenia [20,133]. Complex sleep phenotypes, involving different types of circadian disturbances, have been described in attention-deficit hyperactivity disorder, autism spectrum disorders and some genetic conditions, like the Prader–Willi and Smith–Magenis syndromes [20]. Improving the circadian system’s regulation might be helpful to improve the quality of life of these patients.
7. Parasomnias
Sleep parasomnias are currently defined as undesirable physical experiences occurring during the entry into sleep, within sleep or during arousal from sleep [21]. They may occur in NREM, REM sleep or during the transition from wakefulness to sleep or vice-versa [21].
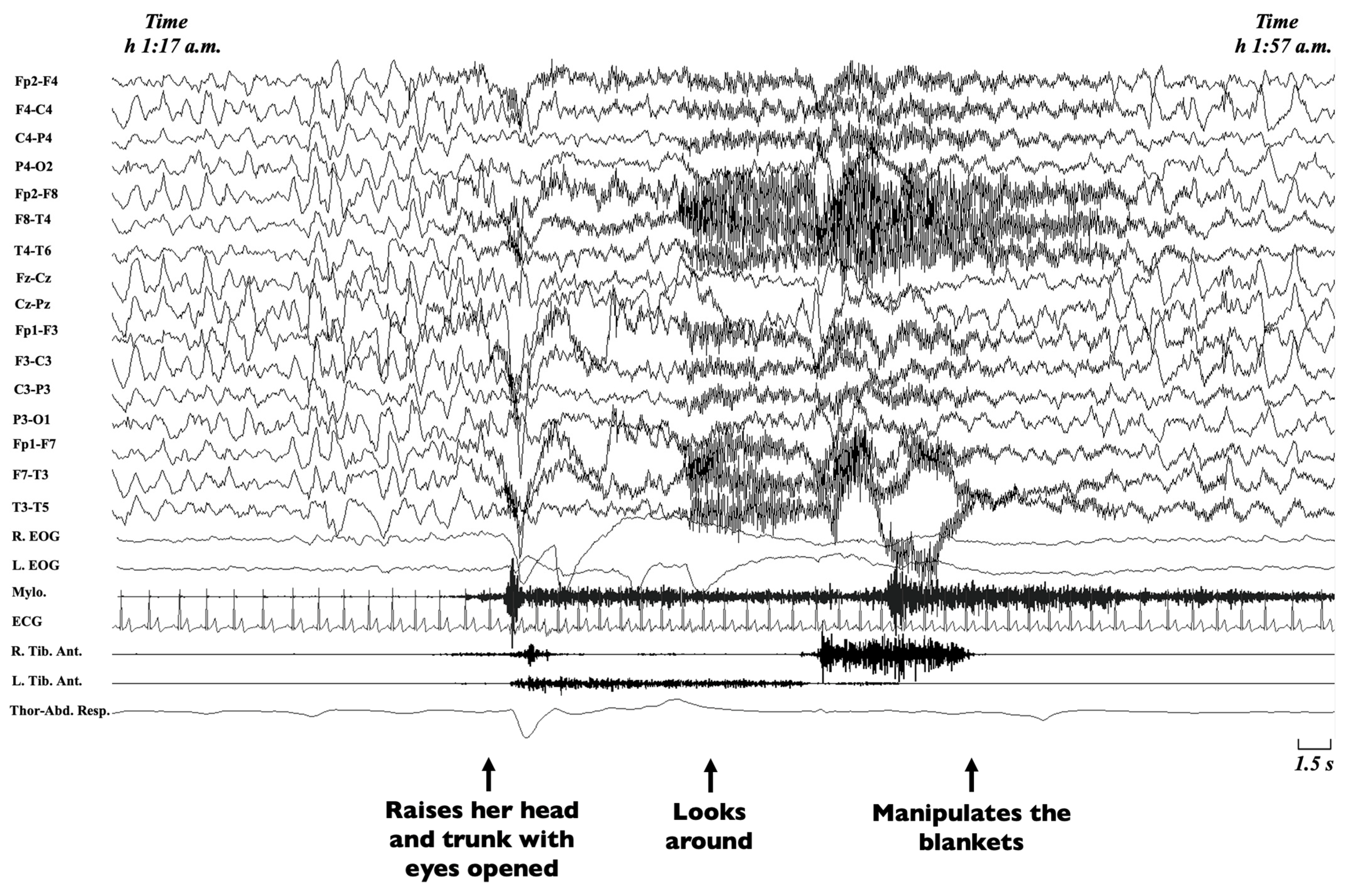
Non-rapid eye movement (NREM) sleep parasomnias are a group of motor manifestations characterised by the occurrence of incomplete awakenings from NREM sleep. This group currently includes the Disorders of Arousal (DoA) and the Sleep-Related Eating Disorder (SRED) [21]. Traditionally, familial recurrence is recognised in SRED, but no systematic genetic studies have been performed [21], while DoA have been more widely studied. DoA are frequent in childhood and adolescence, encompassing three main clinical entities, namely confusional arousals, sleep terrors and sleepwalking [134,135]. Confusional arousals are characterised by a brief awakening from sleep, sitting up in bed, looking around and returning to sleep (Figure 3) [2].
During terrors, children rise abruptly from bed, scream or cry inconsolably, with eyes open and an expression of intense fear [2,136]. In sleepwalking, children may awaken in a different room, wander to their parents’ room or behave inappropriately [2]. To date, a diagnosis is performed on a clinical basis following the ICSD-3 criteria, including the incomplete arousal from sleep, an absent or incongruous response to the external attempts of awakening, a variable or no dream content, amnesia regarding the event and the exclusion of other causes [21]. In atypical cases or to rule out a suspicion of seizures, a VPSG should be performed [137]. Since the earliest descriptions, a strong genetic component has been recognised in DoA [138]. Different studies have shown a higher likelihood of DoA in monozygotic rather than dizygotic twins as well as a higher probability of sleepwalking if at least a parent had presented a DoA in infancy [139,140,141,142,143,144,145,146]. Still, no genes have been identified in family pedigrees, and the most probable mode of inheritance is considered to be multifactorial [141]. The closest identification of a single gene comes from a four-generation family composed of 22 individuals, of which nine were affected by sleepwalking, and in which a linkage to chromosome 20q12-q13.12 was identified. Among the 28 genes from the exonic sequence involved, the Adenosine Deaminase gene (ADA), associated with the quantity of slow wave sleep (SWS), was considered the most likely candidate [147], but no mutation was disclosed. Other studies analysing the relative contribution of genetic and shared and non-shared environmental factors disclosed a double contribution given by genetic factors, on the one hand, accounting for 44%, and non-shared environmental factors for the remaining 56%, on the other [148]. In addition, a higher genetic susceptibility for DoA has been demonstrated in individuals carrying the DQB1*0501 HLA haplotype, a type that is different from the one encountered in narcolepsy [149,150], suggesting a possible involvement of immune-related mechanisms in motor control during sleep. Finally, a specific demonstrated heritability also for homeostatic factors of sleep, together with an EEG trait predisposition, might further account for genetic factors in DoA [9,151].
REM sleep parasomnias include REM Sleep Behaviour Disorder (RBD), Recurrent Isolated Sleep Paralyses and Nightmares. RBD is characterised by vocalisation or complex motor behaviour, mimicking a dream enactment and requiring VPSG evidence of episodes arising from REM sleep or REM sleep without atonia [21]. This parasomnia is currently considered a prodromal phase of alpha-synucleinopathy and occurs typically in adulthood [152]. In paediatric populations, the reports of RBD are mostly anecdotical and related to structural lesions, neurodevelopmental disorders or rare conditions, like the Smith–Magenis and Moebius syndromes [2,153,154,155,156]. More consistently, RBD occurs in paediatric narcolepsy, where it can even be the presenting symptom [157,158].
Sleep paralysis is characterised by the inability to perform voluntary movements at the transition to (hypnagogic) and from (hypnopompic) sleep, lasting seconds or minutes and typically accompanied by an anguishing sensation [21]. When recurrent, they might provoke significant distress and anxiety during sleep time. Other conditions, especially a diagnosis of narcolepsy, have to be excluded [21]. Familial occurrence has been described in some families [159], but, to date, a single twin study disclosed an association with the PER2 circadian gene, which, however, did not survive the Bonferroni correction [160]. Another study failed to identify an association of sleep paralysis with HLA commonly involved in narcolepsy [161].
Nightmares are unpleasant, dysphoric dreams, which lead the individual to awaken in full alertness and report a vivid and detailed recall of the content, without any “dream-enacting” motor activity [162]. To be diagnosed as a disorder, nightmares must be perpetuated and induce a significant impairment in social, occupational or other life aspects of the individual [21]. Nightmares are frequent in children, being an occasional finding in up to 75% of them [21], but persisting as a disorder only in 5% of them, according to a recent meta-analysis [163]. Some twin studies showed a high heritability of nightmares [164,165], but no genetic loci have been identified up to now.
Among other parasomnias, sleep enuresis is characterised by recurrent involuntary voiding during sleep (at least twice a week) for at least 3 months, in children older than 5 years. It further subdivides into primary and secondary, depending on whether the child has never been dry or has been dry for at least 6 months [21]. It is the most frequent urinary symptom in the paediatric field, affecting around 6 million children in the United States [2]. The pathogenesis is complex and probably related to the interplay of multiple factors, such as a high nocturnal production of urine, detrusor overactivity and a reduced arousability from sleep [2,21]. Since the first descriptions, a high heritability has been highlighted in sleep enuresis [166]. Both an autosomal dominant and a recessive mode of inheritance have been described [167]. Some loci were discovered in chromosomes 6, 8, 12, 13 and 22 [167,168,169], involving genes associated with sleep process, urine production and bladder function [169].
Sleep parasomnias, with the exception of adult RBD, are mostly unusual physiological phenomena, which normally do not need pharmacological treatment. Consequently, genetic studies are lacking for this group of disorders, although a clear genetic background is well recognised for some of them. Sleep parasomnias emerge from defective mechanisms controlling the transitions between different sleep and wake stages. Enhancing the knowledge in this particular field could thus help to understand the neurophysiological mechanisms regulating sleep.
8. Sleep-Related Movement Disorders
Sleep-related movement disorders are characterised by simple, stereotyped movements usually affecting sleep or its onset, with the exception of the Restless Leg Syndrome (RLS), which involves deambulation or non-stereotypical movements to reduce leg discomfort [21].
RLS is characterised by: (1) an overwhelming urge to move legs (or other body parts) usually associated with an unpleasant sensation, (2) which begins or worsens at rest, (3) which could be temporarily relieved with movement (4) and occurs predominantly in the evening or night [170]. In children, in addition, the diagnosis of RLS requires the description of discomfort in the child’s own words or two of the following features: (i) a positive family history; (ii) the documentation of periodic limb movements of the sleep (PLMS) index >5/h at VPSG; (iii) sleep disturbance for age [171]. The prevalence rate of definite RLS in childhood is nearly 2%; up to 40% of adults affected by RLS report the onset of symptoms in childhood or adolescence [172,173]. In children, RLS may be idiopathic, with a strong familial recurrence (heritability of around 70% estimated by twin studies [174,175]) with a variable phenotypic expression and the possibility of anticipation [176,177,178]. RLS may be present in conditions such as iron deficiency, Sickle cell disease, Celiac disease, Osgood–Schlatter disease, Diabetes and Thyroid disease [2]. GWAS in RLS identified several risk loci with functions spanning from neurogenesis (MDGA1, MYT1, NTNG1, SEMA6D), cell-junction organisation (PKP4 and SMAD3) and axon guidance (NTNG1 and SEMA6D) to DNA repair/maintenance (APLF, ASTE1, DIS3, PRMT6, and RNF8) and locomotor behaviour (BTBD9, CLN6, HOXB8, and MEIS1) [42,179,180]. Winkelmann et al. were the first to identify the strongest genetic risk factor for RLS in the single nucleotide polymorphisms (SNPs) in the intronics, and rarely coding variants of MEIS1 locus, which encodes for a transcription factor involved in haematopoiesis but also in the neurodevelopment of the proximodistal limb axis [181]. A study specifically targeting childhood-onset RLS confirmed an increased frequency in their index cases of the risk allele in the MEIS1 and the LBXCOR1/MAP2K5 regions, while an association with the third susceptibility region (BTBD9) was excluded [182].
PLMS consist of involuntary recurrent and stereotyped limb movements arising from all sleep stages [65], usually characterised by a dorsiflexion of the foot and lower leg [183]. A higher PLM index could be an independent cause of sleep disturbance with pathological repercussions, representing the Periodic Limb Movement Disorder (PLMD) [21]. Children with PLMD have been reported to present unspecific symptoms such as pain, restless sleep and hyperactivity [184]. The diagnosis of PLMD in children and adolescents requires all the following features: (1) polysomnographic documentation of the PLM index >5 per hour, (2) clinical sleep disturbance, and (3) the absence of another primary sleep disorder (including RLS) [21]. The prevalence of PLMD in paediatric populations has been estimated at around 0.3% [185]. Genetic studies in adults identified some risk loci in PLMD independently of RLS [180,186], but specific data in the paediatric population are lacking.
Sleep bruxism (SB) is a repetitive jaw-muscle activity characterised by involuntary tooth grinding and jaw clenching during sleep. Children or adolescents may complain of transient morning tension headache, jaw muscle pain or jaw lock. Chronic sleep bruxism may provoke tooth wear damage and sleep fragmentation [21,187]. The prevalence in childhood is up to 40% [135]. A tooth-grinding sound during sleep reported by parents, associated with the presence of clinical symptoms/signs of alterations of the stomatognathic system (abnormal tooth wear, hypertrophy of the masseter muscles, discomfort/fatigue or pain in the jaw muscles) allow for a bruxism diagnosis. A video-polysomnography including an EMG recording of masticatory muscles is not routinely required in isolated SB [21]. Twin and familial aggregation studies showed a certain tendency of SB to occur in families [188,189]. The probable genetic component in SB genesis involves personality traits (with a high level of stress and anxiety) [190] and the dopaminergic/serotoninergic systems, as these neurotransmitters regulate the homeostasis of breathing, muscle tone, sleep, stress and reward [191]. Polymorphisms in the gene coding for serotonin receptor 2A (HTR2A) have been associated with SB [192], while SNPs in the dopaminergic pathways could be protective (Dopamine receptors D 2 and 5) or risk factors (Dopamine receptors D3), as they respectively increase or reduce the dopaminergic activity in the central nervous system [193].
Sleep-related rhythmic movements (SRRMs) are repetitive, stereotyped and rhythmic motor behaviours that occur while one is asleep or drowsy, involving large muscle groups. The sleep-related rhythmic movements disorder (SRRMD) may entail significant clinical repercussions (such as sleep disruption, daytime function impairment or body injuries) [21]. SRRMs are characteristic of infants, reaching a prevalence of up to 60%, and usually resolve spontaneously during childhood [194]. A video-polysomnographic recording is required in atypical cases, to exclude possible mimics [194]. Although familial occurrence seems to be rare [195], a genetic predisposition to SRRMs/SRRMD has been proposed in some case reports [196,197,198,199].
Sleep-related movement disorders represent a wide group of manifestations. Many of them show a consistent familial aggregation, even if no specific inheritance patterns and genes have been identified, suggesting complex environmental and genetic interactions (i.e., both iron deficiency and specific genes’ polymorphisms were found to be associated with RLS).
9. Limitations and Conclusions
The genetic predisposition recognised for several sleep disorders has led to a growing body of scientific evidence depicting a complex influence of genetic factors in sleep mechanisms. In addition, highly heritable EEG and sleep traits involved in sleep homeostatic processes have been described and add further complexity to this picture [9,32,200].
Genetic factors play a role of variable weight among the different sleep disorders and show an interplay with the environment that must often be taken into account. From the most recent genetic studies, an intricate picture linking sleep disorders and neuropsychiatric conditions, on the one hand, and metabolic profile, on the other, is emerging [18]. Therefore, expanding the current knowledge on this topic is essential to deepen our understanding of the fascinating variety of sleep functions and their influence on physiological processes, in order to develop possible targeted therapies for these different conditions.
As the main limitation of our paper, we have not performed a quality assessment of the included references, as usually performed for systematic reviews. However, we made a considerable effort to illustrate and synthetise the principal genetic aspects in all paediatric sleep disorders, trying to cover the whole field.
Author Contributions
All authors have seen and approved the manuscript. Conceptualisation, G.M., A.M., G.L. and F.P.; writing—original draft preparation, G.M., A.M. and G.L.; writing—review & editing, A.N., G.D.R., G.M., G.L. and F.P.; supervision, F.P. All authors have read and agreed to the published version of the manuscript.
Funding
The study had no financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
Mainieri, Montini, Loddo, Nicotera and Di Rosa declare that they have no conflicts of interest. Provini reports personal fees from Sanofi, personal fees from Zambon, personal fees from Fidia, personal fees from Bial, personal fees from Eisai Japan and personal fees from Italfarmaco, independent from the submitted work.
Glossary
International Classification of Sleep Disorders—Third Edition (ICSD-3); Genome-Wide Association Studies (GWAS); Restless Leg Syndrome (RLS); Obstructive Sleep Apnea Syndrome (OSAS); Central sleep apnea (CSA); Congenital Central Alveolar Hypoventilation Syndrome (CCHS); Late-Onset Central Hypoventilation with Hypothalamic Dysfunction (ROHHAD); Narcolepsy Type 1 (NT1); Cerebrospinal Fluid (CSF); Narcolepsy Type 2 (NT2); Human Leukocyte Antigen (HLA); Kleine–Levin Syndrome (KLS); Circadian Rhythm Sleep-Wake Disorder (CRSWD); Delayed Sleep-Wake Phase Disorder (DSPD); Variable Number Tandem Repeat (VNTR); Advanced Sleep Phase Disorder (ASPD); Familial Advanced Sleep Phase Disorder (FASPD); Disorders of Arousal (DoA); Sleep-Related Eating Disorder (SRED); REM Sleep Behaviour Disorder (RBD); Single Nucleotide Polymorphisms (SNPs); Periodic Limb Movements of Sleep (PLMS); Periodic Limb Movement Disorder (PLMD); Sleep Bruxism (SB); Sleep-Related Rhythmic Movements (SRRMs); Sleep-Related Rhythmic Movements Disorder (SRRMD).
References
- Anafi, R.C.; Kayser, M.S.; Raizen, D.M. Exploring phylogeny to find the function of sleep. Nat. Rev. Neurosci. 2018, 20, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, S.; Ferber, R.; Kryger, M.; Gozal, D. Principles and Practice of Pediatric Sleep Medicine, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 3–10, 35–42, 99–102, 313–322, 337–347. [Google Scholar]
- Johnson, D.A.; Billings, M.E.; Hale, L. Environmental determinants of insufficient sleep and sleep disorders: Implications for population health. Curr. Epidemiol. Rep. 2018, 5, 61–69. [Google Scholar] [CrossRef]
- Szmyd, B.; Rogut, M.; Białasiewicz, P.; Gabryelska, A. The impact of glucocorticoids and statins on sleep quality. Sleep Med. Rev. 2020, 55, 101380. [Google Scholar] [CrossRef]
- Winkelmann, J.; Kimura, M. Genetics of sleep disorders. Handb. Clin. Neurol. 2011, 99, 681–693. [Google Scholar] [PubMed]
- Garfield, V. Sleep duration: A review of genome-wide association studies (GWAS) in adults from 2007 to 2020. Sleep Med. Rev. 2020, 56, 101413. [Google Scholar] [CrossRef] [PubMed]
- Partinen, M.; Kaprio, J.; Koskenvuo, M.; Putkonen, P.; Langinvainio, H. Genetic and environmental determination of human sleep. Sleep 1983, 6, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.M.; Bianchi, M.T. Mammalian sleep genetics. Neurogenetics 2012, 13, 287–326. [Google Scholar] [CrossRef]
- Franken, P.; Chollet, D.; Tafti, M. The homeostatic regulation of sleep need is under genetic control. J. Neurosci. 2001, 21, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Cirelli, C. The genetic and molecular regulation of sleep: From fruit flies to humans. Nat. Rev. Neurosci. 2009, 10, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Gabryelska, A.; Sochal, M.; Turkiewicz, S.; Białasiewicz, P. Relationship between HIF-1 and circadian clock proteins in obstructive sleep apnea patients—Preliminary study. J. Clin. Med. 2020, 9, 1599. [Google Scholar] [CrossRef]
- Gabryelska, A.; Szmyd, B.; Panek, M.; Szemraj, J.; Kuna, P.; Białasiewicz, P. Serum hypoxia-inducible factor-1α protein level as a diagnostic marker of obstructive sleep apnea. Pol. Arch. Intern. Med. 2019, 130, 158–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chokroverty, S.; Allen, R.P.; Walters, A.S.; Montagna, P. Sleep and Movement Disorders, 2nd ed.; Oxford University Press: Oxford, UK, 2013; pp. 115–117. [Google Scholar]
- Tononi, G.; Cirelli, C. Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 2014, 81, 12–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.M.; Liang, J.; Vlasac, I.; Anderson, S.G.; Bechtold, D.A.; Bowden, J.; Emsley, R.; Gill, S.; Little, M.A.; Luik, A.; et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat. Genet. 2016, 49, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Hammerschlag, A.R.; Stringer, S.; de Leeuw, C.; Sniekers, S.; Taskesen, E.; Watanabe, K.; Blanken, T.F.; Dekker, K.; Lindert, B.H.W.T.; Wassing, R.; et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat. Genet. 2017, 49, 1584–1592. [Google Scholar] [CrossRef]
- Jansen, P.R.; Watanabe, K.; Stringer, S.; Skene, N.; Bryois, J.; Hammerschlag, A.R.; De Leeuw, C.A.; Benjamins, J.S.; Manchado, A.M.; Nagel, M.; et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 2019, 51, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veatch, O.J.; Keenan, B.T.; Gehrman, P.R.; Malow, B.A.; Pack, A.I. Pleiotropic genetic effects influencing sleep and neurological dis-orders. Lancet Neurol. 2017, 16, 158–170. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 2016, 354, 1004–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, R.W.; McClung, C.A. Rhythms of life: Circadian disruption and brain disorders across the lifespan. Nat. Rev. Neurosci. 2018, 20, 49–65. [Google Scholar] [CrossRef]
- American Academy of Sleep Medicine. The International Classification of Sleep Disorders, 3rd ed.; American Academy of Sleep Medicine: Darien, IL, USA, 2014. [Google Scholar]
- Owens, J.A.; Mindell, J.A. Pediatric insomnia. Pediatr. Clin. North. Am. 2011, 58, 555–569. [Google Scholar] [CrossRef]
- Robinson-Shelton, A.; Malow, B.A. Sleep disturbances in neurodevelopmental disorders. Curr. Psychiatry Rep. 2015, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.A.; Moore, M. Insomnia in infants and young children. Pediatr. Ann. 2017, 46, e321–e326. [Google Scholar] [CrossRef] [PubMed]
- Tafti, M.; Maret, S.; Dauvilliers, Y. Genes for normal sleep and sleep disorders. Ann. Med. 2005, 37, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Palagini, L.; Biber, K.; Riemann, D. The genetics of insomnia—Evidence for epigenetic mechanisms? Sleep Med. Rev. 2014, 18, 225–235. [Google Scholar] [CrossRef]
- Lind, M.J.; Gehrman, P.R. Genetic pathways to insomnia. Brain Sci. 2016, 6, 64. [Google Scholar] [CrossRef]
- Lind, M.; Aggen, S.H.; Kirkpatrick, R.M.; Kendler, K.S.; Amstadter, A.B. A longitudinal twin study of insomnia symptoms in adults. Sleep 2015, 38, 1423–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, N.L.; Gehrman, P.R.; Gregory, A.M.; Eaves, L.J.; Silberg, J.L. The heritability of insomnia progression during child-hood/adolescence: Results from a longitudinal twin study. Sleep 2015, 38, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serretti, A.; Benedetti, F.; Mandelli, L.; Lorenzi, C.; Pirovano, A.; Colombo, C.; Smeraldi, E. Genetic dissection of psychopathological symptoms: Insomnia in mood disorders and CLOCK gene polymorphism. Am. J. Med. Genet. 2003, 121, 35–38. [Google Scholar] [CrossRef]
- Wang, C.-C.; Lung, F.-W. The role of PGC-1 and Apoε4 in insomnia. Psychiatr. Genet. 2012, 22, 82–87. [Google Scholar] [CrossRef]
- Viola, A.U.; Archer, S.N.; James, L.M.; Groeger, J.A.; Lo, J.C.; Skene, D.J.; Dijk, D.J. PER3 polymorphism predicts sleep structure and waking performance. Curr. Biol. 2007, 17, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Brower, K.J.; Wojnar, M.; Sliwerska, E.; Armitage, R.; Burmeister, M. PER3 polymorphism and insomnia severity in alcohol de-pendence. Sleep 2012, 35, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brummett, B.H.; Krystal, A.; Ashley-Koch, A.; Kuhn, C.M.; Züchner, S.; Siegler, I.C.; Barefoot, J.C.; Ballard, E.L.; Gwyther, L.P.; Williams, R.B. Sleep quality varies as a function of 5-HTTLPR genotype and stress. Psychosom. Med. 2007, 69, 621–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deuschle, M.; Schredl, M.; Schilling, C.; Wüst, S.; Frank, J.; Witt, S.H.; Schulze, T.G. Association between a serotonin transporter length polymorphism and primary insomnia. Sleep 2010, 33, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Bakish, D.; Ravindran, A.; Hrdina, P.D. MAO-A gene polymorphisms are associated with major depression and sleep dis-turbance in males. Neuroreport 2004, 15, 2097–2101. [Google Scholar] [CrossRef] [PubMed]
- Brummett, B.H.; Krystal, A.D.; Siegler, I.C.; Kuhn, C.; Surwit, R.S.; Züchner, S.; Williams, R.B. Associations of a regulatory polymorphism of monoamine oxidase-A gene promoter (MAOA-uVNTR) with symptoms of depression and sleep quality. Psychosom. Med. 2007, 69, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, H.-J.; Kim, S.C.; Seo, J.; Kang, H.-B.; Choi, J.K. Genetic and metabolic characterization of insomnia. PLoS ONE 2011, 6, e18455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, E.M.; Gehrman, P.R.; Medland, S.E.; Nyholt, D.R.; Heath, A.C.; Madden, P.A.; Chronogen Consortium. A genome-wide association study of sleep habits and insomnia. Am. J. Med. Genet. 2013, 162, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spada, J.; Scholz, M.; Kirsten, H.; Hensch, T.; Horn, K.; Jawinski, P.; Ulke, C.; Burkhardt, R.; Wirkner, K.; Loeffler, M.; et al. Genome-wide association analysis of actigraphic sleep phenotypes in the LIFE Adult Study. J. Sleep Res. 2016, 25, 690–701. [Google Scholar] [CrossRef]
- Amin, N.; Allebrandt, K.V.; Van Der Spek, A.; Müller-Myhsok, B.; Hek, K.; Teder-Laving, M.; Hayward, C.; Esko, T.; Van Mill, J.G.; Mbarek, H.; et al. Genetic variants in RBFOX3 are associated with sleep latency. Eur. J. Hum. Genet. 2016, 24, 1488–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schormair, B.; Zhao, C.; Bell, S.; Tilch, E.; Salminen, A.V.; Pütz, B.; Dauvilliers, Y.; Stefani, A.; Högl, B.; Poewe, W.; et al. Identification of novel risk loci for restless legs syndrome in genome-wide association studies in individuals of European ancestry: A meta-analysis. Lancet Neurol. 2017, 16, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Marcus, C.L.; Brooks, L.J.; Ward, S.D.; Draper, K.A.; Gozal, D.; Halbower, A.C.; Spruyt, K. Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 2012, 130, e714–e755. [Google Scholar] [CrossRef] [Green Version]
- Section on Pediatric Pulmonology; Subcommittee on Obstructive Sleep Apnea Syndrome; American Academy of Pediatrics. Clinical practice guideline: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 2002, 109, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Verhulst, S.; Kaditis, A. Obstructive sleep apnoea in children. Breathe 2011, 7, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Parish, J.M. Genetic and immunologic aspects of sleep and sleep disorders. Chest 2013, 143, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyrum, E.; Balbay, O.; Annakkaya, A.N.; Balbay, E.G.; Silan, F.; Arbak, P. The relationship between obstructive sleep apnea syndrome and apolipoprotein e genetic variants. Respiration 2015, 89, 195–200. [Google Scholar] [CrossRef]
- Ovchinsky, A.; Rao, M.; Lotwin, I.; Goldstein, N.A. The familial aggregation of pediatric obstructive sleep apnea syndrome. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavezzi, A.M.; Casale, V.; Oneda, R.; Gioventù, S.; Matturri, L.; Farronato, G. Obstructive sleep apnea syndrome (OSAS) in children with class III malocclusion: Involvement of the PHOX2B gene. Sleep Breath. 2013, 17, 1275–1280. [Google Scholar] [CrossRef]
- Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C.; Brunet, J.-F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 1999, 399, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Weese-Mayer, D.E.; Berry-Kravis, E.M.; Zhou, L.; Maher, B.S.; Silvestri, J.M.; Curran, M.E.; Marazita, M.L. Idiopathic congenital central hypoventilation syndrome: Analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am. J. Med. Genet. 2003, 123A, 267–278. [Google Scholar] [CrossRef]
- Au, C.T.; Zhang, J.; Cheung, J.Y.F.; Chan, K.C.C.; Wing, Y.K.; Li, A.M. Familial aggregation and heritability of obstructive sleep apnea using children probands. J. Clin. Sleep Med. 2019, 15, 1561–1570. [Google Scholar] [CrossRef]
- Ye, X.H.; Chen, H.; Yu, Q.; Zhu, Q.L. Liver X receptor gene expression is enhanced in children with obstructive sleep apnea-hyperpnoea syndrome and cyclooxygenase-2 (COX-2) is correlated with severity of Obstructive Sleep Apnea-Hypopnea Syndrome (OSAHS). Int. Med. J. Exp. Clin. Res. 2017, 23, 3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, B.; Khalyfa, A.; Spruyt, K.; Kheirandish-Gozal, L.; Capdevila, O.S.; Bhattacharjee, R.; Kim, J.; Keating, B.; Hakonarson, H.; Gozal, D. Fatty-acid binding protein 4 gene polymorphisms and plasma levels in children with obstructive sleep apnea. Sleep Med. 2011, 12, 666–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisgraber, K.H.; Roses, A.D.; Strittmatter, W.J. The role of apolipoprotein E in the nervous system. Curr. Opin. Lipidol. 1994, 5, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Capdevila, O.S.; Kheirandish-Gozal, L.; Crabtree, V.M. APOE epsilon 4 allele, cognitive dysfunction, and obstructive sleep apnea in children. Neurology 2007, 69, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Khalyfa, A.; Capdevila, O.S.; Kheirandish-Gozal, L.; Khalyfa, A.A.; Kim, J. Cognitive function in prepubertal children with obstructive sleep apnea: A modifying role for NADPH oxidase p22 subunit gene polymorphisms? Antioxid. Redox Signal. 2012, 16, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, D.; Dayyat, E.A.; Zhang, S.X.; Wang, Y.; Gozal, D. Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS ONE 2011, 6, e19847. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Capdevila, O.S.; Buazza, M.O.; Serpero, L.D.; Kheirandish-Gozal, L.; Gozal, D. Genome-wide gene expression profiling in children with non-obese obstructive sleep apnea. Sleep Med. 2009, 10, 75–86. [Google Scholar] [CrossRef]
- Khalyfa, A.; Kheirandish-Gozal, L.; Khalyfa, A.A.; Philby, M.F.; Alonso-Álvarez, M.L.; Mohammadi, M.; Gozal, D. Circulating plasma extracellular microvesicle microRNA cargo and endothelial dysfunction in children with obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2016, 194, 1116–1126. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bhattacharjee, R.; Khalyfa, A.; Gozal, L.; Capdevila, O.S.; Wang, Y.; Gozal, D. DNA methylation in inflammatory genes among children with obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2012, 185, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalyfa, A.; Kheirandish-Gozal, L.; Capdevila, O.S.; Bhattacharjee, R.; Gozal, D. Macrophage migration inhibitory factor gene polymorphisms and plasma levels in children with obstructive sleep apnea. Pediatr. Pulmonol. 2012, 47, 1001–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozal, L.; Khalyfa, A.; Gozal, D.; Bhattacharjee, R.; Wang, Y. Endothelial dysfunction in children with obstructive sleep apnea is associated with epigenetic changes in the eNOS gene. Chest 2013, 143, 971–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, M.-S.; Xu, L.; Zheng, J.-S. Association of T lymphocyte immune imbalance and IL-10 gene polymorphism with the risk of obstructive sleep apnea in children with obesity. Sleep Breath. 2017, 177, 1142–1937. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.B.; Brooks, R.; Gamaldo, C.E.; Harding, S.M.; Marcus, C.; Vaughn, B.V. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications; Version 2.6; American Academy of Sleep Medicine: Darien, IL, USA, 2020. [Google Scholar]
- McLaren, A.T.; Bin-Hasan, S.; Narang, I. Diagnosis, management and pathophysiology of central sleep apnea in children. Paediatr. Respir. Rev. 2018, 30, 49–57. [Google Scholar] [CrossRef]
- Marcus, C.L.; Omlin, K.J.; Basinki, D.J.; Bailey, S.L.; Rachal, A.B.; Von Pechmann, W.S.; Keens, T.G.; Ward, S.L.D. Normal polysomnographic values for children and adolescents. Am. Rev. Respir. Dis. 1992, 146, 1235–1239. [Google Scholar] [CrossRef]
- Pillekamp, F.; Hermann, C.; Keller, T.; Von Gontard, A.; Kribs, A.; Roth, B. Factors influencing apnea and bradycardia of prematurity—Implications for neurodevelopment. Neonatology 2006, 91, 155–161. [Google Scholar] [CrossRef]
- Salisbury, E.; Hall, M.H.; Sharma, P.; Boyd, T.; Bednarek, F.; Paydarfar, D. Heritability of apnea of prematurity: A retrospective twin study. Pediatrics 2010, 126, e779–e787. [Google Scholar] [CrossRef]
- Xu, X.-H.; Huang, X.-W.; Qun, L.; Li, Y.-N.; Wang, Y.; Liu, C.; Ma, Y.; Liu, Q.-M.; Sun, K.; Qian, F.; et al. Two functional loci in the promoter of EPAS1 gene involved in high-altitude adaptation of Tibetans. Sci. Rep. 2014, 4, 7465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basang, Z.; Wang, B.; Li, L.; Yang, L.; Liu, L.; Cui, C.; Lanzi, G.; Yuzhen, N.; Duo, J.; Zheng, H.; et al. HIF2A variants were associated with different levels of high-altitude hypoxia among native Tibetans. PLoS ONE 2015, 10, e0137956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kritzinger, F.E.; Al-Saleh, S.; Narang, I. Descriptive analysis of central sleep apnea in childhood at a single center. Pediatr. Pulmonol. 2011, 46, 1023–1030. [Google Scholar] [CrossRef]
- Felix, O.; Amaddeo, A.; Arroyo, J.O.; Zerah, M.; Puget, S.; Cormier-Daire, V.; Baujat, G.; Pinto, G.; Fernandez-Bolanos, M.; Fauroux, B. Central sleep apnea in children: Experience at a single center. Sleep Med. 2016, 25, 24–28. [Google Scholar] [CrossRef] [PubMed]
- White, K.K.; Parnell, S.E.; Kifle, Y.; Blackledge, M.; Bompadre, V. Is there a correlation between sleep disordered breathing and fo-ramen magnum stenosis in children with achondroplasia? Am. J. Med. Genet. 2016, 170, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Ghirardo, S.; Amaddeo, A.; Griffon, L.; Khirani, S.; Fauroux, B. Central apnea and periodic breathing in children with underlying conditions. J. Sleep Res. 2021, e13388. [Google Scholar] [CrossRef]
- Trang, H.; Samuels, M.; Ceccherini, I.; Frerick, M.; Garcia-Teresa, M.A.; Peters, J.; Schoeber, J.; Migdal, M.; Markstrom, A.; Ottonello, G.; et al. Guidelines for diagnosis and management of congenital central hypoventilation syndrome. Orphanet. J. Rare Dis. 2020, 15, 1–21. [Google Scholar] [CrossRef]
- Matera, V.; Bachetti, T.; Puppo, F.; Di Duca, M.; Morandi, F.; Casiraghi, G.; Cilio, M.; Hennekam, R.; Hofstra, R.; Schober, J.; et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset Central Hypoventilation syndrome. J. Med. Genet. 2004, 41, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiel, J.; Laudier, B.; Attie-Bitach, T.; Trang, H.; Pontual, L.; Gener, B.; Trochet, D.; Etchevers, H.; Ray, P.; Simonneau, M.; et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 2003, 33, 459–461. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, A.; Kanai, M.; Kijima, K.; Akaba, K.; Hashimoto, M.; Hasegawa, H.; Hayasaka, K. Molecular analysis of congenital central hypoventilation syndrome. Hum. Genet. 2003, 114, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Todd, E.S.; Weinberg, S.M.; Berry-Kravis, E.M.; Silvestri, J.M.; Kenny, A.S.; Rand, C.M.; Zhou, L.; Maher, B.S.; Marazita, M.L.; Weese-Mayer, D. Facial phenotype in children and young adults with PHOX2B–determined congenital central hypoventilation syndrome: Quantitative pattern of dysmorphology. Pediatr. Res. 2006, 59, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reppucci, D.; Hamilton, J.; Yeh, E.A.; Katz, S.; Al-Saleh, S.; Narang, I. ROHHAD syndrome and evolution of sleep disordered breathing. Orphanet J. Rare Dis. 2016, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Lazea, C.; Sur, L.; Florea, M. ROHHAD (Rapid-onset obesity with hypoventilation, hypothalamic dysfunction, autonomic dysregulation) syndrome—What every pediatrician should know about the etiopathogenesis, diagnosis and treatment: A review. Int. J. Gen. Med. 2021, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Barclay, S.F.; Rand, C.M.; Gray, P.A.; Gibson, W.T.; Wilson, R.J.; Berry-Kravis, E.M.; Ize-Ludlow, D.; Bech-Hansen, N.T.; Weese-Mayer, D.E. Absence of mutations in HCRT, HCRTR1 and HCRTR2 in patients with ROHHAD. Respir. Physiol. Neurobiol. 2016, 221, 59–63. [Google Scholar] [CrossRef]
- Patwari, P.P.; Rand, C.M.; Berry-Kravis, E.M.; Ize-Ludlow, D.; Weese-Mayer, D.E. Monozygotic twins discordant for ROHHAD phenotype. Pediatrics 2011, 128, e711–e715. [Google Scholar] [CrossRef]
- Szymońska, I.; Borgenvik, T.L.; Karlsvik, T.M.; Halsen, A.; Malecki, B.K.; Saetre, S.E.; Malecki, M. Novel mutation-deletion in the PHOX2B gene of the patient diagnosed with neuroblastoma, hirschsprung’s disease, and congenital central hypoventilation syndrome (NB-HSCR-CCHS) cluster. J. Genet. Syndr. Gene Ther. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Gabryelska, A.; Szmyd, B.; Maschauer, E.L.; Roguski, A.; Canham, R.; Morrison, I.; Białasiewicz, P.; Riha, R.L. Utility of measuring CSF hypocretin-1 level in patients with suspected narcolepsy. Sleep Med. 2020, 71, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Wijnans, L.; Lecomte, C.; de Vries, C.; Weibel, D.; Sammon, C.; Hviid, A.; Svanström, H.; Mølgaard-Nielsen, D.; Heijbel, H.; Dahlström, L.A.; et al. The incidence of narcolepsy in Europe: Before, during, and after the influenza A(H1N1)pdm09 pandemic and vaccination campaigns. Vaccine 2013, 31, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, S.; Hartse, K.M.; Walsh, J.K. Characteristics of narcolepsy in preteenaged children. Pediatrics 1990, 85. [Google Scholar]
- Plazzi, G.; Pizza, F.; Palaia, V.; Franceschini, C.; Poli, F.; Moghadam, K.K.; Cortelli, P.; Nobili, L.; Bruni, O.; Dauvilliers, Y.; et al. Complex movement disorders at disease onset in childhood narcolepsy with cataplexy. Brain 2011, 134, 3480–3492. [Google Scholar] [CrossRef]
- Quaedackers, L.; Van Gilst, M.M.; Van Mierlo, P.; Lammers, G.-J.; Dhondt, K.; Amesz, P.; Peeters, E.; Hendriks, D.; Vandenbussche, N.; Pillen, S.; et al. Impaired social functioning in children with narcolepsy. Sleep 2018, 42. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, G.-H.; Sung, S.M.; Jung, D.S.; Pak, K. Prevalence of attention deficit hyperactivity disorder symptoms in narcolepsy: A systematic review. Sleep Med. 2019, 65, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, S.; Krahn, L.E.; Slocumb, N. A putative link between childhood narcolepsy and obesity. Sleep Med. 2004, 5, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Poli, F.; Pizza, F.; Mignot, E.; Ferri, R.; Pagotto, U.; Taheri, S.; Finotti, E.; Bernardi, F.; Pirazzoli, P.; Cicognani, A.; et al. High prevalence of precocious puberty and obesity in childhood narcolepsy with cataplexy. Sleep 2013, 36, 175–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignot, E. Genetic and familial aspects of narcolepsy. Neurology 1998, 50, S16–S22. [Google Scholar] [CrossRef]
- Peyron, C.; Faraco, J.; Rogers, W.; Ripley, B.; Overeem, S.; Charnay, Y.; Nevsimalova, S.; Aldrich, M.; Reynolds, D.; Albin, R.; et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat. Med. 2000, 6, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Hor, H.; Bartesaghi, L.; Kutalik, Z.; Vicário, J.L.; de Andrés, C.; Pfister, C.; Peraita-Adrados, R. A missense mutation in myelin oligodendrocyte glycoprotein as a cause of familial narcolepsy with cataplexy. Am. J. Hum. Genet. 2011, 89, 474–479. [Google Scholar] [CrossRef] [Green Version]
- Degn, M.; Dauvilliers, Y.; Dreisig, K.; Lopez, R.; Pfister, C.; Pradervand, S.; Kornum, B.R.; Tafti, M. Rare missense mutations in P2RY11 in narcolepsy with cataplexy. Brain 2017, 140, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Raizen, D.M.; Mason, T.B.A.; Pack, A.I. Genetic basis for sleep regulation and sleep disorders. Semin. Neurol. 2006, 26, 467–483. [Google Scholar] [CrossRef]
- Ollila, H.M. Narcolepsy type 1: What have we learned from genetics? Sleep 2020, 43, zsaa099. [Google Scholar] [CrossRef] [PubMed]
- Nishino, S.; Okura, M.; Mignot, E. Narcolepsy: Genetic predisposition and neuropharmacological mechanisms: Review article. Sleep Med. Rev. 2000, 4, 57–99. [Google Scholar] [CrossRef] [PubMed]
- European Narcolepsy Network (EU-NN). DQB1 locus alone explains most of the risk and protection in narcolepsy with cataplexy in Europe. Sleep 2014, 37, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Mignot, E.; Lin, L.; Rogers, W.; Honda, Y.; Qiu, X.; Lin, X.; Risch, N. Complex HLA-DR and-DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am. J. Hum. Genet. 2001, 68, 686–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, F.; Faraco, J.; Dong, X.S.; Ollila, H.; Lin, L.; Li, J.; An, P.; Wang, S.; Jiang, K.W.; Gao, Z.C.; et al. Genome wide analysis of narcolepsy in china implicates novel immune loci and reveals changes in association prior to versus after the 2009 H1N1 influenza pandemic. PLoS Genet. 2013, 9, e1003880. [Google Scholar] [CrossRef]
- Orellana, C.; Villemin, E.; Tafti, M.; Carlander, B.; Besset, A.; Billiard, M. Life events in the year preceding the onset of narcolepsy. Sleep 1994, 17, S50–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmayer, J.; Faraco, J.; Lin, L.; Hesselson, S.; Winkelmann, J.; Kawashima, M.; Mignot, E. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat. Genet. 2009, 41, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Mahlios, J.; Mignot, E. Genetic association, seasonal infections and autoimmune basis of narcolepsy. J. Autoimmun. 2013, 43, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnulf, I.; Lin, L.; Gadoth, N.; File, J.; Lecendreux, M.; Franco, P.; Zeitzer, J.; Lo, B.; Faraco, J.H.; Mignot, E. Kleine-Levin syndrome: A systematic study of 108 patients. Ann. Neurol. 2008, 63, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Arnulf, I.; Zeitzer, J.M.; File, J.; Farber, N.; Mignot, E. Kleine–Levin syndrome: A systematic review of 186 cases in the literature. Brain 2005, 128, 2763–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauvilliers, Y.; Mayer, G.; Lecendreux, M.; Neidhart, E.; Peraita-Adrados, R.; Sonka, K.; Billiard, M.; Tafti, M. Kleine-Levin syndrome: An autoimmune hypothesis based on clinical and genetic analyses. Neurology 2002, 59, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Billiard, M.; Jaussent, I.; Dauvilliers, Y.; Besset, A. Recurrent hypersomnia: A review of 339 cases. Sleep Med. Rev. 2011, 15, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Nguyen QT, R.; Groos, E.; Leclair-Visonneau, L.; Monaca-Charley, C.; Rico, T.; Farber, N.; Arnulf, I. Familial Kleine-Levin syndrome: A specific entity? Sleep 2016, 39, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peraita-Adrados, R.; Vicario, J.L.; Tafti, M.; De León, M.G.; Billiard, M. Monozygotic twins affected with Kleine-Levin syndrome. Sleep 2012, 35, 595–596. [Google Scholar] [CrossRef]
- Ambati, A.; Hillary, R.; Leu-Semenescu, S.; Ollila, H.M.; Lin, L.; During, E.H.; Farber, N.; Rico, T.J.; Faraco, J.; Leary, E.; et al. Kleine-Levin syndrome is associated with birth difficulties and genetic variants in the TRANK1 gene loci. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Van Drunen, R.; Eckel-Mahan, K. Circadian rhythms of the hypothalamus: From function to physiology. Clocks Sleep 2021, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, U. Timing to perfection: The biology of central and peripheral circadian clocks. Neuron 2012, 74, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Matenchuk, B.A.; Mandhane, P.J.; Kozyrskyj, A.L. Sleep, circadian rhythm, and gut microbiota. Sleep Med. Rev. 2020, 53, 101340. [Google Scholar] [CrossRef] [PubMed]
- Feder, M.A.; Baroni, A. Just let me sleep in: Identifying and treating delayed sleep phase disorder in adolescents. Child Adolesc. Psychiatr. Clin. 2021, 30, 159–174. [Google Scholar] [CrossRef]
- Archer, S.N.; Carpen, J.D.; Gibson, M.S.; Lim, G.H.; Johnston, J.; Skene, D.; von Schantz, M. Polymorphism in the PER3 promoter associates with diurnal preference and delayed sleep phase disorder. Sleep 2010, 33, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Wulff, K.; Porcheret, K.; Cussans, E.; Foster, R.G. Sleep and circadian rhythm disturbances: Multiple genes and multiple phenotypes. Curr. Opin. Genet. Dev. 2009, 19, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.R.; Huang, A.L.; Ptáček, L.J.; Fu, Y.-H. Genetic basis of human circadian rhythm disorders. Exp. Neurol. 2013, 243, 28–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patke, A.; Murphy, P.J.; Onat, O.E.; Krieger, A.C.; Özçelik, T.; Campbell, S.S.; Young, M.W. Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell 2017, 169, 203.e13–215.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkqvist, J.; Paavonen, J.; Andersson, S.; Pesonen, A.-K.; Lahti, J.; Heinonen, K.; Eriksson, J.; Räikkönen, K.; Hovi, P.; Kajantie, E.; et al. Advanced sleep-wake rhythm in adults born prematurely: Confirmation by actigraphy-based assessment in the Helsinki Study of very low birth weight adults. Sleep Med. 2014, 15, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, A.M.; Storfer-Isser, A.; Rosen, C.; Ievers-Landis, C.E.; Taveras, E.M.; Redline, S. Advanced sleep phase in adolescents born preterm. Behav. Sleep Med. 2013, 12, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toh, K.L.; Jones, C.R.; He, Y.; Eide, E.J.; Hinz, W.A.; Virshup, D.M.; Ptáček, L.J.; Fu, Y.-H. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 2001, 291, 1040–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Padiath, Q.S.; Shapiro, R.E.; Jones, C.R.; Wu, S.C.; Saigoh, N.; Fu, Y.H. Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome. Nature 2005, 434, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Shi, G.; Jones, C.R.; Lipzen, A.; Pennacchio, L.; Xu, Y.; Hallows, W.C.; McMahon, T.; Yamazaki, M.; Ptáček, L.J.; et al. A cryptochrome 2 mutation yields advanced sleep phase in humans. eLife 2016, 5, e16695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hirano, A.; Hsu, P.-K.; Jones, C.R.; Sakai, N.; Okuro, M.; McMahon, T.; Yamazaki, M.; Xu, Y.; Saigoh, N.; et al. A PERIOD3 variant causes a circadian phenotype and is associated with a seasonal mood trait. Proc. Natl. Acad. Sci. USA 2016, 113, E1536–E1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Jones, C.R.; Fujiki, N.; Xu, Y.; Guo, B.; Holder, J.L.; Rossner, M.J.; Nishino, S.; Fu, Y.-H. The transcriptional repressor DEC2 regulates sleep length in mammals. Science 2009, 325, 866–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, A.; Hsu, P.-K.; Zhang, L.; Xing, L.; McMahon, T.; Yamazaki, M.; Ptáček, L.J.; Fu, Y.-H. DEC2 modulates orexin expression and regulates sleep. Proc. Natl. Acad. Sci. USA 2018, 115, 3434–3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, R.; Kavakli, I.H.; Goel, N.; Cardinale, C.J.; Dinges, D.F.; Kuna, S.T.; Maislin, G.; Van Dongen, H.P.; Tufik, S.; HogenEsch, J.B.; et al. A NovelBHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep 2014, 37, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Adan, A.; Archer, S.N.; Hidalgo, M.P.; Di Milia, L.; Natale, V.; Randler, C. Circadian typology: A comprehensive review. Chronobiol. Int. 2012, 29, 1153–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalmbach, D.A.; Schneider, L.; Cheung, J.; Bertrand, S.J.; Kariharan, T.; Pack, A.; Gehrman, P.R. Genetic basis of chronotype in humans: Insights from three landmark GWAS. Sleep 2016, 40. [Google Scholar] [CrossRef]
- Jones, S.E.; Lane, J.M.; Wood, A.R.; Van Hees, V.T.; Tyrrell, J.; Beaumont, R.N.; Jeffries, A.R.; Dashti, H.S.; Hillsdon, M.; Ruth, K.S.; et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, L.; Tremblay, R.E.; Vitaro, F.; Montplaisir, J. Development of parasomnias from childhood to early adolescence. Pediatrics 2000, 106, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit, D.; Touchette, E.; Tremblay, R.E.; Boivin, M.; Montplaisir, J. Dyssomnias and parasomnias in early childhood. Pediatrics 2007, 119, e1016–e1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.K.; Leung, A.A.; Wong, A.H.; Hon, K.L. Sleep terrors: An updated review. Curr. Pediatr. Rev. 2020, 16, 176–182. [Google Scholar] [CrossRef]
- Loddo, G.; Lopez, R.; Cilea, R.; Dauvilliers, Y.; Provini, F. Disorders of arousal in adults: New diagnostic tools for clinical practice. Sleep Sci. Pract. 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.; Hayes, M.; Kirman, B. Somnambulism. Lancet 1942, 239, 186. [Google Scholar] [CrossRef]
- Bakwin, H. Sleep-walking in twins. Lancet 1970, 2, 446–447. [Google Scholar] [CrossRef]
- Abe, K.; Shimakawa, M. Predisposition to sleep-walking. Psychiatr. Neurol. 1966, 152, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Kales, A.; Soldatos, C.R.; Bixler, E.O.; Ladda, R.L.; Charney, D.S.; Weber, G.; Schweitzer, P.K.; Gartner, J.; Langford, A.; O’Brien, A.; et al. Hereditary factors in sleepwalking and night terrors. Br. J. Psychiatry 1980, 137, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Amatomi, M.; Oda, N. Sleepwalking and recurrent sleeptalking in children of childhood sleepwalkers. Am. J. Psychiatry 1984, 141, 800–801. [Google Scholar] [CrossRef]
- Hällström, T. Night terror in adults through three generations. Acta Psychiatr. Scand. 1972, 48, 350–352. [Google Scholar] [CrossRef]
- Hublin, C.; Kaprio, J.; Partinen, M.; Heikkilä, K.; Koskenvuo, M. Prevalence and genetics of sleepwalking: A population-based twin study. Neurology 1997, 48, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hublin, C.; Kaprio, J. Genetic aspects and genetic epidemiology of parasomnias. Sleep Med. Rev. 2003, 7, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Petit, D.; Pennestri, M.H.; Paquet, J.; Desautels, A.; Zadra, A.; Vitaro, F.; Montplaisir, J. Childhood sleepwalking and sleep terrors: A longitudinal study of prevalence and familial aggregation. JAMA Pediatrics 2015, 169, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licis, A.K.; Desruisseau, D.M.; Yamada, K.A.; Duntley, S.P.; Gurnett, C.A. Novel genetic findings in an extended family pedigree with sleepwalking. Neurology 2010, 76, 49–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.H.; Paquet, J.; Petit, M.; Boivin, M.; Tremblay, R.E.; Montplaisir, J.; Pérusse, D. Sleep terrors in children: A prospective study of twins. Pediatrics 2008, 122, e1164–e1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecendreux, M.; Bassetti, C.L.; Dauvilliers, Y.; Mayer, G.; Neidhart, E.; Tafti, M. HLA and genetic susceptibility to sleepwalking. Mol. Psychiatry 2003, 8, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Heidbreder, A.; Frauscher, B.; Mitterling, T.; Boentert, M.; Schirmacher, A.; Hörtnagl, P.; Schennach, H.; Massoth, C.; Happe, S.; Mayer, G.; et al. Not only sleepwalking but NREM parasomnia irrespective of the type is associated with HLA DQB1 05:01. J. Clin. Sleep Med. 2016, 12, 565–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijk, D.-J.; Lockley, S.W. Invited review: Integration of human sleep-wake regulation and circadian rhythmicity. J. Appl. Physiol. 2002, 92, 852–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Högl, B.; Stefani, A.; Videnovic, A. Idiopathic REM sleep behaviour disorder and neurodegeneration—An update. Nat. Rev. Neurol. 2017, 14, 40–55. [Google Scholar] [CrossRef] [PubMed]
- De Barros-Ferreira, M.; Chodkiewicz, J.P.; Lairy, G.C.; Salzarulo, P. Disorganized relations of tonic and phasic events of REM sleep in a case of brain-stem tumour. Electroencephalogr. Clin. Neurophysiol. 1975, 38, 203–207. [Google Scholar] [CrossRef]
- Sheldon, S.H.; Jacobsen, J. REM-sleep motor disorder in children. J. Child Neurol. 1998, 13, 257–260. [Google Scholar] [CrossRef]
- Stores, G. Rapid eye movement sleep behaviour disorder in children and adolescents. Dev. Med. Child Neurol. 2008, 50, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.; Tippmann-Peikert, M.; Slocumb, N.; Kotagal, S. Characteristics of REM sleep behavior disorder in childhood. J. Clin. Sleep Med. 2012, 8, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Nevsimalova, S.; Příhodová, I.; Kemlink, D.; Lin, L.; Mignot, E. REM behavior disorder (RBD) can be one of the first symptoms of childhood narcolepsy. Sleep Med. 2007, 8, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Bin-Hasan, S.; Videnovic, A.; Maski, K. Nocturnal REM sleep without atonia is a diagnostic biomarker of pediatric narcolepsy. J. Clin. Sleep Med. 2018, 14, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Denis, D.; French, C.C.; Gregory, A. A systematic review of variables associated with sleep paralysis. Sleep Med. Rev. 2017, 38, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Denis, D.; French, C.C.; Rowe, R.; Zavos, H.; Nolan, P.; Parsons, M.J.; Gregory, A.M. A twin and molecular genetics study of sleep paralysis and associated factors. J. Sleep Res. 2015, 24, 438–446. [Google Scholar] [CrossRef]
- Dahlitz, M.; Parkes, J.D. Sleep paralysis. Lancet 1993, 341, 406–407. [Google Scholar] [CrossRef]
- Kryger, M.; Roth, T.; Dement, W. Principles and Practice of Sleep Medicine; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1002–1010. [Google Scholar]
- Chen, X.; Ke, Z.L.; Chen, Y.; Lin, X. The prevalence of sleep problems among children in mainland China: A meta-analysis and systemic-analysis. Sleep Med. 2021, 83, 248–255. [Google Scholar] [CrossRef]
- Hublin, C.; Kaprio, J.; Partinen, M.; Koskenvuo, M. Nightmares: Familial aggregation and association with psychiatric disorders in a nationwide twin cohort. Am. J. Med. Genet. 1999, 88, 329–336. [Google Scholar] [CrossRef]
- Coolidge, F.L.; Segal, D.L.; Coolidge, C.M.; Spinath, F.M.; Gottschling, J. Do nightmares and generalized anxiety disorder in childhood and adolescence have a common genetic origin? Behav. Genet. 2010, 40, 349–356. [Google Scholar] [CrossRef]
- Bayoumi, R.A.; Eapen, V.; Al-Yahyaee, S.; Al Barwani, H.S.; Hill, R.S.; Al Gazali, L. The genetic basis of inherited primary nocturnal enuresis: A UAE study. J. Psychosom. Res. 2006, 61, 317–320. [Google Scholar] [CrossRef]
- Fatouh, A.A.; Motawie, A.; Al-Aziz, A.M.A.; Hamed, H.; Awad, M.A.M.; El-Ghany, A.A.; Bassyouni, H.; Shehab, M.; Eid, M. Anti-diuretic hormone and genetic study in primary nocturnal enuresis. J. Pediatr. Urol. 2012, 9, 831–837. [Google Scholar] [CrossRef]
- Arnell, H.; Hjalmas, K.; Jagervall, M.; Lackgren, G.; Stenberg, A.; Bengtsson, B.; Wassen, C.; Emahazion, T.; Anneren, G.; Pettersson, U.; et al. The genetics of primary nocturnal enuresis: Inheritance and suggestion of a second major gene on chromosome 12q. J. Med. Genet. 1997, 34, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, C.S.; Horsdal, H.T.; Rajagopal, V.M.; Grove, J.; Als, T.D.; Kamperis, K.; Nyegaard, M.; Walters, G.B.; Eðvarðsson, V.; Stefánsson, H.; et al. Identification of genetic loci associated with nocturnal enuresis: A genome-wide association study. Lancet Child Adolesc. Health 2021, 5, 201–209. [Google Scholar] [CrossRef]
- Allen, R.P.; Picchietti, D.L.; Garcia-Borreguero, D.; Ondo, W.G.; Walters, A.S.; Winkelman, J.W.; International Restless Legs Syndrome Study Group. Restless legs syndrome/Willis–Ekbom disease diagnostic criteria: Updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria-history, rationale, description, and significance. Sleep Med. 2014, 15, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.P.; Picchietti, D.; Hening, W.; Trenkwalder, C.; Walters, A.S.; Montplaisi, J. Restless legs syndrome: Diagnostic criteria, special considerations, and epidemiology: A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003, 4, 101–119. [Google Scholar] [CrossRef]
- Montplaisir, J.; Boucher, S.; Poirier, G.; Lavigne, G.; Lapierre, O.; Lespérance, P. Clinical, polysomnographic, and genetic characteristics of restless legs syndrome: A study of 133 patients diagnosed with new standard criteria. Mov. Disord. Off. J. Mov. Disord. Soc. 1997, 12, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Picchietti, D.; Allen, R.P.; Walters, A.S.; Davidson, J.E.; Myers, A.; Strambi, L.F. Restless Legs Syndrome: Prevalence and impact in children and adolescents the Peds REST Study. Pediatrics 2007, 120, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, K.A. Asthenia Crurum Paraesthetica (irritable legs)—A new syndrome consisting of weakness, sensation of cold and nocturnal paresthesia in the legs, responding to a certain extent to treatment with Priscol and Doryl—A note on paresthesia in general. Acta Med. Scand. 1944, 118, 197–209. [Google Scholar] [CrossRef]
- Xiong, L.; Jang, K.; Montplaisir, J.; Levchenko, A.; Thibodeau, P.; Gaspar, C.; Turecki, G.; Rouleau, G.A. Canadian restless legs syndrome twin study. Neurology 2007, 68, 1631–1633. [Google Scholar] [CrossRef] [PubMed]
- Trenkwalder, C.; Seidel, V.C.; Gasser, T.; Oertel, W.H. Clinical symptoms and possible anticipation in a large kindred of familial restless legs syndrome. Mov. Disord. 1996, 11, 389–394. [Google Scholar] [CrossRef]
- Winkelmann, J.; Wetter, T.C.; Collado-Seidel, V.; Gasser, T.; Dichgans, M.; Yassouridis, A.; Trenkwalder, C. Clinical characteristics and frequency of the hereditary restless legs syndrome in a population of 300 patients. Sleep 2000, 23, 1–6. [Google Scholar] [CrossRef]
- Winkelmann, J.; Muller-Myhsok, B.; Wittchen, H.-U.; Hock, B.; Prager, M.; Pfister, H.; Strohle, A.; Eisensehr, I.; Dichgans, M.; Gasser, T.; et al. Complex segregation analysis of restless legs syndrome provides evidence for an autosomal dominant mode of inheritance in early age at onset families. Ann. Neurol. 2002, 52, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Didriksen, M.; Nawaz, M.S.; Dowsett, J.; Bell, S.; Erikstrup, C.; Pedersen, O.B.; Stefansson, K. Large genome-wide association study identifies three novel risk variants for restless legs syndrome. Commun. Biol. 2020, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Rye, D.B.; Hicks, A.; Petursson, H.; Ingason, A.; Thorgeirsson, T.; Palsson, S.; Sigmundsson, T.; Sigurdsson, A.P.; Eiriksdottir, I.; et al. A genetic risk factor for periodic limb movements in sleep. N. Engl. J. Med. 2007, 357, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, J.; Schormair, B.; Lichtner, P.; Ripke, S.; Xiong, L.; Jalilzadeh, S.; Fulda, S.; Pütz, B.; Eckstein, G.; Hauk, S.; et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat. Genet. 2007, 39, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Muhle, H.; Neumann, A.; Lohmann-Hedrich, K.; Lohnau, T.; Lu, Y.; Winkler, S.; Waltz, S.; Fischenbeck, A.; Kramer, P.L.; Klein, C.; et al. Childhood-onset restless legs syndrome: Clinical and genetic features of 22 families. Mov. Disord. 2008, 23, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Ferri, R. Two legs, one heart, one sleeping brain. Sleep Med. 2006, 7, 299–300. [Google Scholar] [CrossRef]
- Simakajornboon, N.; Thampratankul, L.; Sharon, D.; Walters, A.S. Restless legs syndrome and periodic limb movement disorder in children and adolescents. Paediatr. Respir. Rev. 2013, 612–623. [Google Scholar] [CrossRef]
- Delrosso, L.M.; Lockhart, C.; Wrede, J.; Chen, M.L.; Samson, M.; Reed, J.; Martin-Washo, S.; Arp, M.; Ferri, R. Comorbidities in children with elevated periodic limb movement index during sleep. Sleep 2019. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.; Winkelmann, J.; Lin, L.; Finn, L.; Peppard, P.; Mignot, E. Periodic leg movements during sleep are associated with poly-morphisms in BTBD9, TOX3/BC034767, MEIS1, MAP2K5/SKOR1, and PTPRD. Sleep 2014, 37, 1535–1542. [Google Scholar] [CrossRef]
- Carra, M.C.; Huynh, N.; Fleury, B.; Lavigne, G. Overview on sleep bruxism for sleep medicine clinicians. Sleep Med. Clin. 2015, 10, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Rintakoski, K.; Hublin, C.; Lobbezoo, F.; Rose, R.J.; Kaprio, J. Genetic factors account for half of the phenotypic variance in liability to sleep-related bruxism in young adults: A nationwide Finnish twin cohort study. Twin Res. Hum. Genet. 2012, 15, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoury, S.; Carra, M.C.; Huynh, N.; Montplaisir, J.; Lavigne, G.J. Sleep bruxism-tooth grinding prevalence, characteristics and familial aggregation: A large cross-sectional survey and polysomnographic validation. Sleep 2016, 39, 2049–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredini, D.; Lobbezoo, F. Role of psychosocial factors in the etiology of bruxism. J. Orofac. Pain 2009, 23. [Google Scholar]
- Oporto, G.; Lagos, J.; Bornhardt, T.; Fuentes, R.; Salazar, L.A. Are there genetic factors involved in bruxism. Int. J. Odontostomatol. 2016, 6, 249–254. [Google Scholar]
- Abe, Y.; Suganuma, T.; Ishii, M.; Yamamoto, G.; Gunji, T.; Clark, G.T.; Tachikawa, T.; Kiuchi, Y.; Igarashi, Y.; Baba, K. Association of genetic, psychological and behavioral factors with sleep bruxism in a Japanese population. J. Sleep Res. 2011, 21, 289–296. [Google Scholar] [CrossRef]
- Oporto, G.H.; Bornhardt, T.; Iturriaga, V.; Salazar, L.A. Single nucleotide polymorphisms in genes of dopaminergic pathways are associated with bruxism. Clin. Oral Investig. 2017, 22, 331–337. [Google Scholar] [CrossRef]
- Gwyther, A.R.; Walters, A.S.; Hill, C.M. Rhythmic movement disorder in childhood: An integrative review. Sleep Med. Rev. 2017, 35, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Mayer, G.; Wilde-Frenz, J.; Kurella, B. Sleep related rhythmic movement disorder revisited. J. Sleep Res. 2007, 16, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Walen, S.R. Jactatio capitis. Acta Paedopsychiatr. 1972, 39, 66–68. [Google Scholar] [PubMed]
- Hayward-Koennecke, H.K.; Werth, E.; Valko, P.O.; Baumann, C.R.; Poryazova, R. Sleep-related rhythmic movement disorder in triplets: Evidence for genetic predisposition? J. Clin. Sleep Med. 2019, 15, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Attarian, H.; Ward, N.; Schuman, C. A multigenerational family with persistent sleep related rhythmic movement disorder (RMD) and insomnia. J. Clin. Sleep Med. 2009, 5, 571–572. [Google Scholar] [CrossRef] [Green Version]
- Prihodova, I.; Skibova, J.; Nevsimalova, S. Sleep-related rhythmic movements and rhythmic movement disorder beyond early childhood. Sleep Med. 2019, 64, 112–115. [Google Scholar] [CrossRef]
- Adamczyk, M.; Ambrosius, U.; Lietzenmaier, S.; Wichniak, A.; Holsboer, F.; Friess, E. Genetics of rapid eye movement sleep in hu-mans. Transl. Psychiatry 2015, 5, e598. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Pathogenetic pathways of upper airway obstruction. Note: On a background influenced by gender, genetics and race, different pathogenetic factors may lead to upper airway obstruction.
Figure 1.
Pathogenetic pathways of upper airway obstruction. Note: On a background influenced by gender, genetics and race, different pathogenetic factors may lead to upper airway obstruction.

Figure 2.
Representative rest activity cycles recorded in a 9-year-old boy with a delayed sleep phase disorder. Note: The actigraphic recording shows a delayed sleep-wake phase disorder with sleep onset times at around 1:30–2:00 a.m. and wake times at 8:00 during the week and at noon during weekends (the black bars indicate wake activity levels recorded at the non-dominant wrist). Downward red arrows indicate the sleep onset, while upward red arrows indicate the awakening. The actigraph model used is Micro MotionloggerWatch (Ambulatory Monitoring, Inc., Ardsley, NY, USA).
Figure 2.
Representative rest activity cycles recorded in a 9-year-old boy with a delayed sleep phase disorder. Note: The actigraphic recording shows a delayed sleep-wake phase disorder with sleep onset times at around 1:30–2:00 a.m. and wake times at 8:00 during the week and at noon during weekends (the black bars indicate wake activity levels recorded at the non-dominant wrist). Downward red arrows indicate the sleep onset, while upward red arrows indicate the awakening. The actigraph model used is Micro MotionloggerWatch (Ambulatory Monitoring, Inc., Ardsley, NY, USA).

Figure 3.
Excerpt of a polysomnographic tracing showing a confusional arousal in a 7-year-old girl. Note: The polysomnographic tracing shows an episode arising from stage-3 NREM sleep during which the patient raises her head and trunk with eyes opened, looks around and manipulates the blankets. The EEG leads before the episode shows diffuse delta waves typical of deep sleep, which persist during the episode, intermingled with movement artifacts. EEG, electroencephalogram (Fp2-F4; F4-C4; C4-P4; P4-O2; Fp2-F8; F8-T4; T4-T6; Fz-Cz; Cz-Pz; Fp1-F3; F3-C3; C3-P3; P3-O1; Fp1-F7; F7-T3; T3-T5); R, right; L, left; EOG, electrooculogram; Mylo., mylohyoideus; Tib. Ant., tibialis anterior muscle; ECG, electrocardiogram; Thor-Abd. Resp., thoraco-abdominal respirogram.
Figure 3.
Excerpt of a polysomnographic tracing showing a confusional arousal in a 7-year-old girl. Note: The polysomnographic tracing shows an episode arising from stage-3 NREM sleep during which the patient raises her head and trunk with eyes opened, looks around and manipulates the blankets. The EEG leads before the episode shows diffuse delta waves typical of deep sleep, which persist during the episode, intermingled with movement artifacts. EEG, electroencephalogram (Fp2-F4; F4-C4; C4-P4; P4-O2; Fp2-F8; F8-T4; T4-T6; Fz-Cz; Cz-Pz; Fp1-F3; F3-C3; C3-P3; P3-O1; Fp1-F7; F7-T3; T3-T5); R, right; L, left; EOG, electrooculogram; Mylo., mylohyoideus; Tib. Ant., tibialis anterior muscle; ECG, electrocardiogram; Thor-Abd. Resp., thoraco-abdominal respirogram.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mainieri, G.; Montini, A.; Nicotera, A.; Di Rosa, G.; Provini, F.; Loddo, G. The Genetics of Sleep Disorders in Children: A Narrative Review. Brain Sci. 2021, 11, 1259. https://doi.org/10.3390/brainsci11101259
AMA Style
Mainieri G, Montini A, Nicotera A, Di Rosa G, Provini F, Loddo G. The Genetics of Sleep Disorders in Children: A Narrative Review. Brain Sciences. 2021; 11(10):1259. https://doi.org/10.3390/brainsci11101259
Chicago/Turabian StyleMainieri, Greta, Angelica Montini, Antonio Nicotera, Gabriella Di Rosa, Federica Provini, and Giuseppe Loddo. 2021. "The Genetics of Sleep Disorders in Children: A Narrative Review" Brain Sciences 11, no. 10: 1259. https://doi.org/10.3390/brainsci11101259
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.