
Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure


Faculty of Pharmacy, Wrocław Medical University, Borowska 211a, 50-556 Wrocław, Poland
*
Author to whom correspondence should be addressed.
Symmetry 2021, 13(10), 1761; https://doi.org/10.3390/sym13101761
Submission received: 30 August 2021
/
Revised: 15 September 2021
/
Accepted: 17 September 2021
/
Published: 22 September 2021
(This article belongs to the Special Issue Exploring Inter- and Intramolecular Interactions as Building Blocks of Larger Systems)
Abstract
:As a result of the synthesis of mefenamic acid with potassium hydroxide, a salt with a polymeric structure is formed. The one-dimensional polymeric structure was studied by single crystal X-ray diffraction. The potassium cation is coordinated to one oxygen atom of the carboxylate group and six water oxygen atoms. Potassium ions are bridged by oxygen atoms of water molecules. The crystal structure was used as an input to QTAIM and NCI approaches to investigate the K-O interactions linking the cation with the water oxygen and carboxylate groups. The weak K-O interactions of the potassium cation and water oxygen atoms were strong enough to form a polymeric structure. The flexibility of the weak interactions is responsible for a novel coordination mode in the potassium mefenamate trihydrate.
1. Introduction
Mefenamic acid is a popular drug from the group of NSAIDs widely used in medicine. Due to the nature of fenamic acids, these drugs exist in a protonated (lipophilic) form (Scheme 1). In plasma, NSAIDs are highly ionized. Mefenamic acid is characterized by a strong analgesic effect and weaker anti-inflammatory, antiviral effect [1,2]. Mefenamic acid can be helpful in managing chronic pain of various etiologies, including neuropathic pain and cancer pain [3]. It may be equivalent or even more effective than other NSAIDs in chronic osteoarthritis, including in elderly patients [3]. However, it should be noted that when used for longer time, it causes serious side effects: skin allergic reactions, gastrointestinal disorders, and nephro-, hepato- and neurotoxicity.
The main problem with the practical use of mefenamic acid as a drug is its low water solubility and low bioaccesibilty, which is connected to the necessary dosage. It can be expected that the salts of fenamic acids with sodium or potassium could improve their solubility in water, which can result in higher bioaccesibility and the reduction of undesirable side effects. For the reasons mentioned above, the salts of mefenamic acid with the bio cations have long been studied [4,5,6]. Also, the crystal structures of mefenamic acid salts with sodium, potassium and calcium have been investigated [7], revealing the formation of polymeric structures. The crystals of the mefenamic acid salts are stable and soluble in water. The coordination numbers of Na+, K+ and Ca2+ are equal to 5, 6 and 7, respectively. Oxygen atoms of the carboxylic group of mefenamic acid are engaged in the interaction with the cation and also in the O–H∙∙∙O, N–H∙∙∙O and C–H∙∙∙O hydrogen bonds.
Since knowledge of the structure is essential to investigate the physicochemical and pharmaceutical properties of the compound, we decided to complete the data on the structure of potassium mefenamate trihydrate obtained by a different method than has been previously used [7]. Another goal of our work is to continue the description of the bonds connecting the potassium cation with mefenamate ion and other surrounding solvent molecules. A similar analysis was previously carried out for sodium fenamate (CSD code-ZOSWIQ) [8] and showed that the Na–O bonds are closed-shell, very weak interactions, and that the interaction of the sodium cation with the oxygen atom of the carboxylate group is comparable to that of the oxygen atoms of the surrounding water molecules. In the case of the potassium salt of mefenamic acid, it is of particular interest to confirm that the coordination of cations can be completed by electrons of the aromatic ring, as previously described (CSD code-NUSTAX) [7].
2. Experimental and Calculation Methods
2.1. Crystal Preparation
Potassium mefenamate trihydrate was obtained according to the analogous procedure as in the case of sodium fenamate [8]. 0.398 g (1.65 mmol) of mefenamic acid (purchased from Sigma-Aldrich) was mixed with 0.100 g (1.79 mmol) of KOH (purchased from P. P. H. ‘STANLAB’ sp. J.). The reaction was carried out at a temperature close to the melting point of mefenamic acid (230 °C). Crystals were obtained through slow evaporation from an aqueous solution.
2.2. X-ray Diffraction
Crystal data of potassium mefenamate trihydrate were collected on Xcalibur Ruby four-circle diffractometer equipped with CCD area detector with Mo Kα (λ = 0.71073 Å) radiation. Structure was solved by direct methods and refined by the full-matrix least-squares method using SHELXL2014 software [9,10]. In supplementary, Table S1 presents the crystal data. Molecular graphics were prepared using DIAMOND program [11].
In the structure of potassium mefenamate trihydrate all H atoms were found in the difference Fourier map. The H atoms of aromatic rings and methyl groups were introduced in positions calculated from geometry, with C–H = 0.95 and Uiso(H) = 1.2Ueq(C) for aromatic rings and C–H = 0.98 Å and Uiso(H) = 1.5Ueq(C) for methyl groups. The positions of the H atoms from the NH groups (N1A–H1NA, N1B–H1NB) were refined freely with Uiso(H) = 1.2Ueq(N). The H atoms of water molecules O1W, O2W, O3W, O4W, O5W and O6W were located in a difference Fourier map, introduced to the solution and refined freely with Uiso(H) = 1.5Ueq(O).
2.3. Computational Details
The molecular structure obtained from the X-ray diffraction data as well as the structure obtained previously [7] were used to generate the wfn file with the Gaussian16 package [12] at the B3LYP/6-311++G** level [13] without further optimization. The most important point related to the interaction analysis was the selection of a crystal fragment containing all interactions with the surrounding atoms of the potassium cation. For such selected crystal fragments, the wave functions were generated. The wfn files were used as an input to generate the electron-density parameters for both potassium mefenamate polymeric structure with the AIMALL program [14]. Similarly, the wfn files were used to produce the NCI plots obtained with the NCI program [15]. Despite the fact that a large fragment of the crystal was analyzed, we preferred to use the wfn instead of xyz files to obtain pictures with more precise localization of the interactions. Cube files delivered by NCI program were an input to VMD program to visualize the interactions.
3. Results and Discussion
3.1. Structure of Potassium Mefenamate—Water (1/3)
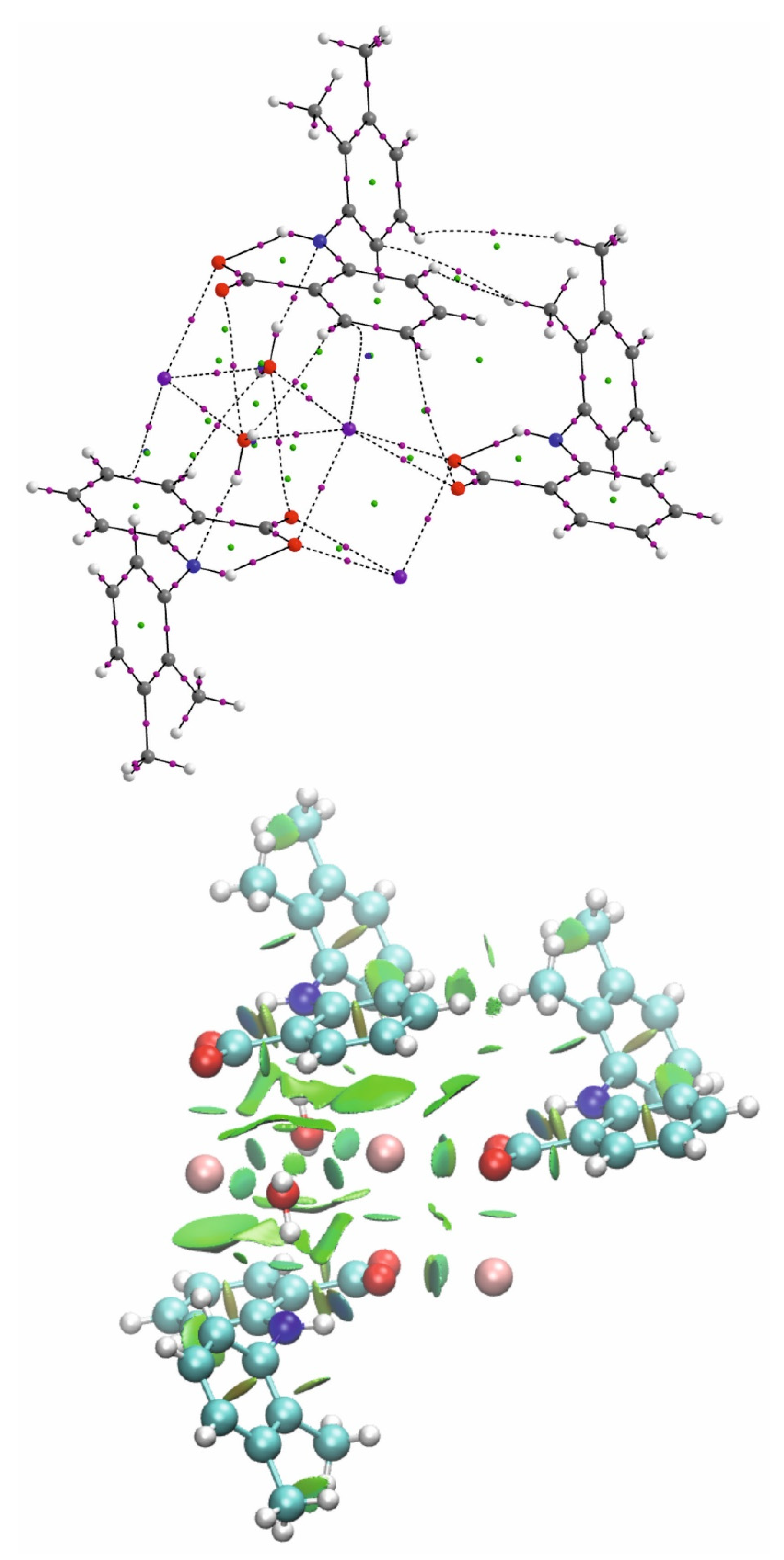
As a result of the synthesis of mefenamic acid with potassium hydroxide, a salt with a polymeric structure of the formula {K(C15H14NO2)(H2O)3} was obtained. Within the coordination polymer, each potassium ion is surrounded by six water molecules, which are potential ligands. The seventh coordination site is occupied by a mefenamate ion bound to the cation through the oxygen atom of the carboxylate group as a monodentate ligand. Considering the possibility of cation binding, the closely located aromatic rings derived from the mefenamate ion should also be taken into account. Therefore, the formation of the K…π interactions is possible, which can complete the eight coordination sphere of potassium ions. In the studied crystal, polymeric chains are present in the structure, in which two adjacent potassium ions are bridged by three oxygen atoms of water molecules (Figure 1). In the asymmetric part of the unit cell, two kinds of potassium ions (K1 and K2) are distinguished, which is related to the disorder of one of the oxygen atoms of the carboxylate group coordinated to K1 ion. The disorder was modeled over two positions (O2A and O21A) with occupancy factors 0.919(10) and 0.081(10), respectively. Both polymeric chains, involving K1 and K2, extend along the [001] direction. The bonds between potassium cations and the oxygen atoms of the carboxylate groups, water molecules and aromatic rings differ slightly for two consecutive cations, vis. K1, K2. The bond lengths and selected angles are presented in Tables S2 and S3 and in repeating themselves form a polymer chain. Except for the interaction between the potassium cations and oxygen atoms, the crystal is stabilized by a network of intermolecular hydrogen bonds O–H⋯N, O–H⋯O. The intramolecular hydrogen bond N–H⋯O is also observed (Table S4). Moreover, the polymeric chains created by K1 and K2 ions can contact each other via the K⋯π interactions mentioned above (Figure 2a,b). Namely, each potassium ion interacts with a mefenamate ion coordinated to the K ion from the adjacent chain. Also, hydrogen bonds such as O2W–H2W1⋯O2Bi, O2W–H2W2⋯O1Bii, O4W–H4W1⋯O1Aiv, O4W–H4W2⋯O3Wv (i = x, y, z − 1; ii = x, −y + 3/2, z − 1/2; iv = x + 1, −y + 3/2, z + 1/2; v = x + 1, y, z) (Table S4) link neighboring chains (Figure 3).
It is worth noting that the structure of the coordination polymer presented in this work differs from the structure of the polymer obtained previously (CSD code-NUSTAX) [7] in terms of composition, the number of coordinated water molecules and the coordination mode of the mefenamate ion. Fundamentally different polymeric structures were obtained by other preparation methods. In the investigated polymer, as outlined above, the potassium cation is surrounded by seven oxygen atoms, one of which belongs to the carboxyl group and the other six to the surrounding water molecules. In addition, the eighth coordination position can be taken by an interacting mefenamate ring from the adjacent polymeric chain. However, in the previously prepared polymer (NUSTAX), the six coordination spheres of the potassium cation consisted of two oxygen atoms from the same carboxylate group (acting as a bidentate ligand), one oxygen atom from the carboxylate group of the next mefenamate ion (acting as a monodentate ligand), two oxygen atoms from two water molecules and the last sixth component coordinating to the potassium ion, which were the electrons from the aromatic ring of mefenamate ion. It is important to mention that in the coordination polymer presented here, the only bridging ligands that connect potassium ions in the polymeric chain are water molecules, while in the structure presented by Kryszynski et al. (CSD code-NUSTAX) [7], two potassium ions are joined alternately by water molecules and then by mefenamate ions. In this way, each mefenamate ion coordinates to one potassium ion as a bidentate ligand and to the other one as a monodentate ligand. The coordination mode of the mefenamate ion to the potassium cation involving two oxygen atoms from the same carboxyl group seems to be a natural method for the bonding of a cation to a carboxylate group, confirmed by theoretical calculations for the salt of fenamic acid with sodium ions [8].
3.2. Analysis of the K—O Bonds
The main problem in the structural analysis is the number of ligands associated with each of the potassium cations, as well as the manner of ligand coordination. Considering only the geometric criteria for the potassium cation, the oxygen atoms and the center of gravity of the aromatic ring, doubts remain as to whether the mentioned ligands are actually bound to the ion. If the geometric criteria are insufficient, additional methods of interaction testing should be applied. The theoretical approach applied to the analysis of interactions used in this paper are the Quantum Theory of Atom in Molecules (QTAIM) [16] and Noncovalent Interaction (NCI) [15]. The analysis of the interactions between the potassium cation and the surrounding ligands has been carried out for both polymers of potassium mefenamate: the polymer investigated in this work and the polymer reported previously (CSD code-NUSTAX) [7].
Regarding QTAIM theory, the main parameter differentiating the interaction is the sign of Laplacian of the electron density at the bond critical point (BCP). Concentration of the electron density between two atoms is characteristic for covalent bonds and is connected with negative sign of Laplacian. The depletion of the electron density at the BCP. which is connected with a positive sign of Laplacian, is typical for hydrogen bonds, van der Waals and ionic interactions. According to Table S5, summarizing the QTAIM parameters for both polymers, the Laplacian of electron density at bond critical points of K…O interaction has a positive sign, which indicates a closed-shell interaction between the potassium cation and the oxygen atoms. Besides the Laplacian, an additional parameter that can be used to classify the bond character is the total energy density H(r) equal to the sum of the potential (V(r)) and kinetic (G(r)) energy density [17,18,19].
Energetic properties of electrons at the critical points can also be investigated in relation to QTAIM theory. The kinetic energy density at a BCP-G(r) is connected with the mobility of the electron density at the BCP [18,19]. G(r), which reflects the pressure exerted by the electrons located at BCP on the other electrons, is always positive. The potential energy density of the electrons at BCP-V(r), which expresses the pressure exerted on the electrons at the BCP by other electrons, is always negative. For negative H(r), the interaction has a covalent contribution which can be described by the covalency degree defined as CD = H(r)/ρ(r). Both parameters, Laplacian of the electron density and covalency degree, can be used to differentiate the interactions; for covalent interactions ∇2ρ(r) < 0, H(r) < 0, for medium-strength interactions with a partial covalent character ∇2ρ(r) > 0, H(r) < 0 and for weak interactions ∇2ρ(r) > 0, H(r) > 0. According to the values of ∇2ρ(r) and H(r) (Table S5), all the interactions of potassium cation can be classified as weak closed-shell interactions.
The QTAIM parameter indicating the strength of the interaction is the value of electron density at BCP (ρ(r)). Characteristic for the investigated interactions of potassium cation is a relatively wide range of electron density at the BCP. The strength of the interaction of the potassium cation with the carboxylate group which should be the strongest is comparable to some interactions with the water oxygen atoms. However, some of the interactions with water oxygen atoms are very weak. The weak interactions include interactions with the π electron cloud. The QTAIM theory expresses these interactions through a reaction pathway between a cation and one of the carbon atoms of the aromatic ring. The electron density values at the critical point of this reaction path are very low, although they are comparable to the values for the cation reaction path with some water molecules. Besides, the strength of the interaction the stability of the bond is also important. The stable interaction is related to low ellipticity of the electron density at the BCP (ε), while high ellipticity is typical for unstable double bond interactions [20,21]. The analysis of the ellipticity values in Table S5 shows that the least stable of the interactions are between the cation and the aromatic ring, including those for the potassium polymeric structure known from the literature, the presence of which is not in doubt due to the geometry [7].
The second parameter related to the stability of the interaction is the shape of the bond path, which cannot be too bent, so the bond length path should be close to the distance between the atoms linked by the bond path. According to this criterion, all cation interactions, apart from interactions with the aromatic ring, are stable.
The analysis of the parameters describing the electron density at the bond critical point is particularly useful in the case of interactions between atoms of considerable distance. According to the geometrical criteria, the distance between the potassium ion and the oxygen atom of 3.3033(15) (K1-O1W) or 3.2332(15) (K2-O5W) Å indicates no interaction. Meanwhile, the presence of a bond path with a low electron density at the critical point but with low ellipticity and low non-linearity indicates a weak but relatively stable interaction.
Another method especially dedicated for investigation of weak interaction is the Noncovalent Interaction (NCI) approach [22,23]. With this method, the visualization of the interactions by reduced density gradients isosurfaces in the real space for the molecule is especially useful for finding and distinguishing attractive, repulsive and dispersive interactions.
In Figure 4 and Figure 5, the QTAIM and NCI diagrams for the investigated potassium cation and mefenamate ion and its analogue investigated previously (NUSTAX) [7]. Presence of the green surface in the NCI diagram confirms the dispersive interactions between potassium-oxygen and potassium π electrons. It is characteristic that according to NCI diagram all interactions are marked as dispersive independent of the distance. Comparison of the results of the QTAIM and NCI methods allows the identification of all interactions of the potassium cation, including the weakest. The nature of the interaction with the oxygen of the carboxylate group, which appears to be the natural binding site of the cation, is similar to the interaction with the oxygen atom of the water located further away from the cation.
The integration of three-dimensional electron density over the basin of the atom delivers another parameter which can be used to describe the bonded atoms. The formation of a bond with another atom changes the electron density in the atomic basin. The numerical integration of the charge density over the atomic basin gives the average electron population of an A atom-N(A), and subtracting N(A) from the nuclear charge Zn gives the net charge of atom: q(A) = Zn-N(A) [24]. Two atoms are bonded if electrons are exchanged between their atomic basins and the QTAIM theory allows the calculation of the quantity of the electrons exchanged between the basin of an atom with the basin of another atom, which is called delocalization index [25] δ(A,B), related to the number of electrons delocalized or shared between the atoms A and B. For covalent bonds, the delocalization index is very high for weak interactions, as in the sodium fenamate investigated previously (ZOSWIQ) [8], where it equals 0.0655, 0.0729, 0.0748, 0.0572 for Na–O(water) and 0.0790 for Na–O(carboxylate group). Another parameter is the localization index λ (A), which gives the number of electrons localized in the atomic basin of atom A. The %λ (A) is the percentage of electron localization in the basin of atom A and it is close to 100% for isolated atoms or atoms engaged in closed-shell interaction. The atomic basin is a topological definition of the atom. In the gradient vector field of the electron density, local zero flux surfaces appear and these surfaces provide a partition of the molecular space into atomic basins. Integration of three-dimensional electron density over the basin of the atom allows the obtaining of the atomic properties.
The parameters resulting from the integration over the atomic basins of selected atoms participating in the interaction with the potassium cation in the polymers of the mefenamic acid complex are summarized in Table S5. All the parameters resulting from the integration of the electron density over the atomic basins confirm that the interaction of the potassium ion with the surrounding atoms is very weak. Only a small percentage of the electrons belonging to the potassium cation basin are exchanged with other atoms and the percentage of electrons located in the pool is about 98. The number of electrons exchanged between the potassium ion and other atoms is less than 0.1 and the percentage of electrons belonging to the potassium cation basin exchanged with other atoms is about 0.2. Despite significant differences in the distances between a cation and the interacting atoms, the parameters describing the electron density at the critical points of bonds between the cation and the surrounded atoms, as well as the parameters describing electron density changes in atomic basins, confirm that all interactions with the cation are very weak. According to the NCI approach, these interactions can be classified as dispersive.
4. Conclusions
- For both known potassium mefenamates, the formation of coordination polymer takes place with the participation of water molecules and the strength of interaction with all oxygen atoms surrounding the cation is similar.
- The weak dispersive interaction may be responsible for the formation of a complex between the central ion and the ligand and the cooperative weak interactions are the driving force building polymeric structures.
- The existence of an interaction between the cation and the ligand is confirmed by the presence of a reaction path, even if the interaction is very weak and not very stable.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/sym13101761/s1, Table S1. Crystallographic data and refinement parameters for potassium mefenamate–water (1/3). Table S2. Bond lengths in potassium mefenamate–water (1/3). Table S3. Selected angles for potassium mefenamate–water (1/3). Table S4. Geometry of the hydrogen bonds in potassium mefenamate–water (1/3). Table S5. Selected QTAIM parameters (in a.u.) for the coordination polymers of potassium mefenamate–water (1/3).
Author Contributions
Conceptualization, M.S.K. and I.M.; methodology, M.S.K. and I.M.; software, M.S.K. and I.M.; validation, M.S.K. and I.M.; formal analysis, M.S.K. and I.M.; investigation, M.S.K. and I.M.; data curation, M.S.K. and I.M.; writing—original draft preparation, M.S.K. and I.M.; writing—review and editing, M.S.K. and I.M.; visualization, M.S.K. and I.M.; funding acquisition, M.S.K. and I.M. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Structural data are available in Cambridge Structural Database with deposition number: 2109559. Calculation data can be obtained from the authors upon request.
Acknowledgments
The authors would like to thank Professor Tadeusz Lis for his valuable help. The Wroclaw Center for Networking and Supercomputing is acknowledged for generous allocations of computer time. This research was financially supported by a grant number D050.21.034 from Wroclaw Medical University.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Rothan, H.A.; Bahrani, H.; Mohamed, Z.; Teoh, T.C.; Shankar, E.M.; Rahman, N.A.; Yusof, R. Mefenamic acid in combination with ribavirin shows significant effects in reducing chikungunya virus infection in vitro and in vivo. Antivir. Res. 2016, 127, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lago, E.M.; Silva, M.P.; Queiroz, T.G.; Mazloum, S.F.; Rodrigues, V.C.; Carnaúba, P.U.; Pinto, P.L.; Rocha, J.A.; Ferreira, L.L.G.; Andricopulo, A.D.; et al. Phenotypic screening of nonsteroidal anti-inflammatory drugs identified mefenamic acid as a drug for the treatment of schistosomiasis. EbioMedicine 2019, 43, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimolai, N. The potential and promise of mefenamic acid. Expert Rev. Clin. Pharm. 2013, 6, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Bani-Jaber, A.; Hamdan, I.; Al-Khalidi, B. Sodium Mefenamate as a Solution for the Formulation and Dissolution Problems of Mefenamic Acid. Chem. Pharm. Bull. 2007, 55, 1136–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafarska, K.; Wolf, W.M. Novel silver complexes with popular non-steroidal anti-inflammatory drugs. Acta Innov. 2016, 21, 51–59. [Google Scholar]
- Topacli, A.; Ide, S. Molecular structures of metal complexes with mefenamic Acid. J. Pharm. Biomed. Anal. 1999, 21, 975–982. [Google Scholar] [CrossRef]
- Kruszynski, R.; Trzesowska-Kruszynska, A.; Majewski, P.; Łukaszewicz, E.; Majewska, K.; Sierański, T.; Lewiński, B. Structure and properties of the sodium, potassium and calcium salts of 2-(2,3-dimethylphenyl)aminobenzoic acid. J. Mol. Struct. 2010, 970, 79–89. [Google Scholar] [CrossRef]
- Krawczyk, M.S.; Majerz, I. The Na—O bond in sodium fenamate. Acta Cryst. 2019, B75, 766–774. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2014. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Keith, T.A. AIMALL; Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Popelier, P.L.A.; Bader, R.F.W. Effect of Twisting a Polypeptide on Its Geometry and Electron Distribution. J. Phys. Chem. 1994, 98, 4473–4481. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.; Woller, T.; Martin-Martinez, F.J.; Contreras-García, J.; Geerlings, P.; De Proft, F. Understanding the Fundamental Role of π/π, σ/σ, and σ/π Dispersion Interactions in Shaping Carbon-Based Materials. Chem. Eur. J. 2014, 20, 4931–4941. [Google Scholar] [CrossRef]
- Wiberg, K.B.; Bader, R.F.W.; Lau, C.D.H. Theoretical analysis of hydrocarbon properties. 1. Bonds, structures, charge concentrations, and charge relaxations. J. Am. Chem. Soc. 1987, 109, 1001–1012. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis Model and Beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
Scheme 1.
Mefenamic acid.

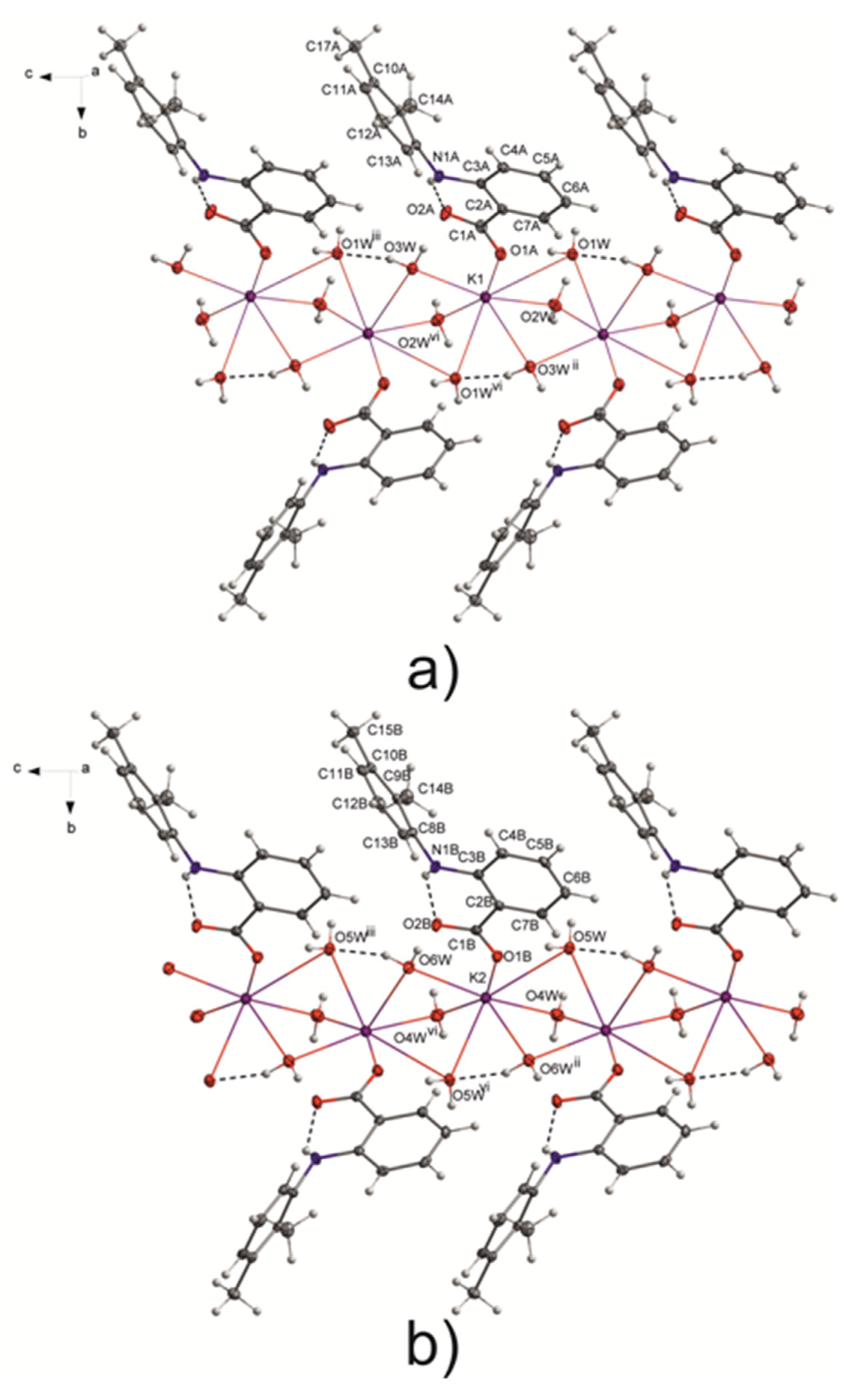
Figure 1.
Polymeric structure of potassium mefenamate–water (1/3). (a) Polymeric chain formed with K1 ions; (b) Polymeric chain created with K2 ions. Symmetry codes: ii = iii = x, y, z + 1; vi = x, −y + 3/2, z + 1/2. In the pictures the component O21A with a lower occupancy factor and the K⋯π interactions were omitted for clarity.
Figure 1.
Polymeric structure of potassium mefenamate–water (1/3). (a) Polymeric chain formed with K1 ions; (b) Polymeric chain created with K2 ions. Symmetry codes: ii = iii = x, y, z + 1; vi = x, −y + 3/2, z + 1/2. In the pictures the component O21A with a lower occupancy factor and the K⋯π interactions were omitted for clarity.

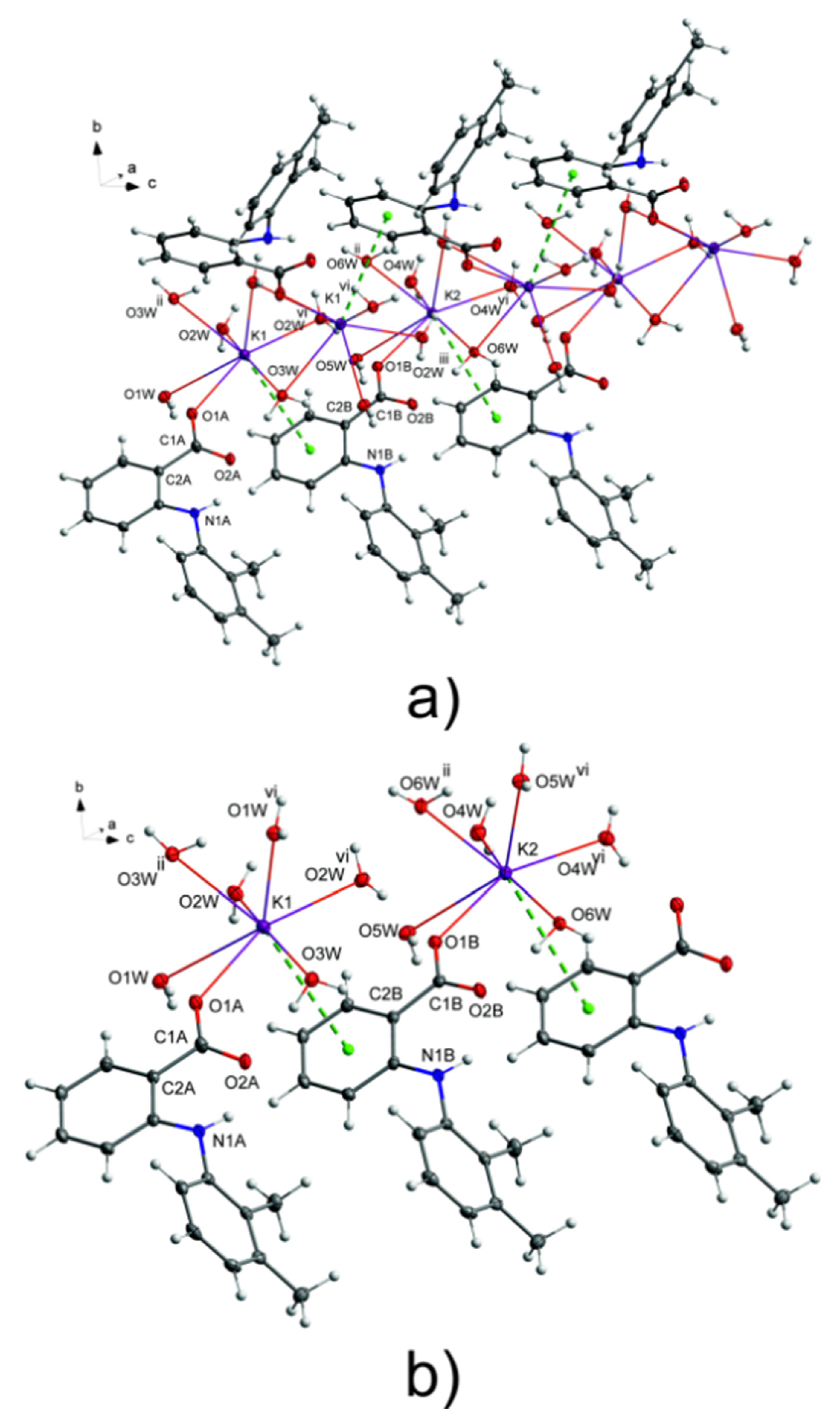
Figure 2.
(a) A fragment of a packing diagram for potassium mefenamate–water (1/3); (b) A fragment of a chain showing coordination spheres of K1 and K2 ions connected via K⋯π interactions with adjacent mefenamate ions. K⋯π interactions are marked by green dashed lines. The component O21A with a lower occupancy factor of a disordered carboxylate oxygen atom (O21, O21A) was omitted for clarity. Symmetry codes: ii = x, −y + 3/2, z − 1/2, iii = x, y, z + 1; vi = x, −y + 3/2, z + 1/2.
Figure 2.
(a) A fragment of a packing diagram for potassium mefenamate–water (1/3); (b) A fragment of a chain showing coordination spheres of K1 and K2 ions connected via K⋯π interactions with adjacent mefenamate ions. K⋯π interactions are marked by green dashed lines. The component O21A with a lower occupancy factor of a disordered carboxylate oxygen atom (O21, O21A) was omitted for clarity. Symmetry codes: ii = x, −y + 3/2, z − 1/2, iii = x, y, z + 1; vi = x, −y + 3/2, z + 1/2.

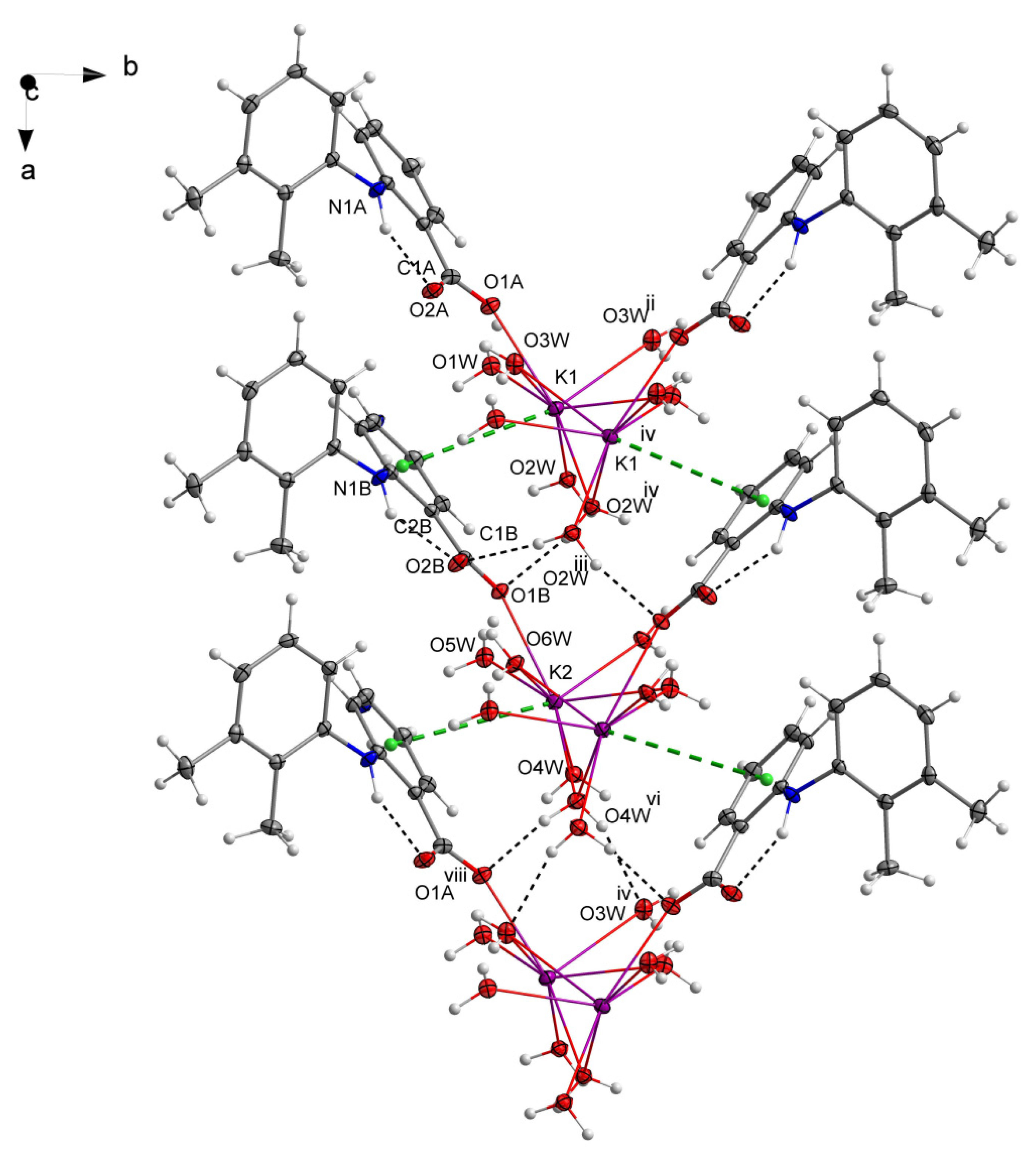
Figure 3.
Hydrogen bond interactions between neighboring polymeric chains. Symmetry codes: ii = x, −y + 3/2, z − 1/2; iii = x, y, z + 1; iv = x + 1, −y + 3/2, z + 1/2; vi = x, −y + 3/2, z + 1/2; viii = x + 1, y, z + 1. The component O21A with a lower occupancy factor of a disordered carboxylate oxygen atom (O21, O21A) was omitted for clarity.
Figure 3.
Hydrogen bond interactions between neighboring polymeric chains. Symmetry codes: ii = x, −y + 3/2, z − 1/2; iii = x, y, z + 1; iv = x + 1, −y + 3/2, z + 1/2; vi = x, −y + 3/2, z + 1/2; viii = x + 1, y, z + 1. The component O21A with a lower occupancy factor of a disordered carboxylate oxygen atom (O21, O21A) was omitted for clarity.

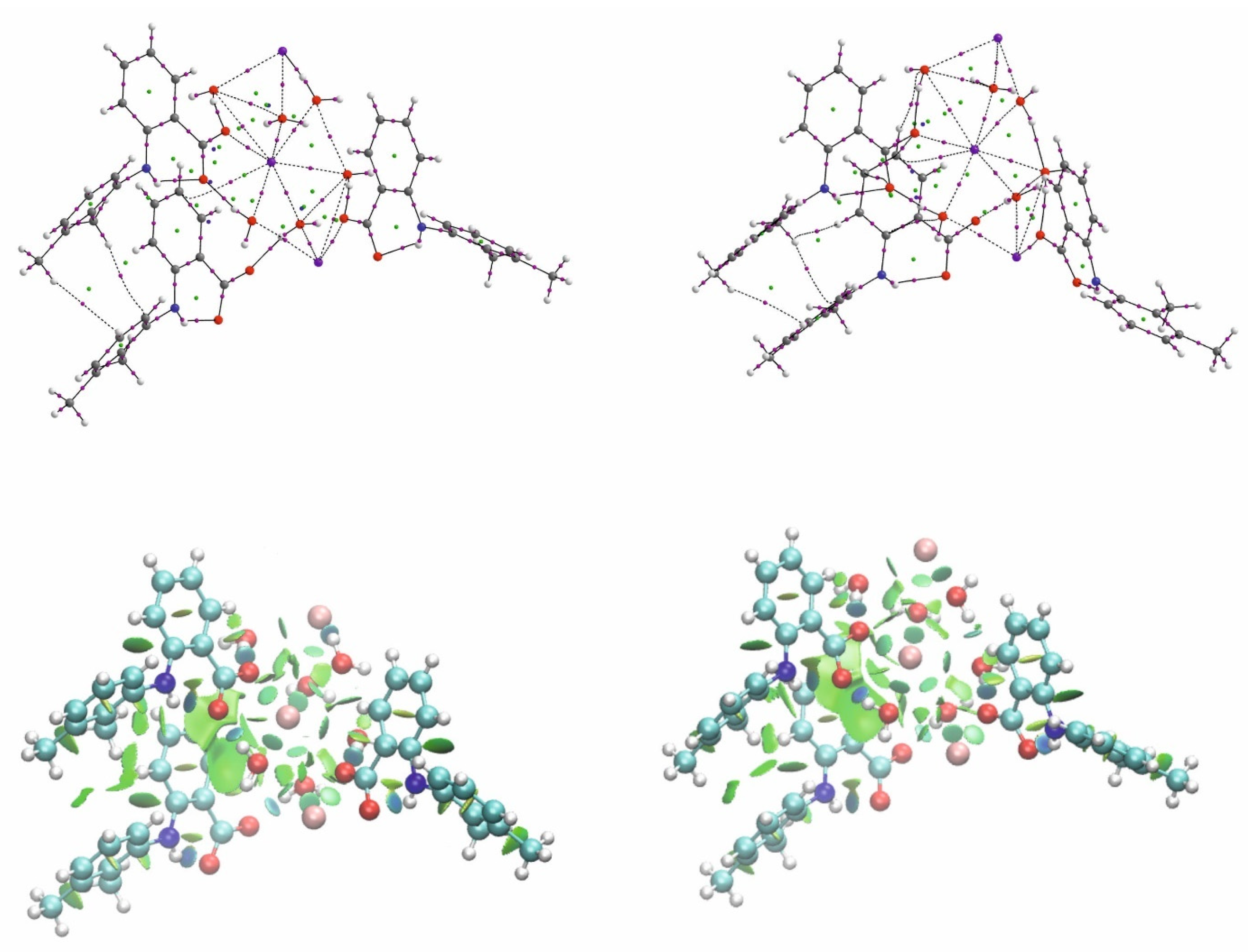
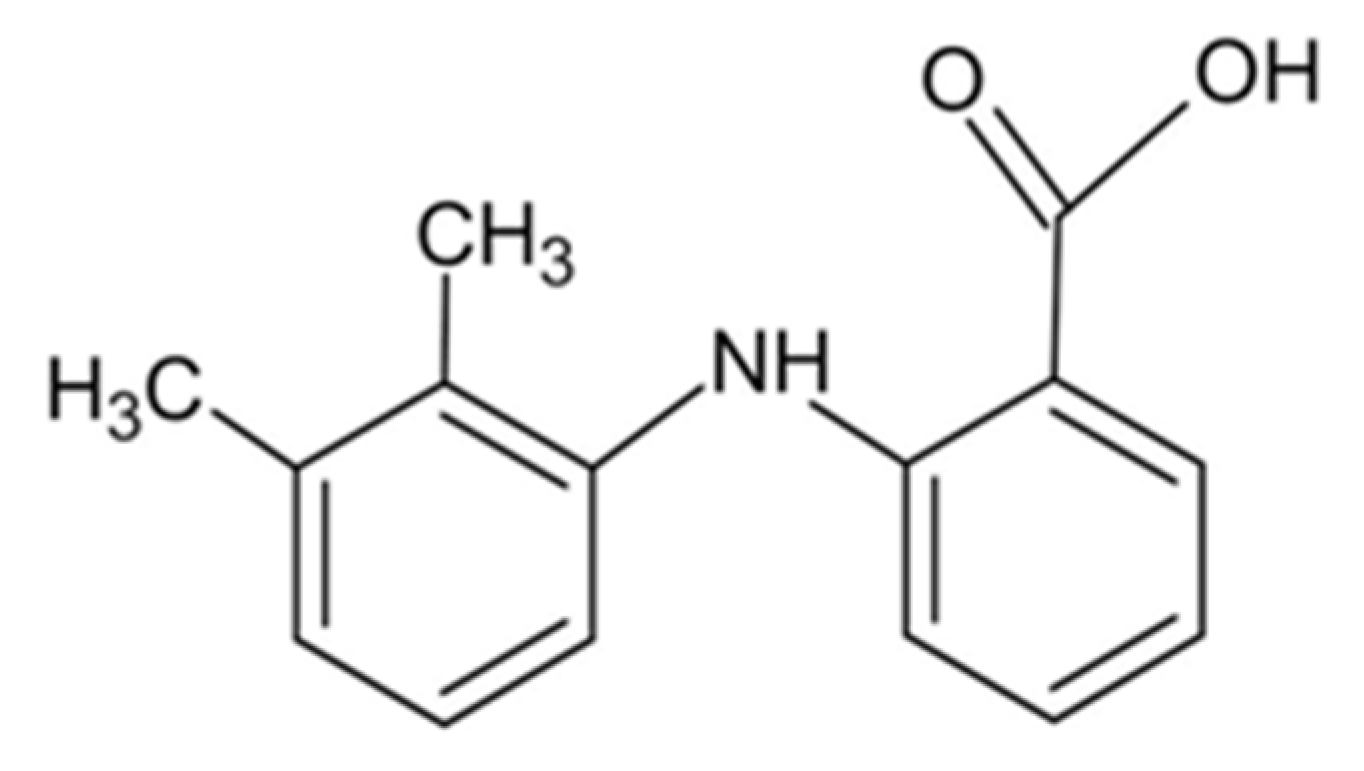
Figure 4.
Comparison of QTAIM and NCI diagram for the investigated potassium mefenamate trihydrate. Left-K1 and K2-right. Carbon, hydrogen, nitrogen, oxygen and potassium atoms are marked as grey, white, blue, red and pink dots, respectively. Red, yellow and green dots represent bond-critical points-BCPs, ring-critical points-RCPs and cage-critical points-CCP, respectively. NCI isosurfaces are blue for attractive, red for repulsive and green for the intermediate interactions.
Figure 4.
Comparison of QTAIM and NCI diagram for the investigated potassium mefenamate trihydrate. Left-K1 and K2-right. Carbon, hydrogen, nitrogen, oxygen and potassium atoms are marked as grey, white, blue, red and pink dots, respectively. Red, yellow and green dots represent bond-critical points-BCPs, ring-critical points-RCPs and cage-critical points-CCP, respectively. NCI isosurfaces are blue for attractive, red for repulsive and green for the intermediate interactions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Krawczyk, M.S.; Majerz, I. Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure. Symmetry 2021, 13, 1761. https://doi.org/10.3390/sym13101761
AMA Style
Krawczyk MS, Majerz I. Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure. Symmetry. 2021; 13(10):1761. https://doi.org/10.3390/sym13101761
Chicago/Turabian StyleKrawczyk, Marta S., and Irena Majerz. 2021. "Novel Coordination Mode in the Potassium Mefenamate Trihydrate Polymeric Structure" Symmetry 13, no. 10: 1761. https://doi.org/10.3390/sym13101761
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.