
Abstract
Long-term stability is an essential requirement for perovskite solar cells to be commercially viable. Encapsulating 3D perovskites with 2D perovskite structures is an effective strategy for improving resistance to moisture. However, long-chain alkylammonium cation-based 2D perovskites have been rarely studied in solar cells. Here, we study three different alkyl chain length organic cation-based 2D perovskite coatings for 3D perovskites. The 2D perovskite incorporated solar cells show significant improvement in solar cell stability with limited compromise in solar cell efficiency, with the longest alkyl chain length sample showing only a 20% drop in power conversion efficiency after 6 months at a relative humidity of 25-80%, and could be completely immersed in water for a few minutes before degradation started. The 2D perovskite coating also mitigated non-radiative recombination in the light-absorbing 3D perovskite, leading to an enhancement in the open circuit voltage. These findings suggest that long-chain alkylammonium cation based 2D perovskites can improve the environmental stability of 3D based perovskites without significant losses to device performance.
Similar content being viewed by others


Introduction
Organic–inorganic hybrid perovskites gained worldwide attention as a light-absorbing material in solar cells due to their large light-absorption coefficient1, long carrier-diffusion length2,3, and have achieved certified power-conversion efficiencies (PCEs) above 25% for single-junction solar cell devices4. The stability of the hybrid perovskites, especially those based on the methylammonium (MA) and formamidinium (FA) cations, is a major concern these days to commercialize the hybrid perovskite solar cell technology5,6. There is significant improvement observed over the years in the operational stability of the solar cell devices. However, most of the reported high-efficiency cells still fail to fulfill the industry standards, one such problem is accelerated aging over long-term operating conditions of the solar cell5, where the perovskite structure undergoes irreversible degradation and decomposes back to the precursors due to hydrolysis under ambient conditions7,8.
The two-dimensional (2D) structured hybrid perovskites have recently got attention as alternatives with potential applications in both light-emitting diodes and photovoltaics9. Recent developments in the 2D perovskite have shown significant improvement in the stability of the hybrid perovskites10. The 2D perovskites consist of a bulky organic ligand molecule sandwiched between the inorganic layers of corner-shared PbI6 octahedra9,11. The hydrophobic nature of organic cation helps in repelling the water from entering into the structure and prevents degradation of the perovskite, leading to enhanced stability under harsh moist conditions10,12,13. However, the presence of bulky organic cations in 2D structure hampers the charge transport, and as a result of this, the solar cell devices based on 2D perovskite materials have lower efficiencies compared with the pure three-dimensional (3D) perovskite-based solar cells12,14,15. Due to these reasons, 2D perovskites are not usually used as primary absorbers in the active layers of PSCs.
Recently, a lot of efforts have been focused on embedding 2D perovskites in 3D perovskite structure to achieve improved device stability without compromising the photovoltaic efficiencies. Recent studies suggest that the presence of bulky organic cation increases the formation energy of the 2D perovskite relative to the 3D perovskite and this further improves the thermal stability of perovskite and ion migration within the material16,17. Due to these features, the 2D perovskite-based devices show improved operating lifetime under ambient conditions. Stacked 3D/2D heterostructures consisting of a thin layer of 2D perovskites atop a light-absorbing 3D perovskite have achieved PCEs more than 20% with superior ambient stability compared with 3D perovskite-based solar cells18,19,20,21. In addition to this, the 3D/2D architecture is also used in tandem solar cells to improve the crystallinity and photovoltaic properties of the wide-band-gap top cells. The addition of a small percentage of PEAI and Pb(SCN)2 in the perovskite precursor was found to enhance the crystal growth, leading to improved carrier mobility22. However, the search for a water-repellent 2D perovskite material that can extend the moisture stability without significantly lowering the solar cell efficiency is still an ongoing effort.
Different alkylammonium ligand-based cations have been explored in 2D perovskites9,10,11,23,24,25,26,27,28,29,30. Usually the short-chain-length cations are used in 2D perovskite to minimize the inter-quantum-well distance to ease the charge transfer31,32. Whereas larger organic cations increase this distance and hamper the charge transport and thus reduce the device efficiency. Compared with 3D perovskites, 2D perovskites containing long-chain organic cations possess superior environmental stability, but the long-chain cations have yet to be explored in solar cells.
While synthesis of 3D perovskite through solvent evaporation of the surface of the 3D perovskite end up with a large number of defects, due to formation of lead halide impurities and the presence of dangling bonds33,34, and passivation of these defects is extremely important to achieve high performance in PSCs34. In PSCs, 2D perovskites such as (PEA)2PbI418,35, (BA)2PbI419,20,36,37, and organic salts38,39,40,41, have been mostly employed to passivate such defects. To the best of our knowledge, the long alkylammonium cations used in this work (tetradecylamine, hexadecylamine, and octadecylamine) have not been explored in solar cells although there are some reports of using them as capping ligands in nanocrystals42,43,44. There are also some reports available where salts of alkylamine cations are mixed/doped with 3D perovskite and used as a passivating layer on 3D perovskite to improve the stability of the PSCs against moisture21,39,40,41,45,46. A recent study shows the effect of different alkyl chain lengths of long-chained alkylammonium iodide salts on the 3D/2D-structured solar cells. Both the PCE and moisture stability were found to improve after post treatment with organic salts47.
Moreover, water-stable 2D perovskites have very rarely been reported in the literature. Passarelli et al.15 have demonstrated 2D perovskite [(aromatic O-linker–NH3)2PbI4] that resists water even after a brief but complete immersion. This water tolerance is due to the ether-linked pyrene ligands that π–π stack within the structure. However, there are no reports of using this perovskite as an encapsulation layer in solar cells and the PCE values measured of pristine 2D perovskite devices are about 1.38%. There are few examples of 2D perovskites, which show prolonged stability under harsh humid conditions, are reported21,26 and further research in this direction is needed.
Inspired by the water stability of 2D perovskites from our previous work13, we were curious whether the stability of the 2D materials could be imparted to 3D materials through alloying. In this study, we aimed to use highly moisture resistive alkylammonium cation-based lead iodide perovskites (CnH2n+1NH3)2PbI4 where n = 14, 16, and 18 (from here onward denoted as C14, C16, and C18), as an encapsulating layer on the top of the light-absorbing FA/MA cation-based 3D perovskite. While using such a long-chain organic cation, it is challenging to retain the PCEs compared with 3D perovskite. In the complete solar cell, we achieved a champion efficiency of ca. 17% with an open-circuit voltage (Voc) of 1.13 V. We observe inhibition of the nonradiative recombination after encapsulation of the 3D perovskite with 2D perovskite due to the passivation of surface defects, which leads to an increase of the Voc in 3D@2D heterostructures compared with 3D only devices. Our work demonstrates that the use of a long-carbon-chain organic cation in 3D@2D heterostructures is possible without compromising the performance of the perovskite solar cell.
Results and discussion
Structural and optical characterizations of 3D@2D perovskite films
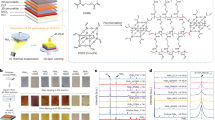
In C14–C16-based 2D perovskites, the alkylammonium ligands are interdigitated in the crystal structure (Fig. 1a) and held together by Van der Waals interactions. The presence of long alkylammonium cations increases their desorption energy compared with the smaller cations like MA25, as a result, a more stabilized structure, which is even stable for days when completely immersed in water, is achieved13. We took advantage of this water-repellent property and used these 2D perovskites in PSCs. We obtain the 3D@2D architecture by spin-coating a diluted solution of 2D perovskite in tetrahydrofuran (THF) on top of 3D perovskite film followed by annealing at 100 °C for 10–15 mins. For 2D perovskite coating, 10 mg of 2D perovskite per milliliter THF was used, unless otherwise mentioned, further details can be found in “Methods” section. X-ray diffraction (XRD) patterns of bare (FA0.85MA0.15)Pb(I0.85Br0.15)3 and 2D perovskite (C14, C16, and C18) coated (FA0.85MA0.15)Pb(I0.85Br0.15)3 are shown in Fig. 1b As observed from the XRD patterns, the coating of 2D perovskite does not seem to have any impact on the crystallinity of the light-absorbing 3D perovskite. The magnified XRD patterns show a notably suppressed impurity peak (at 2ϴ = 12.45), corresponding to the PbI2 impurity phase in 3D perovskite, after coating of the 2D perovskite layer. The PbI2 could be a site for charge recombination and thus removing it could possibly help in reducing surface-defect states present in the 3D perovskite. The XRD patterns of the 2D perovskite- (10 mg/ml) coated 3D perovskite thin films do not show any diffraction peaks corresponding to the 2D perovskite, this could be due to the formation of a very thin layer or trace amount of 2D perovskite on the top of 3D perovskite, which makes it difficult to prove the existence of 2D perovskite on the top of 3D perovskite within the detection limit of the characterization tools used here. To prove the existence of 2D perovskite on the top of 3D perovskite layer, we varied the concentration of 2D perovskite in THF (15 and 20 mg/ml) and observed clear diffraction peaks at lower 2ϴ degrees (see Supplementary Fig. 1), however, this paper does not show direct evidence for the presence of 2D layer on top of 3D perovskite for 5 and 10 mg/ml concentration of 2D perovskite. When we compared the diffraction-peak positions in 3D@C18 thin films with that of the pure 2D perovskite films (see Supplementary Fig. 1), we observed a shift (0.64 degrees) in the diffraction peaks toward lower 2ϴ angle in 3D@C18 perovskite films. This could be either due to the presence of mixed phase or the formation of an entirely new phase because of the interaction of 3D and C18 perovskite. To answer this question, we did a separate experiment where we tried to substitute FA cations into the pure C18 perovskite. After the addition of formamidinium iodide (FAI) salt into C18 + THF solution, the color of the solution changed from yellow to orange-red to brown with increasing the concentration of FAI salt (see Supplementary Fig. 2a), and brown-colored precipitate was obtained for higher FAI concentration. We then prepared thin films out of these solutions and measured XRD pattern and the absorption spectra. The diffraction peaks observed in FA substituted C18 perovskite matched exactly with the diffraction peaks observed in 3D@C18 films at lower angles (Fig. 1c and Supplementary Fig. 3). This new phase has red-shifted absorption edge compared with pure C18 perovskite (see Supplementary Fig. 4). This experiment elucidates that the FA cation in 3D films interacts with the 2D perovskite and forms an entirely new phase that has even larger cell dimensions compared with pure C18 perovskite. We further confirmed that by simply spin-coating C18-iodide salts on the top of 3D perovskite layer does not lead to the formation of this new phase.

a Schematic of the crystal structure of 2D perovskites at room temperature, b X-ray diffraction (XRD) patterns of 3D-only and 2D perovskite-coated thin films, the panel on the right shows the enlarged view of the XRD patterns at lower angles, c XRD patterns of thin films of FA-substituted C18 2D perovskites spin-coated on MeTiO2 layer along with the XRD patterns of pure 2D perovskite and 3D@C18 (20 mg/mL) perovskite thin films for comparison. d Ultraviolet–visible (UV–Vis) absorption spectra, the inset shows a small red shift in the absorption-band edge of 3D perovskite after 2D perovskite coating and e photoluminescence (PL)-emission spectra of pure 3D perovskite and 3D@2D perovskite thin films.
The UV–Vis absorption spectra (Fig. 1d) and PL spectra (Fig. 1e) show that the 2D perovskite coating does not significantly alter the absorption or PL-emission wavelengths of 3D perovskite. The absorption spectra show a marginal redshift in the band edge of 3D perovskite after 2D perovskite coating, otherwise, there is no significant change in the absorption spectra of light-absorbing perovskite layer. The Tauc plot (see Supplementary Fig. 5) calculated from the UV–Vis absorption spectra shows an optical bandgap of 1.60 eV for FA/MA films and 1.59 eV for 2D-coated perovskites, respectively, indicating no significant change in the bandgap of the 3D perovskite after 2D perovskite coating. The steady-state PL profiles were investigated on pure 3D- and 2D-coated perovskite as shown in Fig. 1e. The PL intensity was enhanced significantly after coating of 2D perovskites onto (FA0.85MA0.15)Pb(I0.85Br0.15)3 film. The enhancement of PL intensity could be attributed to the passivation of surface states by 2D perovskites35,40,48. XRD results also support this observation where the PbI2 impurities in 3D perovskite were completely suppressed after 2D perovskite coating. The improvement in the perovskite thin-film quality after introducing the 2D perovskite layer motivated us to further investigate the role of 2D perovskites in solar cell devices.
Performance of 3D@2D perovskite solar cells
We investigated the influence of 2D perovskite layer on the photovoltaic performance of 3D perovskite fabricated using the device architecture given in Fig. 2a. The current density–voltage (J–V) curves of pure 3D and 2D perovskite coated thin films were recorded under standard 1-sun air mass (AM) 1.5 G-illumination. J–V curves of the representative 3D-only and 3D@C18 devices are given in Fig. 2b. For representative solar cells, we obtained an efficiency of 16.79% for 3D@2D (C18)-based PSCs with an open-circuit voltage (Voc) of 1.13 V, a fill factor (FF) of 0.74, and a short-circuit current (Jsc) of 20.01 mA·cm−2. Under similar conditions, the pure 3D perovskites showed a PCE of 18.19% with a Voc of 1.09 V, FF of 0.76, and a Jsc of 21.98 mA·cm−2 (Fig. 2b).

a Schematic illustration of 2D-coated perovskite solar cell (PSC) with regular configuration, b current-voltage characteristic of 3D-only and C18 perovskite-coated PSCs, c comparison of the current–voltage characteristic of 3D-only and 2D perovskite-coated PSCs. Photovoltaic parameters determined from the J–V characteristic of a relevant number (50 in total) of 3D-only and C18 perovskite-coated PSCs: d PCE, open-circuit voltage (Voc), short-circuit current density (Jsc) and fill factor (FF). e The statistical distribution of PCE values of 3D-only and C14, C16, and C18 perovskite-coated PSCs. f Effect of variation in concentration of C18 perovskite per ml THF on PCE values of PSCs. g Steady-state photocurrent density and PCE of the 3D-only and C18-coated PSC monitored at a maximum powerpoint under 1-sun AM 1.5-G illumination. h Incident photon-to-electron-conversion efficiency (IPCE) spectra and the corresponding integrated Jsc of 3D-only and C18 perovskite-coated PSCs.
The J–V curves of the 3D-only and 2D perovskite-coated PSCs show similar Jsc for C14 and C16 perovskite (Fig. 2c) coating, whereas C18 perovskite shows a slightly lower Jsc, but an increase of about 30–40 mV in the Voc was observed. We observed negligible hysteresis in both 3D-only and 2D perovskite-coated PSCs (see Supplementary Fig. 6). Further, the J–V curves are not affected by the scan speed during J–V measurements of both 3D-only and 2D perovskite-coated PSCs, suggesting minimal ion migration in these devices (see Supplementary Fig. 7)49,50. In order to check the reproducibility of the results, we fabricated 50 such devices and carried out the J–V measurements on them. Figure 2d shows the statistics of PCE, Voc, Jsc, and FF indicating good reproducibility of the photovoltaic parameters within a batch of 50 cells. The average photovoltaic parameters (PCE: 17.16 ± 0.67 and 16.11 ± 0.74, Voc: 1.08 ± 0.008 and 1.12 ± 0.012, Jsc: 21.51 ± 0.42 and 19.43 ± 0.70 and FF: 0.73 ± 0.02 and 0.70 ± 0.024 for 3D only 3D@C18 devices, respectively) indicate a small deviation in the photovoltaic parameters. The enhancement in the Voc values of the 2D perovskite coated solar cell performance has been observed previously for 3D@2D devices and attributed to the reduction in the surface trap states that could lead to the charge trapping and accumulation18,21,51.
We used C14-, C16-, and C18-alloyed perovskites in the solar cells, the PCEs of the respective devices are given in Fig. 2e. The 3D@C14-based devices show PCE values close to 3D-only devices, whereas for 3D@C16 and 3D@C18 devices, slightly lower PCE values were obtained, this may be linked to the length of the alkyl chain. From our previous study13, we know that C18-based 2D perovskites are more stable against moisture compared with C14- and C16-based perovskites. Though we obtain slightly higher PCE values in case of C14- and C16-coated PSCs compared with C18-based PSCs, but due to the higher stability of C18 perovskite, we primarily studied the performance of the 3D@C18-based PSCs. We also optimized the concentration of 2D perovskite in THF to achieve a maximum power output from 3D@2D PSCs. Figure 2f illustrates for lower concentrations (5 and 10 mg/ml) of C18 perovskite in THF, the efficiency values were close to 3D-only devices, whereas for higher concentrations (15 and 20 mg/ml), the PCE values dropped significantly, which could be attributed to the formation of thicker 2D perovskite overlayer on the top of the 3D perovskite and hampering the charge transport across the perovskite- and hole-transporting material (HTM) interface.
The steady-state photocurrent-density measurements show comparable Jsc and PCE values extracted from J–V curves (Fig. 2g). Both 3D and 3D@C18 devices show stable Jsc (21.3 mA·cm−2 and 20.0 mA·cm−2 for 3D only and 3D@C18) and PCE (17.9% and 16.7% for 3D only and 3D@C18) when monitored at maximum powerpoint for about an hour. To identify the major factors responsible for the drop in the PCE values in 2D perovskite-coated cells, we compared the incident photon-to-electron-conversion efficiency (IPCE) spectra of 3D-only and 2D perovskite-coated cells. The C18-coated PSC shows slightly lower photon-conversion efficiency compared with the 3D device, particularly in the 666–800-nm wavelength range (Fig. 2h), which could be a contributing factor for the drop in the current density in 2D perovskite-coated cells. We then compared the optical absorption spectra of the films with similar configuration, but without HTM and Au coating. Surprisingly, we do not observe any significant change in the absorption spectra (Fig. 1c). To further resolve this conflict, we measured the absorption spectra in the same way (by facing FTO toward incident light) as we did the IPCE measurements. We then compared the absorption spectra recorded from the front and backside of the films (see Supplementary Fig. 8), but the absorption spectra were almost identical without any noticeable difference. The integrated Jsc values for 3D- and 2D-coated devices were calculated to be 20.04 mA·cm−2 and 19.7 mA·cm−2, respectively. The integrated Jsc values estimated from the IPCE spectra are in good agreement with the Jsc values extracted from the J–V curves. From this, we can rule out the difference in light-harvesting efficiency as the main problem for the differences in photocurrent.
Characterization of 3D@2D perovskite solar cells
We used time-resolved photoluminescence (TRPL) technique to assess the perovskite films’ quality after coating them with 2D perovskites, TRPL plots of samples with the structure glass/FTO/mp-TiO2/perovskite layer/without and with 2D perovskite layer are shown in Fig. 3a. The films coated with 2D perovskites exhibit carrier lifetimes longer than those of 3D-only films (see Supplementary Table 1) when decay curves were fitted with bi-exponential function. The relatively faster component (τ1) is assigned to the defect-related states such as charge-carrier trapping, vacancies, or interstitial defects, whereas the slower component (τ2) is attributed to the free carrier radiative recombination. After 2D perovskite coating the proportion of the faster component decreased and the slower component increased, which indicates reduced defect density and enhanced charge-carrier recombination. This gain in the lifetime values indicates that the carrier recombination is significantly suppressed upon the addition of a top 2D perovskite layer, steady-state PL measurements also complement this observation where an enhancement in the PL intensity was observed after 2D perovskite coating (Fig. 1e). The enhancement in the Voc values arises from the suppression of the nonradiative carrier recombination at the perovskite/HTM interface, which is well supported by the longer carrier lifetimes in 3D@2D thin films21.

a Time-resolved PL decay curves of the 3D-only and 2D perovskite-coated thin films monitored at 766 nm under 450-nm excitation. b Current–voltage curves of 3D-only and 2D perovskite-coated devices measured at room temperature under 1-sun illumination, the IV plots of 3D@C14 and 3D@C16 are smoothened to have a better clarity. c Nyquist plots of 3D-only and 2D perovskite-coated PSC devices measured at a bias of 0.8 V under dark conditions and the inset shows a circuit diagram of an equivalent circuit model that was used for fitting the Nyquist plots.
To know the effect of 2D perovskite coating on the charge transport properties, we carried out conductivity measurements both in the dark (see Supplementary Fig. 9) and under 1-sun illumination (Fig. 3b). The dark-conductivity values of 3D-only and 2D perovskite-coated devices are in a similar range, as tabulated in Supplementary Table 2, indicating that coating of 2D perovskite does not hamper the charge transport. Whereas under illumination, the conductivity values of 3D@2D devices are one order of magnitude lower compared with 3D-only devices.
We further carried out the electrochemical-impedance spectroscopy (EIS) to probe into the charge-transporting process in 3D-only and 2D perovskite-coated PSC devices. The Nyquist plot shown in Fig. 3c and Supplementary Fig. 10 shows two distinguishable arcs in the low- and high-frequency regions. The arc in the low-frequency region is associated with the charge-recombination resistance (Rrec), whereas the arc in the high-frequency region corresponds to the charge transport (Rct) between perovskite and charge-transporting layers52,53. The comparison of Nyquist plots of the 3D-only and 2D-coated devices reveals significant difference in both the arcs. The increased arc radius in the low-frequency region and reduced arc radius in the high frequency region corresponds to an increased recombination resistance and improved charge transfer across the interface of the perovskite/charge-selective layers. The obtained EIS curves were fitted with simple-capacitance model and constant-phase element (CPE). The EIS curves simulated using the CPE model showed best fit to the experimental EIS curves, although the parameters extracted from the two different models are nearly similar. The Rrec values of 2D-coated devices are higher than 3D-only devices (see Supplementary Table 3), which further confirms lower recombination/ defect density and thus gives rise to an increase in the Voc values54. This behavior is again consistent with the increase of the carrier lifetime observed in time-resolved PL measurements.
Morphology, hydrophobicity of perovskite films, and device architecture
The scanning electron microscopy (SEM) images of pure 3D perovskite- (Fig. 4a, b) and 2D perovskite- (Fig. 4c, d, and Supplementary Fig. 11) coated thin films show that the film morphology gets less affected by the 2D perovskite coating as the grain-size distribution is almost identical in both films. However, the surface texture of the 2D perovskite-coated thin films appears rougher compared with the thin film of pure 3D perovskite. When 2D perovskite (in THF) is coated on the top of the 3D perovskite, it does not influence the grain growth of the 3D perovskite.

Scanning electron microscopy (SEM) images of (a, b) pure 3D and (c, d) C18 perovskite-coated 3D perovskite film, respectively. Contact angle made by a water droplet on the surface of thin films of (e) pure 3D perovskites and (f) C14-, (g) C16-, (h) C18-coated 3D perovskite measured after stabilizing the water droplet for 10 s. Cross-sectional SEM images of (i) pure 3D and (j) 3D@2D PSCs.
In order to check the water-repellent behavior of the 2D perovskite-coated thin films we performed contact-angle measurements on 3D@2D perovskite thin films and made a comparison of the contact angle on pure 3D perovskite film. The 2D perovskite coating dramatically enhanced the hydrophobicity of the films. The pure 3D perovskite shows a contact angle of 52 °C (Fig. 4e), whereas the C14, C16, and C18 perovskite-coated films show 84 °C, 88 °C, and 104 °C contact angles (Fig. 4f–h), respectively. We also measured the contact angle on the thin films of pure 2D perovskites (see Supplementary Fig. 12), the measurements show contact angles of 84 °C, 104 °C, and 111 °C for C14, C16, and C18 perovskites, respectively. From Fig. 4f–h, it is quite evident that the contact angle made by the water droplet is highly dependent on the length of the alkylammonium cation. We further performed the dynamic contact-angle measurements to check the variation of the contact angle with time. Supplementary Fig. 13 illustrates a variation in contact angle within the first 10 s and after which it remains constant until the next 50 s, indicating that the hydrophobicity of the surface remains intact with time even after exposure to water.
We further looked at the cross section (Fig. 4i, j) of the pure 3D and 2D perovskite-coated solar cell devices. In 2D perovskite-coated solar cells, we were expecting an additional overlayer on the top of the 3D perovskite layer, but we could not observe any such layer (for 10 mg of C18 in 1 ml of THF) in the cross-sectional SEM, this suggests that the 2D perovskite blends quite well with the 3D perovskite and forms a homogeneous single layer. From cross-sectional SEM, it is difficult to estimate the increase in the thickness of the 3D perovskite layer after 2D perovskite coating. Overall, the cross-sectional SEM images of pure 3D- and 2D-coated perovskite look identical. The estimated thickness of the perovskite layer is in the 300–400-nm range, which is the average grain size of the perovskite.
To demonstrate the potential of these 2D perovskites as the encapsulating layer in solar cells, we monitored changes in the XRD patterns of 3D-only and 3D@2D perovskite thin films with time by exposing thin films to ambient conditions with relative humidity (RH) 25–80%. There is a significant increase in the diffraction intensity of the peaks corresponding to the PbI2 phase observed in 3D-only thin films, as shown in Fig. 5a. Whereas the films encapsulated with 2D perovskites do not show the emergence of any impurity peak at all within this period (Fig. 5b and Supplementary Fig. 14). All three 2D perovskites act as a good encapsulating layer and prevent 3D perovskite from moisture. To further examine the stability of the devices coated with 2D perovskite and stored under ambient conditions, we monitored the PCE values of 3D-only and 2D-coated 3D perovskites under the identical conditions of RH 25–80%. As shown in Fig. 5c, the 3D-only devices show more than 60% drop in the PEC values after six months of storage under ambient condition, whereas C16- and C18-coated devices show less than 20% drop in the efficiency within this period, indicating an efficient encapsulation offered by the 2D perovskite layer. The variation in the PCE values during the first few days after fabrication is usually observed and ascribed to the stabilization of the device interface with time and occurs due to the interdiffusion of HTM and Au electrode inside the perovskite layer55,56,57.

XRD patterns of (a) 3D-only and (b) 3D@C18 thin films illustrating the evolution of degradation in 3D-only perovskite thin films and 2D perovskite prevents this degradation when monitored over a period of two months and stored under an ambient atmosphere with a relative humidity of 25-80%. c Variation in PCE of nonencapsulated 3D-only PSCs and C14, C16, and C18 perovskite-coated PSCs kept under ambient atmosphere (RH 25–80%, 10–25 °C, in the dark). d Photograph of 3D-only (on the left) and C18 perovskite-coated 3D perovskite (on the right) thin films after immersion in water for 3 min.
Stability of the perovskite films and solar cells
The XRD measurements on 3D-only and 2D-coated thin films further support the stability trend observed in 3D@2D PSCs in solar cell stability measurements. However, the observed drop in the PCE values is not only due to the degradation of the light-absorbing 3D perovskite layer alone but also contributed by the moisture/O2, photoinduced degradation of the HTM (Spiro-OMeTAD)58,59. We believe that the stability of the solar cell devices can be further improved by replacing moisture-resistant HTMs. The PCEs of the devices coated with 2D perovskite are close to that of the 3D-only devices, with much improved stability, confirming the advantage of using long alkylammonium cation-based 2D perovskite as an encapsulating layer. This is also in accordance with the trend of improved film quality as deduced from the XRD and PL measurements.
To further investigate the stability, we immersed 3D-only and 2D perovskite-coated thin films in water and monitored how quickly degradation takes place in these thin films. To make it clearer, we have recorded a video (see Supplementary Movie 1) while performing this test. As observed from the video, the pure 3D perovskite film degrades immediately after dropping water onto the film, whereas in the case of 2D perovskite-coated film the degradation initiated after few minutes in water (see Supplementary Fig. 15). This video illustrates the capability of 2D perovskite as an encapsulating layer. Figure 5d, shows complete degradation in 3D perovskite films and stable 2D perovskite-coated thin films, a photo was captured after leaving the films completely immersed in water for about 3 min. This suggests that the new phase, which is produced after the interaction, and 3D and 2D perovskites, is also highly water-repellent in nature.
It is known in the perovskite community that the 2D halide perovskites are versatile in composition tuning11,27, and the crystal structure of these systems is quite open for metal ion doping either on A-cation or Pb sites60. The new crystallographic phase observed in this study is as a result of interaction of FA cations from 3D perovskite layer with that of the 2D perovskite layer. From a future research-direction perspective, it will be exciting to explore the interaction of FA/MA cations with 2D perovskite and to study other possible metal ion doping in 2D perovskite to tune the optical properties and achieve enhanced optoelectronic performance out of the long alkylammonium cation-based 2D perovskite.
Conclusion
In summary, we demonstrated excellent photovoltaic performance with a PCE ca. 17% and superior stability under the ambient conditions for a prolonged time in 2D perovskite-coated PSCs. The 2D perovskite coating also suppresses PbI2 impurity phase and aids improvement in the Voc by 30 mV, owing to the surface-trap-state passivation, which is quite well supported by steady-state PL and EIS measurements. Cross-sectional SEM measurements show that 2D perovskite does not form an overlayer on the top of 3D perovskite, but blends very well with the 3D perovskite. 2D perovskite coating improves the hydrophobicity of the thin films, which helps in fabricating moisture-tolerant PSCs. The 2D perovskite materials presented here not only encapsulate the 3D perovskite but also act as a passivation layer and significantly reduce the recombination losses. Furthermore, 2D perovskite coating does not seem to affect the film quality and charge transport of the light-absorbing 3D perovskite layer. We anticipate that these results will help in further exploring other long alkyl chain cation-based 2D perovskites to develop stable solar cell devices in the near future, the current study also paves a new avenue to use long organic cation-based 2D perovskites for high-performance optoelectronic devices.
Methods
Materials
Lead iodide (PbI2, 99.99%) and lead bromide (PbBr2, 99.999%) were purchased from TCI chemicals. Formamidinium iodide (FAI, 99.999%) and FK 209 (>95%) Co(III) were purchased from Dyenamo. TiO2 paste (DSL 30NR-D) was purchased from Dyesol. Lithium bis-(trifluoromethylsulfonyl)imide (LiTFSI, 99%) salt was purchased from Ionic Liquids Technologies. Methylammonium bromide (MABr, anhydrous, >99%), N, N-Dimethylformamide (DMF, anhydrous, 99.8%) and Dimethyl sulfoxide (DMSO, anhydrous, ≥99.9%), Tetrahydrofuran (THF, anhydrous, ≥99.9%), Acetonitrile (anhydrous, 99.8%), chlorobenzene (anhydrous, 99.8%), isopropanol (anhydrous, 99.5%), Titanium(IV) isopropoxide (97%), Acetylacetone (>99%), and 4-tert-Butylpyridine (TBP, 96%) were purchased from Sigma Aldrich. Ethanol (98%) was purchased from Fisher Scientific. All the chemicals were used as received without any further purification.
Synthesis of 2D perovskites
The synthesis protocol given in ref. 13 is followed for preparation of (C14H29NH3)2PbI4, (C16H33NH3)2PbI4, and (C18H37NH3)2PbI4 2D perovskites. The brief description of 2D perovskite crystal synthesis is given below.
For synthesis of 2D perovskite crystals, 2 mmol of PbI2 was dissolved in 2 ml of hydroiodic (HI) acid. In the second step, 4 mmol of C14H29NH2/C16H33NH2/C18H37NH2 was dissolved in the mixture of 5 ml of HI and 50–75 ml of ethanol, with continuous stirring for 30 mins. This mixture was then heated to 60 °C and PbI2 in HI was then dropwise added under continuous stirring. The reaction mixture was refluxed for nearly 45 mins and slowly cooled to room temperature at 5 °C/hour cooling rate. Needle-shape single crystals were obtained by washing the obtained crystals with diethyl ether and finally drying under vacuum.
3D perovskite deposition
FAI (375.86 mg), PbI2 (1058.02 mg), MABr (45.36 mg), and PbBr2 (148.64 mg) were dissolved in 2 ml of solvent (DMF:DMSO, 4:1) to form a perovskite precursor solution. The perovskite precursor solution was left for continuous stirring overnight inside the glovebox. For perovskite-film deposition, a 70-μl perovskite-precursor solution was spin-coated at 4500 rpm for 30 sec, after 20 sec 100 μl of chlorobenzene was dropped onto the film to achieve a rapid crystallization of perovskite. The film was then annealed on a hot plate at 100 °C for 30–40 min.
2D perovskite deposition
For deposition of 2D perovskite on the top of 3D perovskite film, 10 mg of 2D perovskite crystals were dissolved in 1 ml of THF under continuous stirring at room temperature. About 120 μl of 2D perovskite solution was dropped onto the 3D perovskite film while spinning at 4000 rpm for 30 sec, and finally the film was annealed at 100 °C for 10–15 min.
Solar cell fabrication
Fluorine-doped tin oxide (FTO) substrates with a size 1.5 × 2.5 cm were used for solar cell fabrication. In the first step, the FTO substrates were patterned using etching method (zinc powder and 2 M HCl solution). Patterned FTO substrates were then cleaned with a diluted RBS solution in deionized water, acetone, and ethanol under continuous sonication for about 30 min in each step. Afterward, the dried substrates were treated under a UV-ozone lamp for 15–20 min, to oxidize and remove organic residuals present on the surface of the FTO. The compact TiO2 layer was deposited by spray pyrolysis using 0.2 M titanium (IV) isoporoxide and 2 M actyl acetone dissolved in isopropanol. Several cycles of the titanium (IV) isoporoxide/actyl acetone solution were sprayed onto the preheated (450 °C) FTO substrates and annealed for 15–20 min.
For the mesoporous TiO2 (MeTiO2)layer, TiO2 paste (Dyesol 30NR-D) was suspended in ethanol (in w/w = 1/5.5 ratio) by sonicating the mixture for 45 mins. The solution was then spin-coated onto the compact TiO2 layer at 4500 rpm for 30 s followed by heating at 80 °C for 20 min. The substrates were then annealed at 450 °C for 1 h in the furnace. The as-prepared substrates were further cleaned by UV-Ozone cleaner for 20 min. The perovskite layer was deposited inside the glovebox using the aforementioned spin-coating methods.
For the hole-transporting layer (HTL), a conventionally doped spiro-OMeTAD was used. A hole-transport layer (HTL) was spin-coated onto the perovskite layer at 4000 rpm for 30 sec. The 85.8-mg spiro-OMeTAD, 35 μL of Li-TFSI stock solution (0.6 M in acetonitrile), 33.8 μL of TBP, and 10 μL of FK209 stock solution (0.2 M in acetonitrile) were dissolved in 921.2 μL of chlorobenzene solvent. At last, an approx. 80-nm Au layer was deposited on the HTL by thermally evaporating gold at 2 × 10−5 bar pressure.
Characterization
X-ray diffraction (XRD) patterns were measured on X’Pert PRO, PANalytical using CuKα radiation (λ = 1.5406 Å) to analyze the purity of the prepared samples. The ultraviolet–visible (UV–Vis) absorption spectra were recorded on a Varian Cary 300 Bio UV–Vis spectrophotometer. The steady-state photoluminescence (PL) spectra were recorded on a Varian Cary Eclipse fluorescence spectrophotometer. The time-resolved photoluminescence (TRPL) measurements were carried out on a home-built laser setup as described in ref. 61. A Thorlabs NPL45C laser (450 nm) operating at 5 MHz was used as an excitation source coupled with a Siglent SDG 1032X waveform generator. A Thorlabs photomultiplier tube (PMTSS2) was used for detecting the photon count from the sample. The surface and cross-sectional SEM images were recorded using the Ultra high-resolution FE-SEM (Hitachi S-4800) instrument. Contact angles were determined with a PGX + Pocket Goniometer. For contact-angle measurements, approx. 3 µL of deionized water was dropped on the surface of perovskite thin film, and images were immediately collected with the help of a camera coupled to Goniometer. The electrochemical-impedance spectroscopy (EIS) measurements were carried out on Autolab PGstat12 potentiostat in 100-mHz to 1-MHz frequency range under applied-bias potential.
J–V measurements were carried out using a standard AM 1.5 G solar simulator (Newport-91160) coupled with a sourcemeter. The incident light (1000 W·m−2) from the solar simulator was calibrated using a reference Si solar cell. A Keithley 2400 sourcemeter was used for the J–V measurements of solar cells using a black mask with an area of 0.126 cm2 and all the scans were recorded with a scan speed of 22.7 mV·s−1 at room temperature and under ambient conditions, unless otherwise mentioned.
IPCE measurements were performed on a program-controlled Keithley multimeter (model 2700) coupled with a light source ASB-XE-175 and monochromator (Spectral Products CM110). The IPCE setup was calibrated with a certified silicon solar cell (Fraunhofer ISE) before the actual measurements.
Conductivity measurements
For conductivity measurements, devices with glass/MeTiO2/perovskite/Au structure were made. A thicker layer of MeTiO2 (DSL 30 NR-D to absolute ethanol, w/w = 1:3) was used for spin-coating the perovskite layer.
The electrical conductivities of different materials were determined using two-probe method and the conductivity values were calculated using the following equation:
where σ is conductivity of the material, L is channel length (2 mm), μ is channel width (10 mm), and d is film thickness. R is film resistance estimated by reciprocal of the slope, and the slope was obtained by fitting a least-squares fit of the current–voltage data in a linear region.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Data that support the findings within this work are available from the Swedish National Data Service at the following https://doi.org/10.5878/wa8r-cq65 or from the corresponding author upon reasonable request.
References
Hoke, E. T. et al. Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chemical Science 6, 613–617 (2015).
Johnston, M. B. & Herz, L. M. Hybrid perovskites for photovoltaics: charge-carrier recombination, diffusion, and radiative efficiencies. Acc. Chem. Res. 49, 146–154 (2016).
Shi, D. et al. Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science 347, 519 (2015).
NREL photovoltaic efficiency chart. https://www.nrel.gov/pv/assets/pdfs/cell-pv-eff-emergingpv.20200922.pdf (2020).
Correa-Baena, J.-P. et al. Promises and challenges of perovskite solar cells. Science 358, 739 (2017).
Li, N., Niu, X., Chen, Q. & Zhou, H. Towards commercialization: the operational stability of perovskite solar cells. Chem. Soc. Rev. 49, 8235–8286 (2020).
Leguy, A. M. A. et al. Reversible hydration of CH3NH3PbI3 in films, single crystals, and solar cells. Chem. Mater. 27, 3397–3407 (2015).
Kim, M. et al. Moisture resistance in perovskite solar cells attributed to a water-splitting layer. Commun. Mater. 2, 6 (2021).
Mao, L., Stoumpos, C. C. & Kanatzidis, M. G. Two-dimensional hybrid halide perovskites: principles and promises. J. Am. Chem. Soc. 141, 1171–1190 (2019).
Zhang, F. et al. Advances in two-dimensional organic–inorganic hybrid perovskites. Energy Environ. Sci. 13, 1154–1186 (2020).
Grancini, G. & Nazeeruddin, M. K. Dimensional tailoring of hybrid perovskites for photovoltaics. Nat. Rev. Mater. 4, 4–22 (2019).
Kim, B. & Seok, S. I. Molecular aspects of organic cations affecting the humidity stability of perovskites. Energy Environ. Sci. 13, 805–820 (2020).
Kore, B. P. & Gardner, J. M. Water-resistant 2D lead(ii) iodide perovskites: correlation between optical properties and phase transitions. Mater. Adv. 1, 2395–2400 (2020).
Safdari, M. et al. Layered 2D alkyldiammonium lead iodide perovskites: synthesis, characterization, and use in solar cells. J. Mater. Chem. A 4, 15638–15646 (2016).
Passarelli, J. V. et al. Enhanced out-of-plane conductivity and photovoltaic performance in n = 1 layered perovskites through organic cation design. J. Am. Chem. Soc. 140, 7313–7323 (2018).
Zhang, F. et al. Colloidal synthesis of air-stable CH3NH3PbI3 quantum dots by gaining chemical insight into the solvent effects. Chem. Mater. 29, 3793–3799 (2017).
Quan, L. N. et al. Ligand-stabilized reduced-dimensionality perovskites. J. Am. Chem. Soc. 138, 2649–2655 (2016).
Lee, J.-W. et al. 2D perovskite stabilized phase-pure formamidinium perovskite solar cells. Nat. Commun. 9, 3021 (2018).
Cho, K. T. et al. Water-repellent low-dimensional fluorous perovskite as interfacial coating for 20% efficient solar cells. Nano Lett. 18, 5467–5474 (2018).
Zhou, T. et al. Highly efficient and stable solar cells based on crystalline oriented 2D/3D hybrid perovskite. Adv. Mater. 31, 1901242 (2019).
Liu, Y. et al. Ultrahydrophobic 3D/2D fluoroarene bilayer-based water-resistant perovskite solar cells with efficiencies exceeding 22%. Sci. Adv. 5, eaaw2543 (2019).
Kim, D. H. et al. Bimolecular additives improve wide-band-gap perovskites for efficient tandem solar cells with CIGS. Joule 3, 1734–1745 (2019).
Adnan, M., Dehury, K. M., Kanaujia, P. K. & Vijaya Prakash, G. Real-time dynamic evolution monitoring of laser-induced exciton phase flips in 2D hybrid semiconductor (C12H25NH3)2PbI4. J. Appl. Phys. 128, 023104 (2020).
Ahmad, S. et al. In situ intercalation dynamics in inorganic–organic layered perovskite thin films. ACS Appl. Mater. Interfaces 6, 10238–10247 (2014).
Proppe, A. H. et al. Photochemically cross-linked quantum well ligands for 2D/3D perovskite photovoltaics with improved photovoltage and stability. J. Am. Chem. Soc. 141, 14180–14189 (2019).
Jana, A., Ba, Q. & Kim, K. S. Compositional and dimensional control of 2D and quasi-2D lead halide perovskites in water. Adv. Functi. Mater. 29, 1900966 (2019).
Smith, M. D., Connor, B. A. & Karunadasa, H. I. Tuning the luminescence of layered halide Perovskites. Chem. Rev. 119, 3104–3139 (2019).
Billing, D. G. & Lemmerer, A. Synthesis, characterization and phase transitions of the inorganic–organic layered perovskite-type hybrids [(CnH2n+1NH3)2PbI4] (n = 12, 14, 16 and 18). New J. Chem. 32, 1736–1746 (2008).
Mitzi, D. B. Synthesis, crystal structure, and optical and thermal properties of (C4H9NH3)2MI4 (M = Ge, Sn, Pb). Chem. Mater. 8, 791–800 (1996).
Saparov, B. & Mitzi, D. B. Organic–inorganic perovskites: structural versatility for functional materials design. Chem. Rev. 116, 4558–4596 (2016).
Zheng, K. et al. Inter-phase charge and energy transfer in Ruddlesden–Popper 2D perovskites: critical role of the spacing cations. J. Mater. Chem. A 6, 6244–6250 (2018).
Proppe, A. H. et al. Spectrally resolved ultrafast exciton transfer in mixed perovskite quantum wells. J. Phys. Chem. Lett. 10, 419–426 (2019).
Meggiolaro, D., Mosconi, E. & De Angelis, F. Formation of surface defects dominates ion migration in lead-halide perovskites. ACS Energy Lett. 4, 779–785 (2019).
Wang, F., Bai, S., Tress, W., Hagfeldt, A. & Gao, F. Defects engineering for high-performance perovskite solar cells. npj Flex. Electron. 2, 22 (2018).
Chen, P. et al. In situ growth of 2D perovskite capping layer for stable and efficient perovskite solar cells. Adv. Funct. Mater. 28, 1706923 (2018).
Grancini, G. et al. One-Year stable perovskite solar cells by 2D/3D interface engineering. Nat. Commun. 8, 15684 (2017).
Zhang, T. et al. Bication lead iodide 2D perovskite component to stabilize inorganic α-CsPbI3 perovskite phase for high-efficiency solar cells. Sci. Adv. 3, e1700841 (2017).
Zheng, X. et al. Defect passivation in hybrid perovskite solar cells using quaternary ammonium halide anions and cations. Nat. Energy 2, 17102 (2017).
Cho, Y. et al. Mixed 3D–2D passivation treatment for mixed-cation lead mixed-halide perovskite solar cells for higher efficiency and better stability. Adv. Energy Mater. 8, 1703392 (2018).
Jiang, Q. et al. Surface passivation of perovskite film for efficient solar cells. Nat. Photonics 13, 460–466 (2019).
Liu, G. et al. Introduction of hydrophobic ammonium salts with halogen functional groups for high-efficiency and stable 2D/3D perovskite solar cells. Adv. Funct. Mater. 29, 1807565 (2019).
Teunis, M. B., Johnson, M. A., Muhoberac, B. B., Seifert, S. & Sardar, R. Programmable colloidal approach to hierarchical structures of methylammonium lead bromide perovskite nanocrystals with bright photoluminescent properties. Chem. Mater. 29, 3526–3537 (2017).
Zhang, F. et al. Brightly luminescent and color-tunable colloidal CH3NH3PbX3 (X = Br, I, Cl) quantum dots: potential alternatives for display technology. ACS Nano 9, 4533–4542 (2015).
Liu, X.-D. et al. Solution-phase growth of organolead halide perovskite nanowires and nanoplates assisted by long-chain alkylammonium and solvent polarity. Mater. Lett. 206, 75–79 (2017).
Alharbi, E. A. et al. Atomic-level passivation mechanism of ammonium salts enabling highly efficient perovskite solar cells. Nat. Commun. 10, 3008 (2019).
Zhu, H. et al. Tailored amphiphilic molecular mitigators for stable perovskite solar cells with 23.5% efficiency. Adv. Mater. 32, 1907757 (2020).
Kim, H. et al. Optimal interfacial engineering with different length of alkylammonium halide for efficient and stable perovskite solar cells. Adv. Energy Mater. 9, 1902740 (2019).
Hangoma, P. M. et al. 2D perovskite seeding layer for efficient air-processable and stable planar perovskite solar cells. Adv. Funct. Mater. 30, 2003081 (2020).
van Reenen, S., Kemerink, M. & Snaith, H. J. Modeling anomalous hysteresis in perovskite solar cells. J. Phys. Chem. Lett. 6, 3808–3814 (2015).
Belisle, R. A. et al. Interpretation of inverted photocurrent transients in organic lead halide perovskite solar cells: proof of the field screening by mobile ions and determination of the space charge layer widths. Energy Environ. Sci. 10, 192–204 (2017).
Gharibzadeh, S. et al. Record open-circuit voltage wide-bandgap perovskite solar cells utilizing 2D/3D perovskite heterostructure. Adv. Energy Mater. 9, 1803699 (2019).
Liu, Z. et al. In situ observation of vapor-assisted 2D–3D heterostructure formation for stable and efficient perovskite solar cells. Nano Lett. 20, 1296–1304 (2020).
Liu, D., Yang, J. & Kelly, T. L. Compact layer free perovskite solar cells with 13.5% efficiency. J. Am. Chem. Soc. 136, 17116–17122 (2014).
Yangi, L. et al. A novel 2D perovskite as surface “patches” for efficient flexible perovskite solar cells. J. Mater. Chem. A 8, 7808–7818 (2020).
Sanehira, E. M. et al. Influence of electrode interfaces on the stability of perovskite solar cells: reduced degradation using MoOx/Al for hole collection. ACS Energy Lett. 1, 38–45 (2016).
Bi, E. et al. Diffusion engineering of ions and charge carriers for stable efficient perovskite solar cells. Nat. Commun. 8, 15330 (2017).
Cacovich, S. et al. Gold and iodine diffusion in large area perovskite solar cells under illumination. Nanoscale 9, 4700–4706 (2017).
Sanchez, R. S. & Mas-Marza, E. Light-induced effects on Spiro-OMeTAD films and hybrid lead halide perovskite solar cells. Sol. Energy Mater. Sol. Cells 158, 189–194 (2016).
Hawash, Z., Ono, L. K. & Qi, Y. Recent advances in spiro-MeOTAD hole transport material and its applications in organic–inorganic halide perovskite solar cells. Adv. Mater. Interfaces 5, 1700623 (2018).
Zhou, Y., Chen, J., Bakr, O. M. & Sun, H.-T. Metal-doped lead halide perovskites: synthesis, properties, and optoelectronic applications. Chem. Mater. 30, 6589–6613 (2018).
Leandri, V. et al. Excited-state dynamics of [Ru(bpy)3]2+ thin films on sensitized TiO2 and ZrO2. ChemPhysChem 20, 618–626 (2019).
Acknowledgements
We would like to thank Prof. Eva Blomberg, for helping us with the contact-angle measurements. Mr. Allan Starkholm is kindly acknowledged for assisting us with EIS measurements. B. K. and J. G. gratefully acknowledge the support from the Swedish government through the research initiative “STandUP for ENERGY”, the Swedish Foundation for Strategic Research (SSF: RMA15-0130), and the Olle Engqvist Foundation (2017/192). M. S. acknowledges financial support from the Swedish research council for sustainable development-Formas (2017-01134).
Funding
Open access funding provided by Royal Institute of Technology.
Author information
Authors and Affiliations
Contributions
B.K. and J.G. conceived and supervised the project. W.Z. helped in the solar cell device fabrication part. B.K. did most of the experimental work. B.H. performed the SEM characterization and M.S. helped in the IPCE measurements. The paper was drafted and revised by B.K. and J.G. with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: John Plummer. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kore, B.P., Zhang, W., Hoogendoorn, B.W. et al. Moisture tolerant solar cells by encapsulating 3D perovskite with long-chain alkylammonium cation-based 2D perovskite. Commun Mater 2, 100 (2021). https://doi.org/10.1038/s43246-021-00200-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-021-00200-8
This article is cited by
-
Mixed perovskites (2D/3D)-based solar cells: a review on crystallization and surface modification for enhanced efficiency and stability
Advanced Composites and Hybrid Materials (2023)
