
Cell-Adapted Mutations and Antigenic Diversity of Influenza B Viruses in Missouri, 2019–2020 Season

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Virus Isolation in MDCK-hCK Cell Lines Showed the Highest Virus Recovery Rate
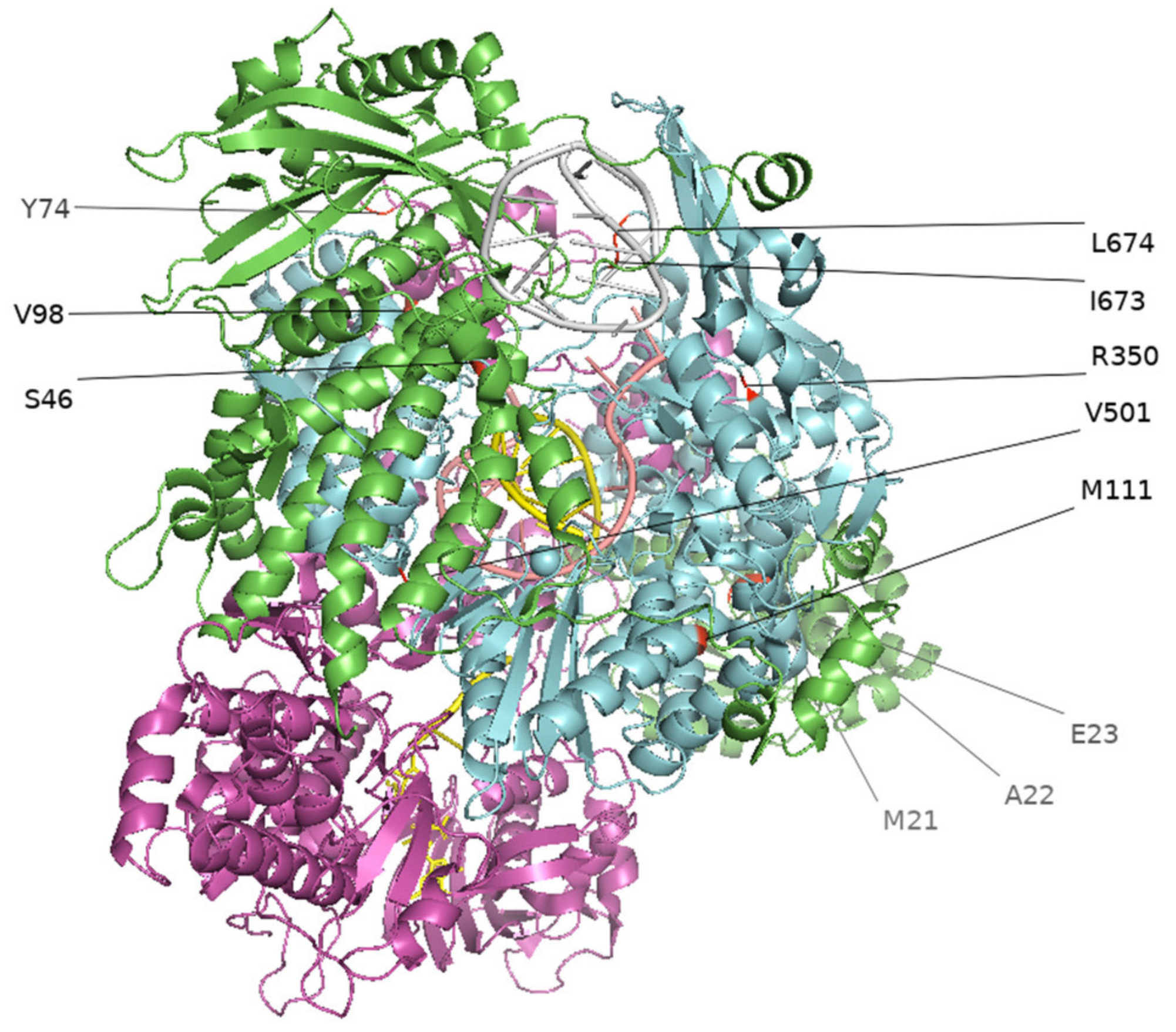
3.2. Culture-Adapted Mutations Were Identified in IBVs
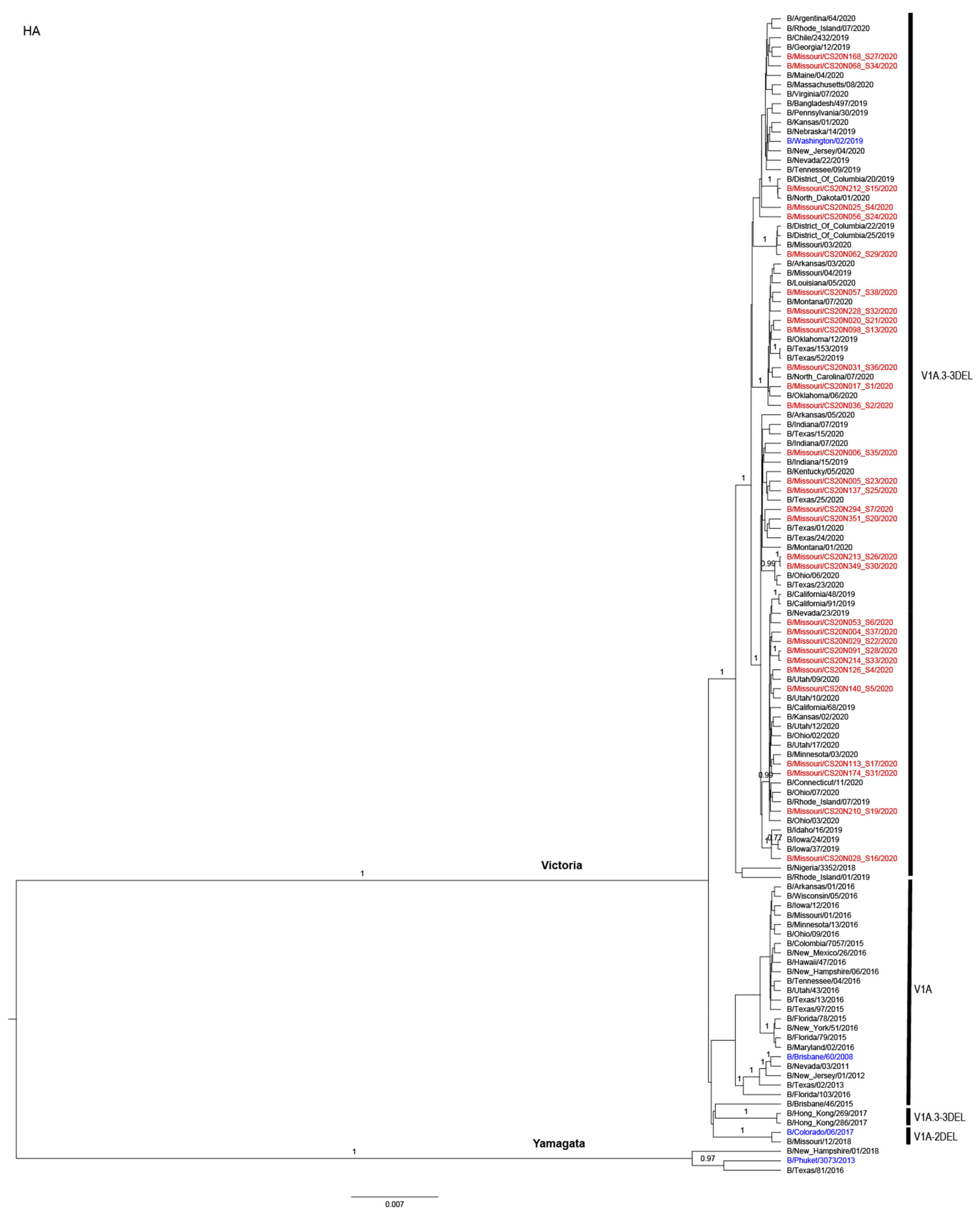
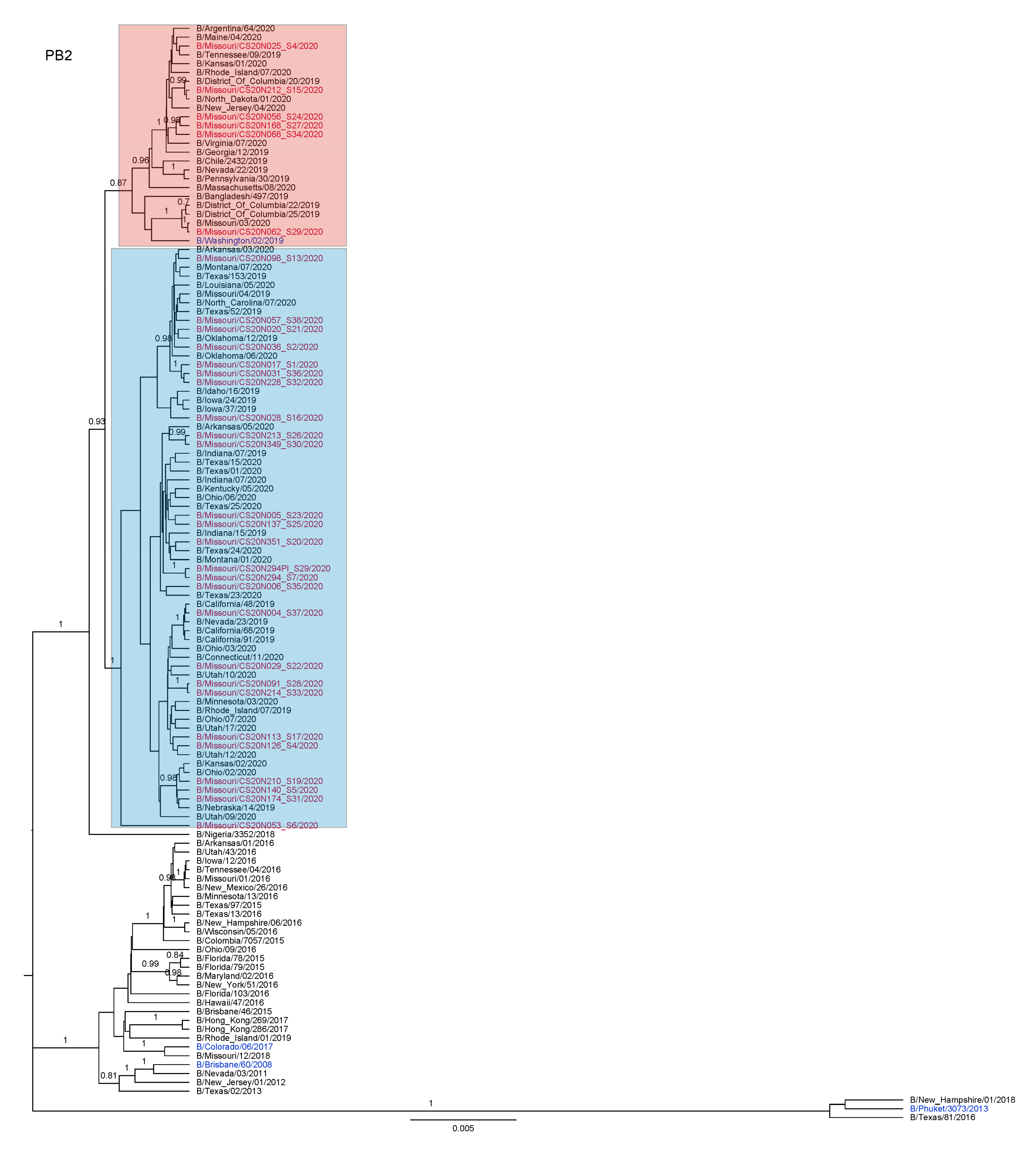
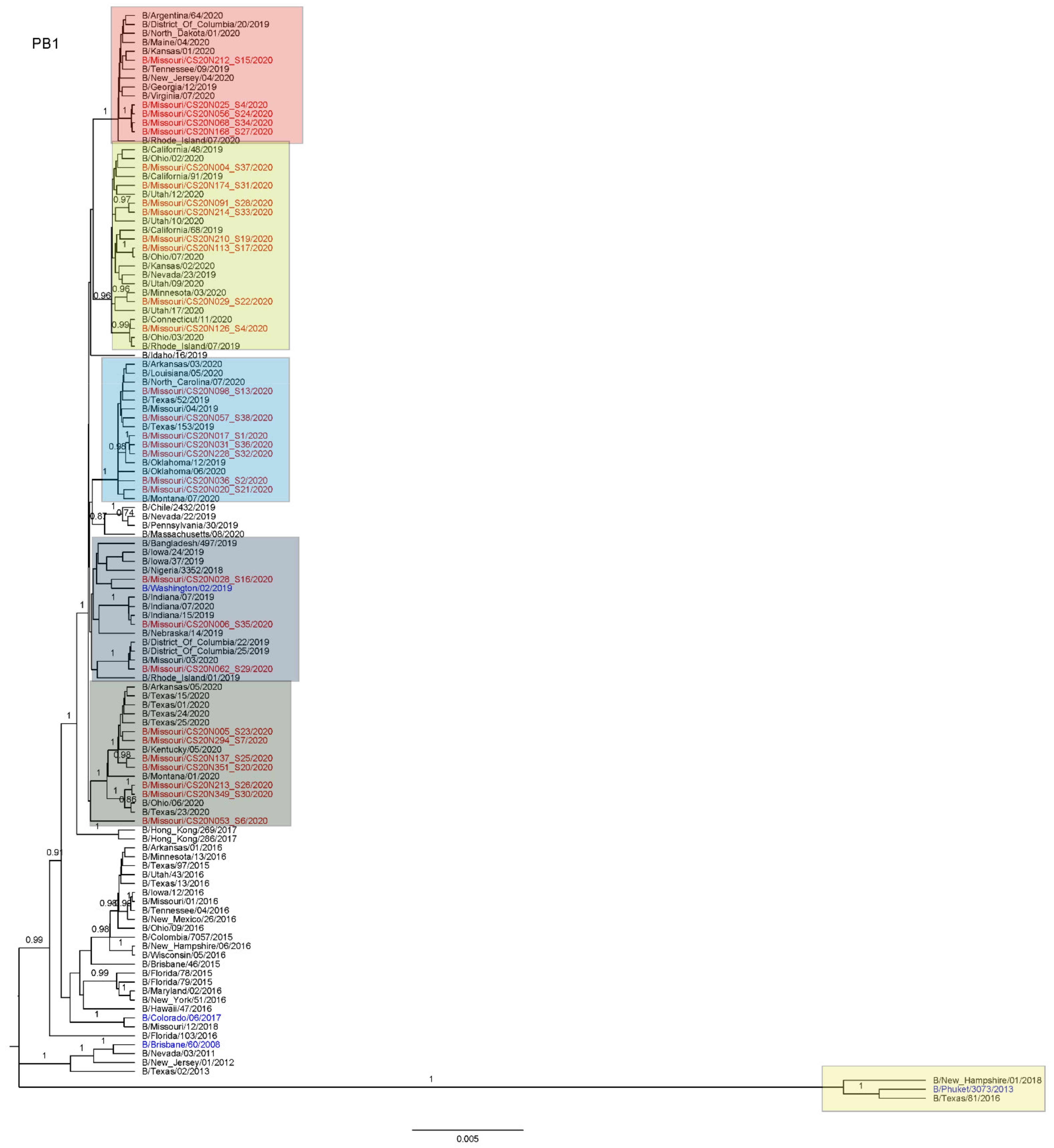
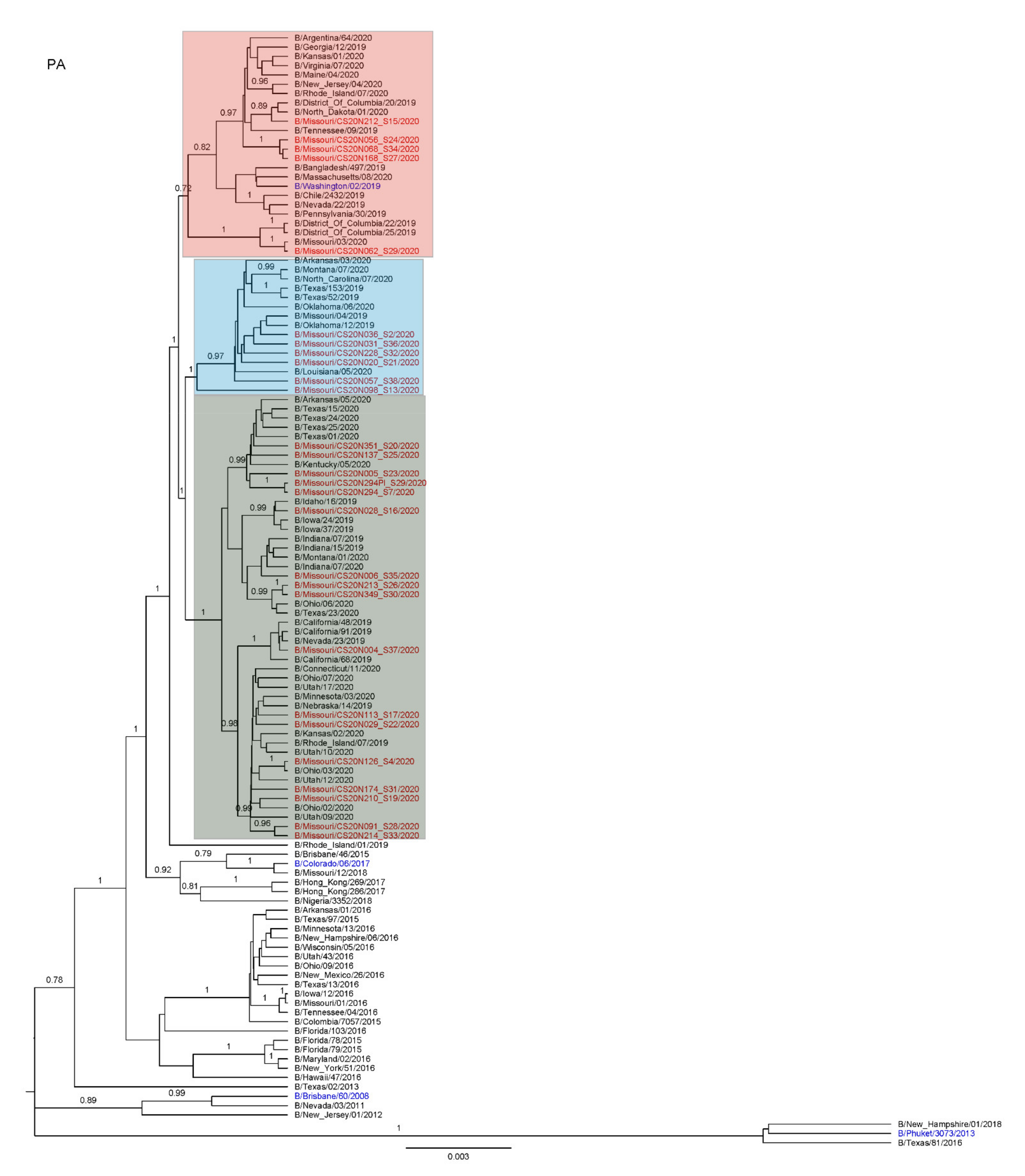
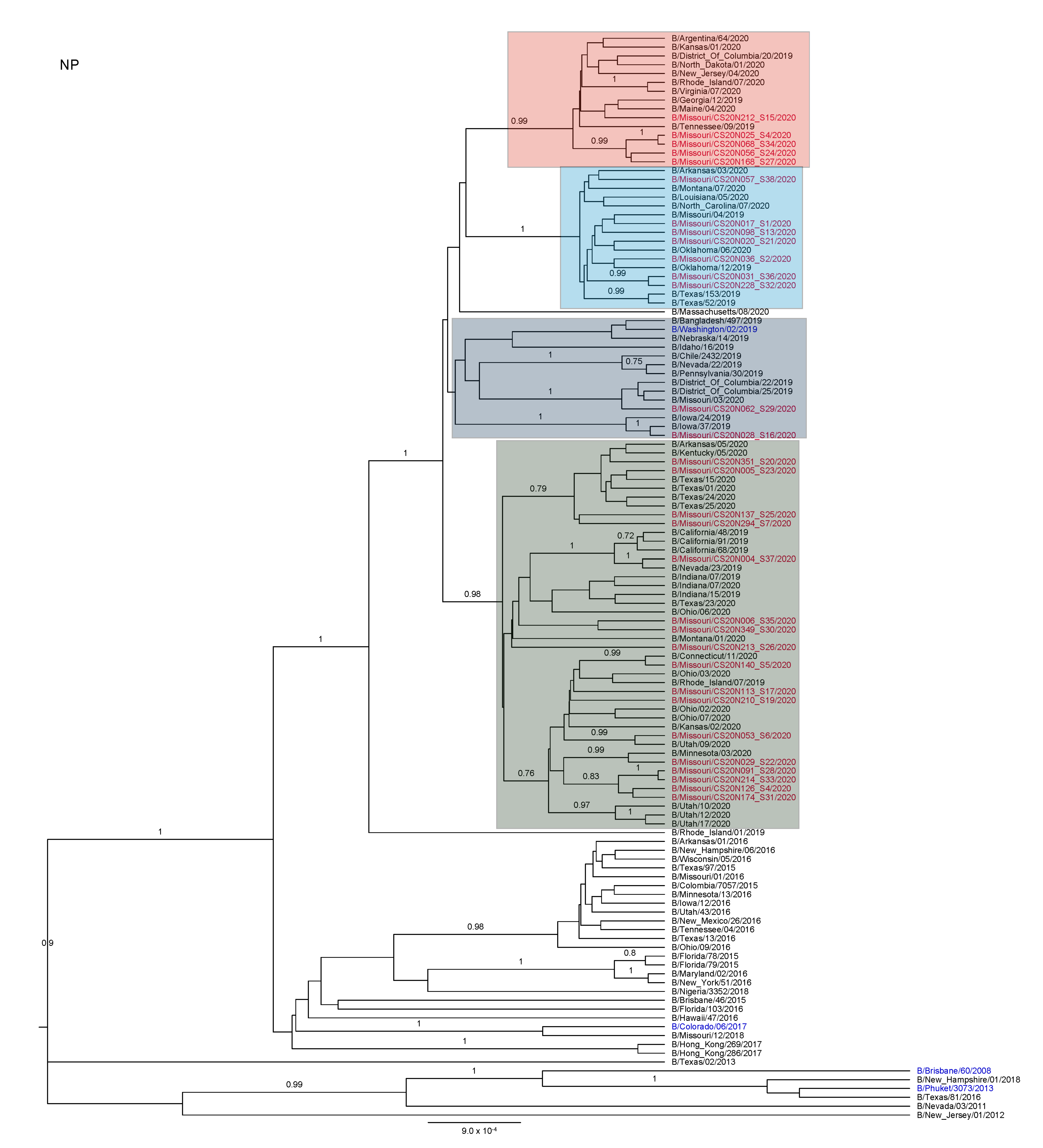
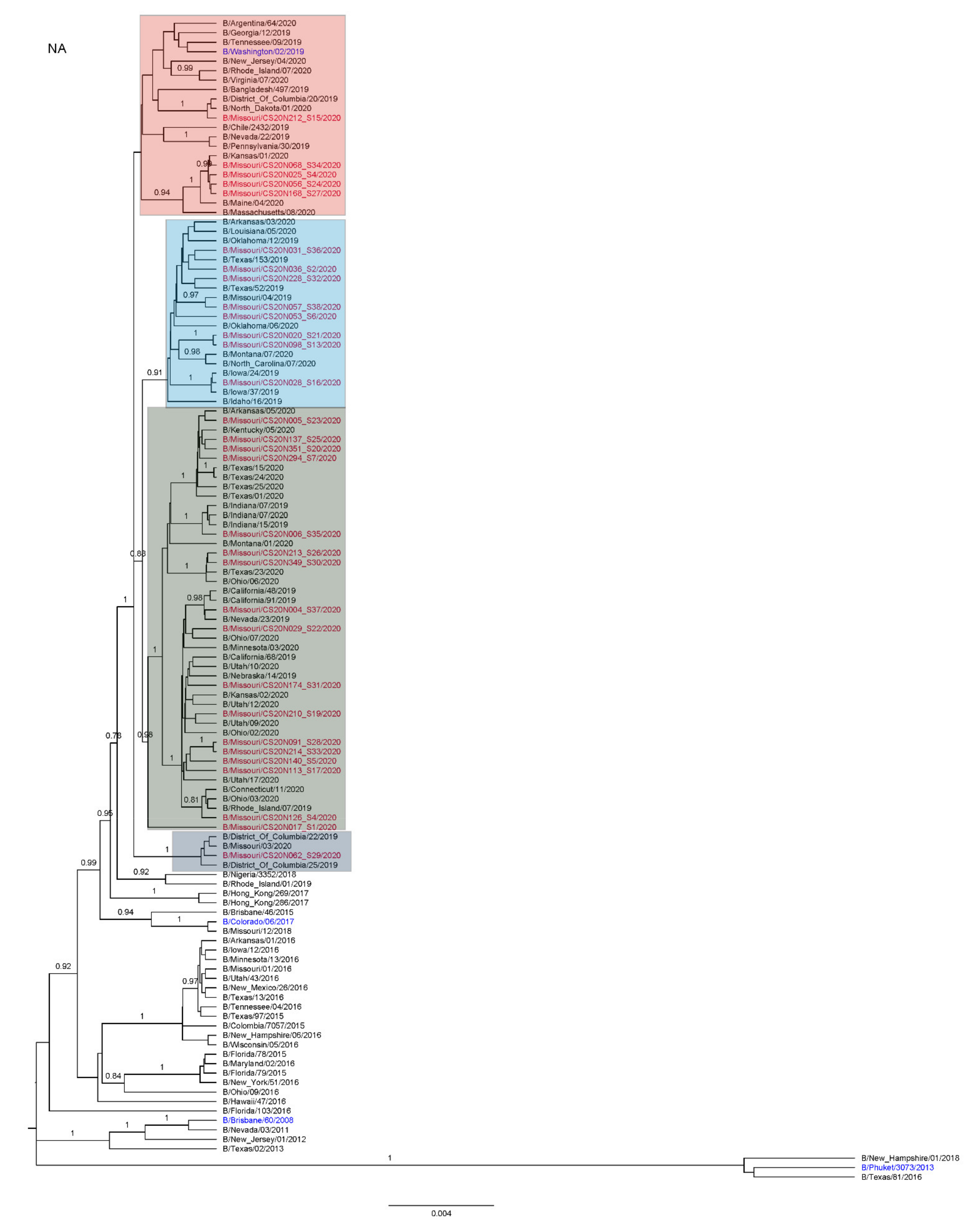
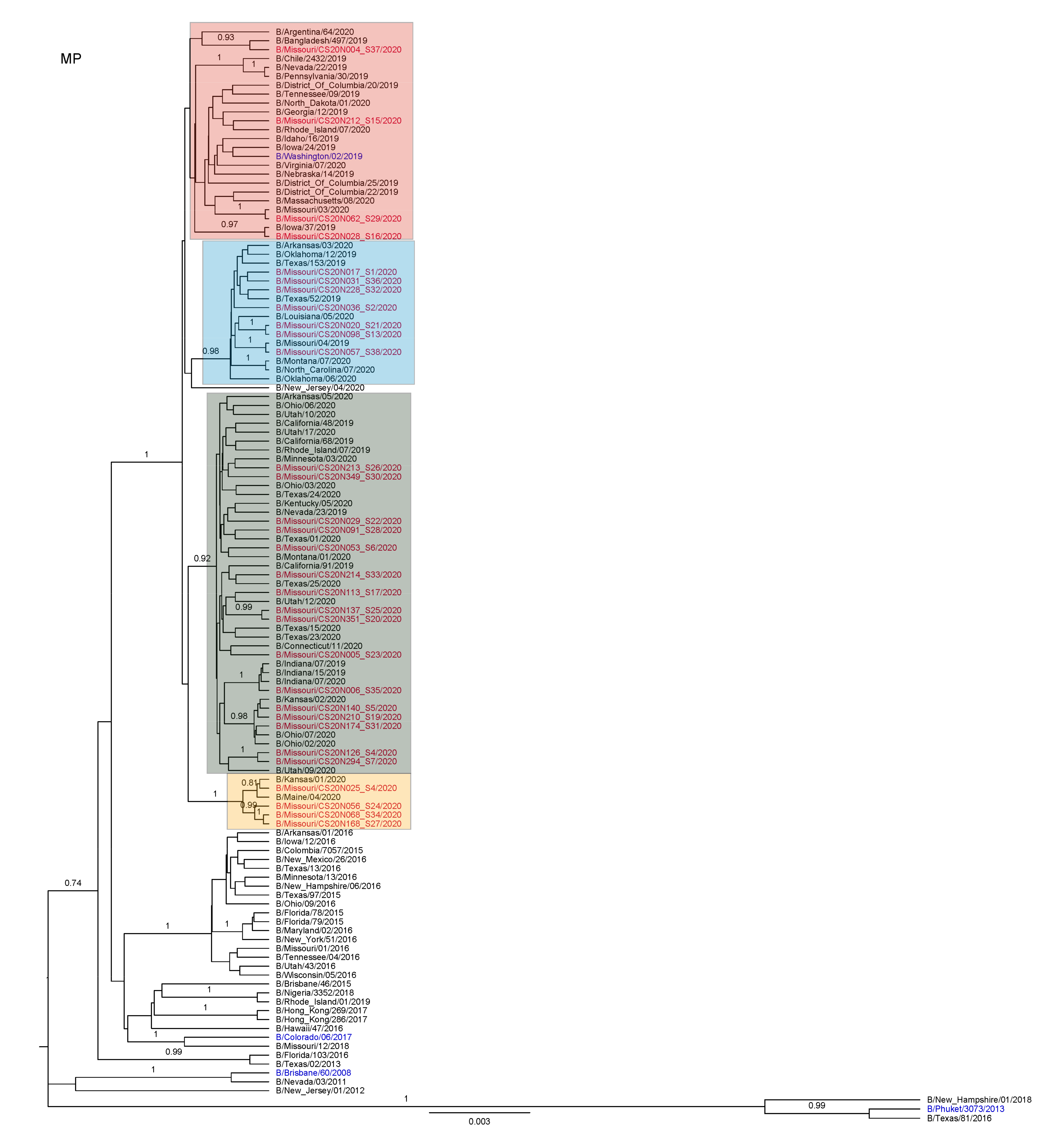
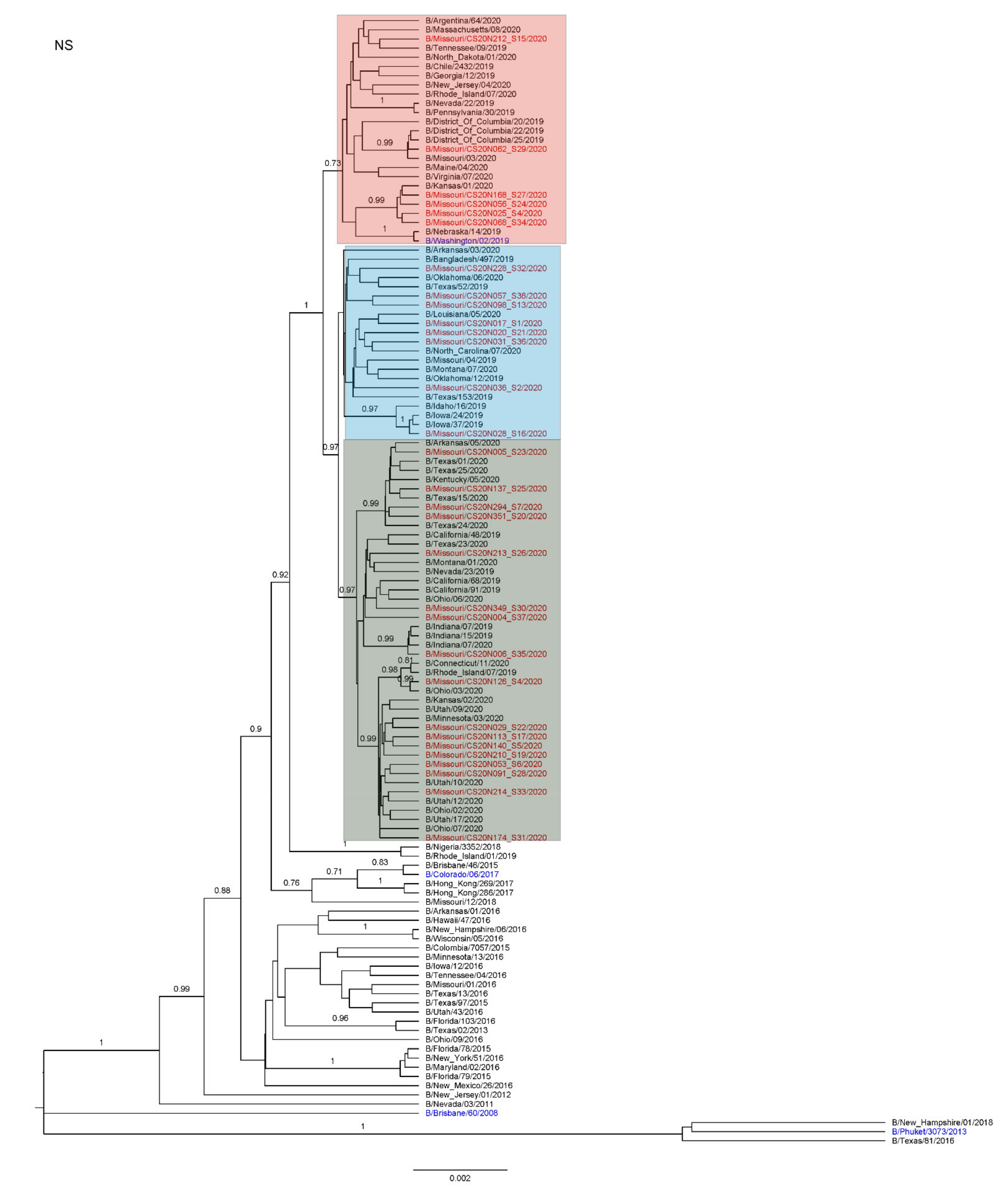
3.3. Antigenic and Genetic Diversity of Missourian IBV in the 2019–2020 Influenza Season
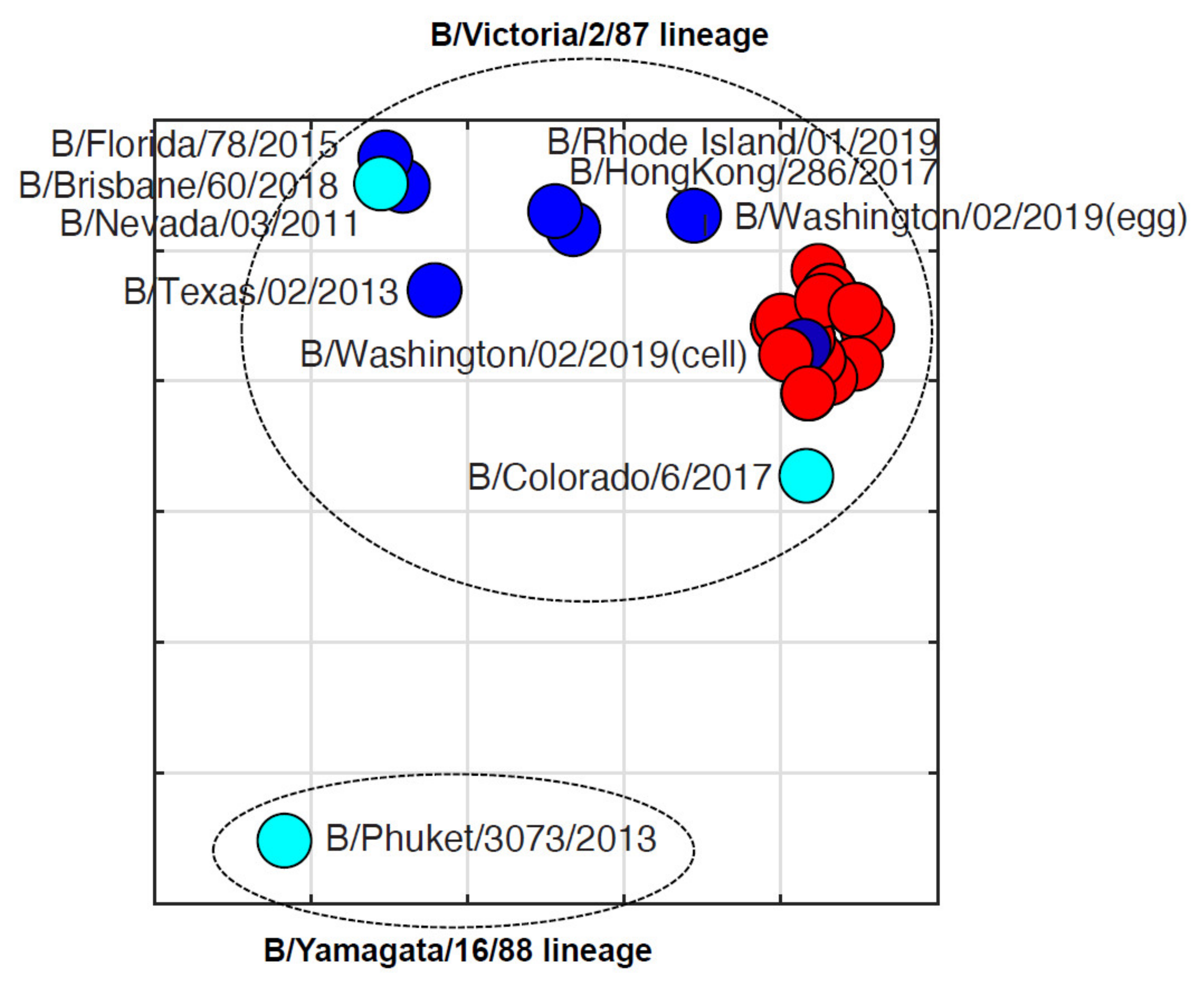
3.4. Antigenic Diversity of Missourian IBVs in the 2019–2020 Influenza Season
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A








References
- World Health Organization. Burden of Disease, Global Influenza Programme. Available online: https://www.who.int/teams/global-influenza-programme/surveillance-and-monitoring/burden-of-disease (accessed on 16 June 2021).
- Medina, R.A.; Stertz, S.; Manicassamy, B.; Zimmermann, P.; Sun, X.; Albrecht, R.; Uusi-Kerttula, H.; Zagordi, O.; Belshe, R.B.; Frey, S.E.; et al. Glycosylations in the Globular Head of the Hemagglutinin Protein Modulate the Virulence and Antigenic Properties of the H1N1 Influenza Viruses. Sci. Transl. Med. 2013, 5, 187ra70. [Google Scholar] [CrossRef] [Green Version]
- Young, J.F.; Desselberger, U.; Palese, P. Evolution of human influenza a viruses in nature: Sequential mutations in the genomes of new H1N1 isolates. Cell 1979, 18, 73–83. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Swine influenza A (H1N1) infection in two children—Southern California, March-April 2009. Morb. Mortal. Wkly. Rep. 2009, 58, 400–402. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention. Update: Novel influenza A (H1N1) virus infections—Worldwide, May 6, 2009. Morb. Mortal. Wkly. Rep. 2009, 58, 453–458. [Google Scholar] [PubMed]
- Wentworth, D.; McGregor, M.W.; Macklin, M.D.; Neumann, V.; Hinshaw, V.S. Transmission of Swine Influenza Virus to Humans after Exposure to Experimentally Infected Pigs. J. Infect. Dis. 1997, 175, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.K. National influenza experience in Hong Kong, 1968. Bull. World Heal. Organ. 1969, 41, 349–351. [Google Scholar] [PubMed]
- Francis, T., Jr. A NEW TYPE OF VIRUS FROM EPIDEMIC INFLUENZA. Science 1940, 92, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Kanegae, Y.; Sugita, S.; Endo, A.; Ishida, M.; Senya, S.; Osako, K.; Nerome, K.; Oya, A. Evolutionary pattern of the hemagglutinin gene of influenza B viruses isolated in Japan: Cocirculating lineages in the same epidemic season. J. Virol. 1990, 64, 2860–2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Li, J.; Cai, R.; Wang, X.; Gu, Z.; Yu, H.; Fang, B.; Chen, L.; Wang, C. Age- and sex-specific excess mortality associated with influenza in Shanghai, China, 2010–2015. Int. J. Infect. Dis. 2020, 98, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Rebaza, A.; Dela Cruz, C.S. When “B” becomes “A”: The emerging threat of influenza B virus. Eur. Respir. J. 2019, 54, 1901325. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Influenza-associated pediatric deaths—United States, September 2010-August 2011 (2011). Morb. Mortal. Wkly. Rep. 2011, 60, 1233–1238. [Google Scholar] [PubMed]
- Tran, D.; Vaudry, W.; Moore, D.; Bettinger, J.; Halperin, S.A.; Scheifele, D.; Jadvji, T.; Lee, L.; Mersereau, T.; for the members of the Canadian Immunization Monitoring Program Active. Hospitalization for Influenza A Versus, B. Pediatrics 2016, 138, e20154643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Regional Office for Europe. How Pandemic Influenza Emerges. Available online: https://www.euro.who.int/en/health-topics/communicable-diseases/influenza/pandemic-influenza/how-pandemic-influenza-emerges. (accessed on 20 June 2021).
- Vijaykrishna, D.; Holmes, E.C.; Joseph, U.; Fourment, M.; Sum, Y.C.; Halpin, R.; Lee, R.T.; Deng, Y.M.; Gunalan, V.; Lin, X.; et al. The contrasting phylodynamics of human influenza B viruses. Elife 2015, 4, e05055. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Pybus, O.G.; Nelson, M.I.; Viboud, C.; Taubenberger, J.K.; Holmes, E.C. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453, 615–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Holmes, E.C. The Evolutionary Dynamics of Human Influenza B Virus. J. Mol. Evol. 2008, 66, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-F.; Chang, C.-F.; Chi, C.-Y.; Wang, H.-C.; Wang, J.-R.; Su, I.-J. Characterization of glycan binding specificities of influenza B viruses with correlation with hemagglutinin genotypes and clinical features. J. Med. Virol. 2012, 84, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Velkov, T. The specificity of the influenza B virus hemagglutinin receptor binding pocket: What does it bind to? J. Mol. Recognit. 2013, 26, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.; Fukuda, K.; Uyeki, T.M.; Cox, N.J.; Bridges, C.B. Prevention and control of influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morb. Mortal. Wkly. Rep. Recomm. Rep. 2005, 54, 1–41. [Google Scholar]
- Ferdinands, J.M.; Gaglani, M.; Martin, E.T.; Middleton, D.; Monto, A.S.; Murthy, K.; Silveira, F.P.; Talbot, H.K.; Zimmerman, R.; Alyanak, E.; et al. Prevention of Influenza Hospitalization Among Adults in the United States, 2015-2016: Results From the US Hospitalized Adult Influenza Vaccine Effectiveness Network (HAIVEN). J. Infect. Dis. 2019, 220, 1265–1275. [Google Scholar] [CrossRef]
- Influenza Research Database. World Health Organization Recommendations for Composition of Influenza Vaccines. 2021. Available online: https://www.fludb.org/brc/vaccineRecommend.spg?decorator=influenza (accessed on 20 June 2021).
- Youil, R.; Su, Q.; Toner, T.J.; Szymkowiak, C.; Kwan, W.S.; Rubin, B.; Petrukhin, L.; Kiseleva, I.; Shaw, A.R.; DiStefano, D. Comparative study of influenza virus replication in Vero and MDCK cell lines. J. Virol. Methods 2004, 120, 23–31. [Google Scholar] [CrossRef]
- Gambaryan, A.; Robertson, J.; Matrosovich, M. Effects of Egg-Adaptation on the Receptor-Binding Properties of Human Influenza A and B Viruses. Virology 1999, 258, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Gambaryan, A.; Tuzikov, A.; Piskarev, V.; Yamnikova, S.; Lvov, D.; Robertson, J.; Bovin, N.; Matrosovich, M. Specification of Receptor-Binding Phenotypes of Influenza Virus Isolates from Different Hosts Using Synthetic Sialylglycopolymers: Non-Egg-Adapted Human H1 and H3 Influenza A and Influenza B Viruses Share a Common High Binding Affinity for 6′-Sialyl(N-acetyllactosamine). Virology 1997, 232, 345–350. [Google Scholar] [CrossRef]
- Gambaryan, A.; Marinina, V.; Tuzikov, A.; Bovin, N.; Rudneva, I.; Sinitsyn, B.; Shilov, A.; Matrosovich, M. Effects of Host-Dependent Glycosylation of Hemagglutinin on Receptor-Binding Properties of H1N1 Human Influenza A Virus Grown in MDCK Cells and in Embryonated Eggs. Virology 1998, 247, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Govorkova, E.; Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gerdil, C.; Fanget, B.; Webster, R. Selection of Receptor-Binding Variants of Human Influenza A and B Viruses in Baby Hamster Kidney Cells. Virology 1999, 262, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, A.; Bartolomei-Corsi, O.; Zakrzewska, K.; Corcoran, T.; Newman, R.; Robertson, J.S.; Yates, P.; Oxford, J.S. The haemagglutinins of influenza A (H1N1) viruses in the ‘O’ or ‘D’ phases exhibit biological and antigenic differences. Epidemiol. Infect. 1993, 111, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaram, S.; Boikos, C.; Gelone, D.K.; Gandhi, A. Influenza vaccines: The potential benefits of cell-culture isolation and manufacturing. Ther. Adv. Vaccines Immunother. 2020, 8, 2515135520908121. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, M.A.; Flannery, B.; Chung, J.R.; O’Halloran, A.; Garg, S.; Belongia, E.A.; Gaglani, M.; Zimmerman, R.K.; Jackson, M.L.; Monto, A.S.; et al. Us Influenza Vaccine Effectiveness Network tIHSN, the Assessment Branch ISDCfDC, Prevention. Effects of Influenza Vaccination in the United States During the 2017–2018 Influenza Season. Clin. Infect. Dis. 2019, 69, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Blanton, L.; Elal, A.I.A.; Alabi, N.; Barnes, J.; Biggerstaff, M.; Brammer, L.; Budd, A.P.; Burns, E.; Cummings, C.N.; et al. Update: Influenza Activity in the United States During the 2018–2019 Season and Composition of the 2019–2020 Influenza Vaccine. Morb. Mortal. Wkly. Rep. 2019, 68, 544–551. [Google Scholar] [CrossRef]
- Flannery, B.; Chung, J.R.; Thaker, S.N.; Monto, A.S.; Martin, E.T.; Belongia, E.A.; McLean, H.Q.; Gaglani, M.; Murthy, K.; Zimmerman, R.K.; et al. Interim Estimates of 2016-17 Seasonal Influenza Vaccine Effectiveness—United States, February 2017. Morb. Mortal Wkly. Rep. 2017, 66, 167–171. [Google Scholar] [CrossRef]
- McLean, H.Q.; Thompson, M.G.; Sundaram, M.E.; Kieke, B.A.; Gaglani, M.; Murthy, K.; Piedra, P.A.; Zimmerman, R.K.; Nowalk, M.P.; Raviotta, J.M.; et al. Influenza Vaccine Effectiveness in the United States During 2012–2013: Variable Protection by Age and Virus Type. J. Infect. Dis. 2014, 211, 1529–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disease Burden of Influenza. Available online: https://www.cdc.gov/flu/about/burden/index.html (accessed on 20 June 2021).
- Takada, K.; Kawakami, C.; Fan, S.; Chiba, S.; Zhong, G.; Gu, C.; Shimizu, K.; Takasaki, S.; Sakai-Tagawa, Y.; Lopes, T.J.S.; et al. A humanized MDCK cell line for the efficient isolation and propagation of human influenza viruses. Nat. Microbiol. 2019, 4, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Matrosovich, T.; Carr, J.; Roberts, N.A.; Klenk, H.-D. Overexpression of the α-2,6-Sialyltransferase in MDCK Cells Increases Influenza Virus Sensitivity to Neuraminidase Inhibitors. J. Virol. 2003, 77, 8418–8425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, D.Y.; Barr, I.G.; Mosse, J.A.; Laurie, K.L. MDCK-SIAT1 Cells Show Improved Isolation Rates for Recent Human Influenza Viruses Compared to Conventional MDCK Cells. J. Clin. Microbiol. 2008, 46, 2189–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-C.; Kappes, M.A.; Chen, M.-C.; Lin, C.-C.; Wang, T.T. Distinct susceptibility and applicability of MDCK derivatives for influenza virus research. PLoS ONE 2017, 12, e0172299. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza. World Health Organization. 2011. Available online: https://apps.who.int/iris/bitstream/handle/10665/44518/9789241548090_eng.pdf (accessed on 24 June 2021).
- Barnett, J.L.; Yang, J.; Cai, Z.; Zhang, T.; Wan, X.-F. AntigenMap 3D: An online antigenic cartography resource. Bioinformatics 2012, 28, 1292–1293. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Zhang, T.; Wan, X.-F. A Computational Framework for Influenza Antigenic Cartography. PLoS Comput. Biol. 2010, 6, e1000949. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, T.; Wan, X.-F. Concepts and applications for influenza antigenic cartography. Influ. Other Respir. Viruses 2011, 5, 204–207. [Google Scholar] [PubMed]
- Hoffmann, E.; Mahmood, K.; Yang, C.-F.; Webster, R.G.; Greenberg, H.; Kemble, G. Rescue of influenza B virus from eight plasmids. Proc. Natl. Acad. Sci. USA 2002, 99, 11411–11416. [Google Scholar] [CrossRef] [Green Version]
- Langat, P.; Raghwani, J.; Dudas, G.; Bowden, T.; Edwards, S.; Gall, A.; Bedford, T.; Rambaut, A.; Daniels, R.S.; Russell, C.A.; et al. Genome-wide evolutionary dynamics of influenza B viruses on a global scale. PLoS Pathog. 2017, 13, e1006749. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Lin, X.; Wang, W.; Halpin, R.A.; Bera, J.; Stockwell, T.B.; Barr, I.G.; Wentworth, D.E. Universal Influenza B Virus Genomic Amplification Facilitates Sequencing, Diagnostics, and Reverse Genetics. J. Clin. Microbiol. 2014, 52, 1330–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kechin, A.; Boyarskikh, U.; Kel, A.; Filipenko, M. cutPrimers: A New Tool for Accurate Cutting of Primers from Reads of Targeted Next Generation Sequencing. J. Comput. Biol. 2017, 24, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- World Health Organization Regional Office for Europe. WHO Releases Recommendations for the 2019–2020 Northern Hemisphere Seasonal Influenza Vaccine. 2019. Available online: https://www.euro.who.int/en/health-topics/communicable-diseases/influenza/news/news/2019/3/who-releases-recommendations-for-the-2019–2020-northern-hemisphere-seasonal-influenza-vaccine (accessed on 16 July 2021).
- Centers for Disease Control and Prevention. Estimated Influenza Illnesses, Medical Visits, Hospitalizations, and Deaths in the United States—2019–2020 Influenza Season. 2021. Available online: https://www.cdc.gov/flu/about/burden/2019-2020.html (accessed on 20 June 2021).
- Tenforde, M.W.; Kondor, R.J.G.; Chung, J.R.; Zimmerman, R.K.; Nowalk, M.P.; Jackson, M.L.; Jackson, L.A.; Monto, A.S.; Martin, E.T.; Belongia, E.A.; et al. Effect of Antigenic Drift on Influenza Vaccine Effectiveness in the United States—2019–2020. Clin. Infect. Dis. 2020, ciaa1884. [Google Scholar] [CrossRef] [PubMed]
- Dawood, F.S.; Chung, J.R.; Kim, S.S.; Zimmerman, R.K.; Nowalk, M.P.; Jackson, M.L.; Jackson, L.A.; Monto, A.S.; Martin, E.T.; Belongia, E.A. Interim Estimates of 2019–2020 Seasonal Influenza Vaccine Effectiveness—United States, February 2020. Morb. Mortal Wkly. Rep. 2020, 69, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Owusu, D.; Hand, J.; Tenforde, M.W.; Feldstein, L.R.; DaSilva, J.; Barnes, J.; Lee, G.; Tran, J.; Sokol, T.; Fry, A.M.; et al. Early Season Pediatric Influenza B/Victoria Virus Infections Associated with a Recently Emerged Virus Subclade—Louisiana, 2019. Morb. Mortal Wkly. Rep. 2020, 69, 40–43. [Google Scholar] [CrossRef]
- Xie, H.; Xiang, R.; Wan, H.J.; Plant, E.P.; Radvak, P.; Kosikova, M.; Li, X.; Zoueva, O.; Ye, Z.; Wan, X.F. Reduced Influenza B–Specific Postvaccination Antibody Cross-reactivity in the B/Victoria Lineage–Predominant 2019/20 Season. Clin. Infect. Dis. 2021, 72, e776–e783. [Google Scholar] [CrossRef]
- Hu, W.; Gruner, W.E.; DeMarcus, L.S.; Thervil, J.W.; Kwaah, B.; Fries, A.C.; Sjoberg, A.P.; Robbins, A.S. Influenza Surveillance Trends and Influenza Vaccine Effectiveness Among Department of Defense Beneficiaries During the 2019–2020 Influenza Season. MSMR 2021, 28, 2–8. [Google Scholar] [PubMed]
- McAuley, J.; Zhang, K.; McCullers, J.A. The Effects of Influenza A Virus PB1-F2 Protein on Polymerase Activity Are Strain Specific and Do Not Impact Pathogenesis. J. Virol. 2010, 84, 558–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-M.; Chen, G.-W.; Velu, A.B.; Dash, S.; Han, Y.-J.; Tsao, K.-C.; Shih, S.-R. Circulating pattern and genomic characteristics of influenza B viruses in Taiwan from 2003 to 2014. J. Formos. Med. Assoc. 2016, 115, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Sage, V.; Kormuth, K.A.; Nturibi, E.; Lee, J.M.; Frizzell, S.A.; Myerburg, M.M.; Bloom, J.D.; Lakdawala, S.S. Cell-Culture Adaptation of H3N2 Influenza Virus Impacts Acid Stability and Reduces Airborne Transmission in Ferret Model. Viruses 2021, 13, 719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cheng, F.; Lu, M.; Tian, X.; Ma, J. Crystal Structure of Unliganded Influenza B Virus Hemagglutinin. J. Virol. 2008, 82, 3011–3020. [Google Scholar] [CrossRef] [Green Version]
- Gatherer, D. Passage in egg culture is a major cause of apparent positive selection in influenza B hemagglutinin. J. Med. Virol. 2009, 82, 123–127. [Google Scholar] [CrossRef]
- Zost, S.; Parkhouse, K.; Gumina, M.; Kim, K.; Perez, S.D.; Wilson, P.C.; Treanor, J.J.; Sant, A.J.; Cobey, S.; Hensley, S.E. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 2017, 114, 12578–12583. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.W.; Kim, Y.H.; Jung, H.U.; Jeong, O.S.; Hong, E.J.; Kim, H.; Lee, J.I. Comparison of antigenic mutation during egg and cell passage cultivation of H3N2 influenza virus. Clin. Exp. Vaccine Res. 2020, 9, 56–63. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Samples | CCL34 P1 | CCL34 P2 | SIAT1 P1 | hCK P1 |
|---|---|---|---|---|---|
| IAV (H1) | 108 | 39 (42.12%) | 76 (70.37%) | 77 (71.30%) | 95 (87.96%) |
| IBV | 38 | 22 (57.89%) | 24 (63.16%) | 22 (57.89%) | 31 (81.58%) |
| Total | 146 | 61 (41.78%) | 100 (68.49%) | 99 (67.80%) | 127 (86.98%) |
| Sample | Protein | Position | Clinical Swab (Nucleotide) | Isolate (Nucleotide) | Amino Acid e | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Swab (Proportion Reads) a | Reads (Total) b | Reads (Position) c | % Reads at Position d | Isolate (Proportion Reads) a | Reads (Total) b | Reads (Position) c | % Reads at Position d | Position | Swab | Isolate | |||
| CS20N017 | PB2 | 136 | A (0.99)/T (0.01) | 1,574,718 | 26224 | 1.67 | T (0.99) | 1,149,216 | 17,538 | 1.53 | 46 | T | S |
| PB2 | 2181 | T (0.98)/C (0.02) | 1,574,718 | 7075 | 0.45 | C (0.99)/T (0.01) | 1,149,216 | 54,788 | 4.77 | 727 | N | N | |
| PB1 | 2160 | G (1.00) | 1,574,718 | 47919 | 3.04 | A (1.00) | 1,149,216 | 43,524 | 3.79 | 720 | R | R | |
| CS20N025 | HA | 1741 | C (0.79)/T (0.16)/N (0.05) | 1,588,004 | 45 | 0.00 | T (1.00) | 182,972 | 14,112 | 7.71 | 581 | R | C |
| CS20N036 | PB1 | 2016 | T (0.97)/A (0.03) | 1,940,088 | 39 | 0.00 | C (0.50)/T (0.46)/A (0.04) | 1,468,024 | 42,152 | 2.87 | 672 | S | S |
| PB1 | 2020 | T (1.00) | 1,940,088 | 41 | 0.00 | A (0.53) | 1,468,024 | 43,968 | 3.00 | 674 | L | Q | |
| CS20N091 | NP | 419 | A (0.96)/N (0.03)/T(0.01) | 2,161,170 | 1615 | 0.07 | C (0.76)/A (0.24) | 177,412 | 4283 | 2.41 | 140 | E | A |
| CS20N137 | PB2 | 293 | T (0.96)/N (0.03/A(0.01) | 2,078,362 | 336 | 0.02 | G (0.57)/T (0.43) | 216,996 | 514 | 0.24 | 98 | V | G |
| CS20N140 | PB2 | 2097 | T(0.55)/C(0.44) | 1,939,620 | 5992 | 0.31 | C (0.98)/T (0.02) | 1,057,874 | 76,928 | 7.27 | 699 | L | L |
| CS20N212 | PB1 | 1048 | A (0.96)/N (0.03)/C (0.01) | 1,927,990 | 230 | 0.01 | G (0.62)/A (0.38) | 142,724 | 148 | 0.10 | 350 | R | G |
| PB1 | 1502 | T (0.94)/G (0.02)/N (0.02)/A(0.01)/C (0.01) | 1,927,990 | 238 | 0.01 | C (0.69)/T (0.31) | 142,724 | 580 | 0.41 | 501 | V | A | |
| CS20N214 | PB1 | 255 | A (0.77)/G (0.13)/N (0.08)/C (0.01)/T (0.01) | 1,779,338 | 1811 | 0.10 | G (0.56)/A (0.43)/T (0.01) | 180,974 | 59,503 | 32.88 | 85 | L | L |
| CS20N294 | PB1 | 335 | A (0.99)/C (0.01) | 1,495,964 | 650 | 0.04 | G (0.56)/A (0.43) | 1,599,890 | 12,662 | 0.79 | 112 | E | G |
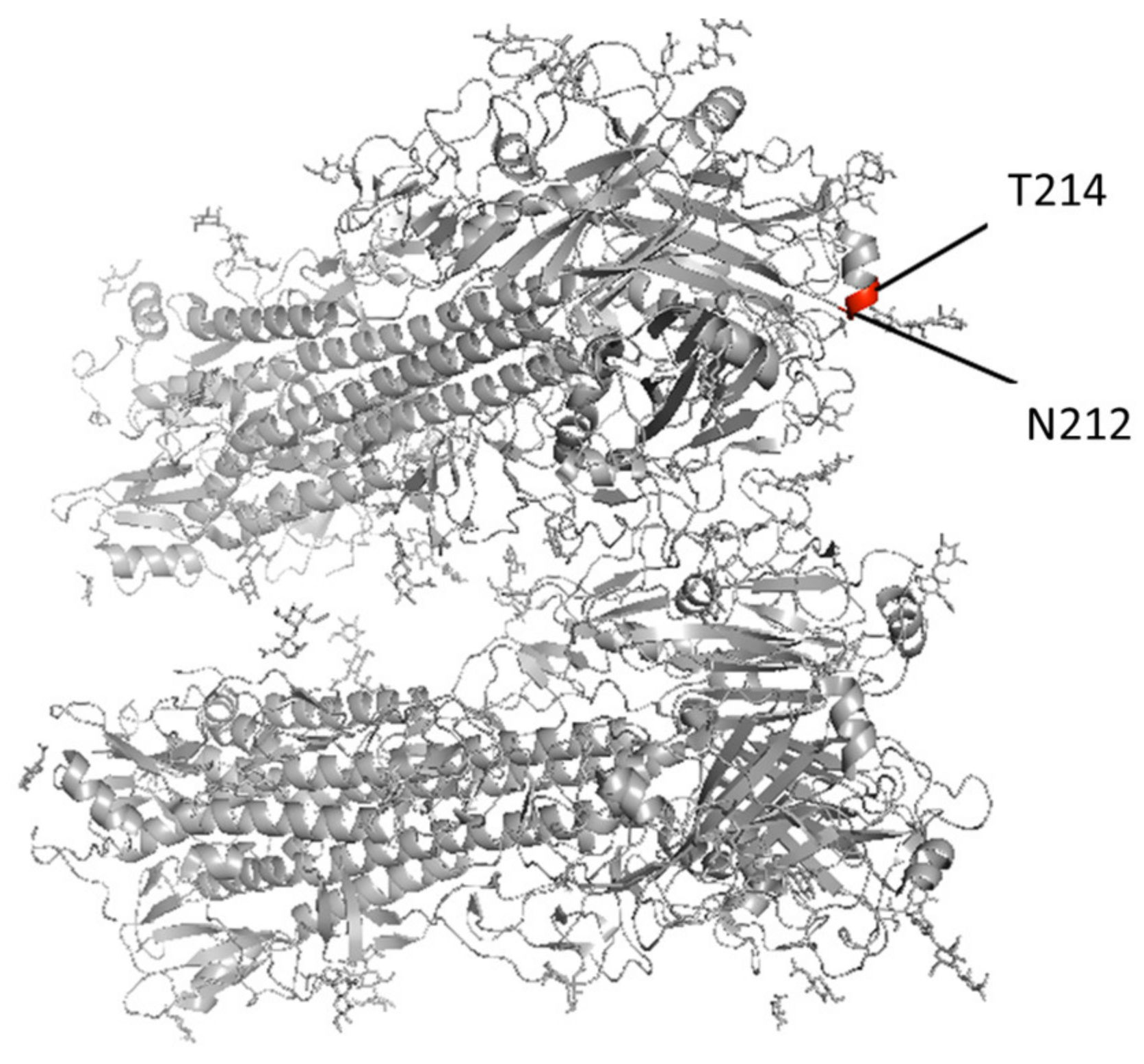
| Protein | Strain | Position | Original | Cell | Egg |
|---|---|---|---|---|---|
| HA | B/North Carolina/17/2019/EPI1693897 | 143 | E | K | |
| B/Florida/78/2015/EPI721064 | 156 | G | G | R | |
| B/Mozambique/413/2016/EPI854592 | 179 | D | Y | ||
| B/Pennsylvania/19/2016/EPI807677 | 212 | N | N | K | |
| B/Michigan/18/2016/EPI807685 | 212 | N | N | T | |
| B/Indiana/08/2016/EPI807669 | 212 | N | N | D | |
| B/Florida/94/2016/EPI872715 | 212 | N | N | S | |
| B/Hawaii/26/2018/EPI1315023 | 212 | N | D | ||
| B/Florida/79/2015/EPI745107 | 212 | N | Y | ||
| B/Utah/18/2018/EPI1249578 | 212 | N | T | ||
| B/Montana/34/2018/EPI1386557 | 212 | N | D | ||
| B/Iowa/14/2017/EPI1025486 | 212 | T | T | N | |
| B/Guatemala/35/2018/EPI1249639 | 212 | N | S | ||
| B/Guatemala/109/2018/EPI1312932 | 212 | N | S | ||
| B/Florida/97/2017/EPI1138021 | 212 | N | K | ||
| B/Alabama/02/2017/EPI980739 | 212 | N | N | S | |
| B/Lebanon/16/2020/EPI1840705 | 212 | N | N | S | |
| B/Pennsylvania/60/2016/EPI892343 | 214 | T | I | ||
| B/Hawaii/55/2017/EPI1089741 | 214 | T | N | ||
| B/Costa Rica/5521/2016/EPI908149 | 214 | T | A | ||
| B/El Salvador/697/2018/EPI1357148 | 214 | T | A | ||
| B/Louisiana/16/2019/EPI1574236 | 214 | T | T | N | |
| B/Hong Kong/269/2017/EPI1141721 | 214 | T | T | I | |
| B/Minnesota/39/2017/EPI1026300 | 236 | I | T | ||
| B/Ulaabaatar/1868/2017/EPI1089764 | 433 | N | D | ||
| B/Ulaabaatar/1834/2017/EPI1053102 | 433 | D | N | ||
| B/Ulaabaatar/1767/2017/EPI1053094 | 433 | N | D | ||
| B/Wisconsin/22/2018/EPI1312153 | 528 | D | G | ||
| NA | B/North Carolina/17/2019/EPI1693896 | 19 | L | I | |
| B/North Carolina/16/2019/EPI1664425 | 19 | I | L | ||
| B/Wisconsin/45/2016/EPI834520 | 154 | L | K | ||
| B/North Carolina/17/2019/EPI1693896 | 284 | S | G | ||
| B/North Carolina/16/2019/EPI1664425 | 284 | G | S | ||
| B/Ulaabaatar/1868/2017/EPI1089763 | 310 | T | S | ||
| B/Nevada/23/2019/EPI1688225 | 321 | D | N | ||
| B/Colombia/9962/2016/EPI908031 | 391 | D | E | ||
| B/Bangladesh/8018/2016/EPI880659 | 395 | A | D | ||
| B/North Carolina/17/2019/EPI1693896 | 400 | V | I | ||
| B/Arizona/83/2017/EPI1147826 | 401 | M | V | ||
| B/California/18/2017/EPI1009716 | 435 | G | R | ||
| B/Guyane/310/2020/EPI1807176 | 450 | M | M | V | |
| NB | B/North Carolina/17/2019/EPI1693896 | 21 | I | N | |
| B/North Carolina/16/2019/EPI1664425 | 21 | N | I | ||
| NEP | B/Oregon/11/2020/EPI1840666 | 30 | S | S | L |
| B/Iowa/14/2017/EPI1011596 | 79 | D | D | N | |
| NP | B/Bangladesh/5006/2017/EPI1094384 | 33 | R | K | |
| B/Ulaabaatar/1868/2017/EPI1089757 | 239 | A | T | ||
| NS1 | B/North Carolina/17/2019/EPI1693891 | 62 | T | A | |
| B/North Carolina/16/2019/EPI1664420 | 62 | A | T | ||
| B/Ulaabaatar/1868/2017/EPI1089758 | 115 | T | M | ||
| B/Ulaabaatar/1834/2017/EPI1053096 | 115 | M | T | ||
| B/Ulaabaatar/1767/2017/EPI1089797 | 115 | T | M | ||
| PA | B/Bangladesh/8018/2016/EPI880656 | 57 | E | K | |
| B/New Jersey/09/2017/EPI1011540 | 393 | I | I | T | |
| PB1 | B/North Carolina/17/2019/EPI1693895 | 378 | L | S | |
| B/North Carolina/16/2019/EPI1664424 | 378 | S | L | ||
| PB2 | B/Florida/97/2017/EPI1138018 | 263 | V | I | |
| B/Macedonia/421/2020/EPI1806732 | 379 | S | S | C | |
| B/Bangladesh/5006/2017/EPI1094388 | 391 | A | T |
| Virus a | Clade b | Ferret Reference Sera c | ||||||
|---|---|---|---|---|---|---|---|---|
| B/Nevada/ 03/2011 | B/Texas/ 02/2013 | B/Brisbane/ 60/2008 | B/Colorado/ 6/2017 | B/Washington/ 02/2019 (Egg) | B/Washington/ 02/2019 (Cell) | B/Wisconsin/1/2010 | ||
| B/Nevada/03/2011 | V1A | 1280 | 320 | 640 | 20 | 160 | 80 | 10 |
| B/New Jersey/01/2012 | V1A | 1280 | 640 | 320 | 40 | 40 | 40 | <10 |
| B/Texas/02/2013 | V1A | 320 | 320 | 160 | 40 | 40 | 40 | <10 |
| B/Florida/78/2015 | V1A | 1280 | 1280 | 640 | 80 | 80 | 80 | 20 |
| B/Brisbane/60/2018 | V1A | 640 | 320 | 1280 | 40 | 160 | 40 | 10 |
| B/Colorado/06/2017 | V1A-2DEL | 40 | 20 | 0 | 320 | 40 | 160 | <10 |
| B/Hong Kong/286/2017 | V1A.3-3DEL | 320 | 160 | 160 | 80 | 320 | 160 | 10 |
| B/Washington/02/2019 (egg) | V1A.3-3DEL | 320 | 80 | 80 | 80 | 640 | 640 | <10 |
| B/Washington/02/2019 (cell) | V1A.3-3DEL | 80 | 40 | 10 | 80 | 320 | 640 | <10 |
| B/Rhode Island/01/2019 | V1A.3-3DEL | 320 | 160 | 320 | 40 | 320 | 320 | 10 |
| B/Phuket/3073/2013 | Y3 | 10 | <10 | <10 | <10 | <10 | <10 | 1280 |
| CS20N004 | V1A.3-3DEL | 80 | 40 | 10 | 160 | 320 | 640 | <10 |
| CS20N005 | 80 | 40 | 10 | 160 | 320 | 640 | <10 | |
| CS20N006 | 80 | 80 | 20 | 160 | 640 | 1280 | <10 | |
| CS20N017 | 80 | 40 | 10 | 160 | 320 | 640 | <10 | |
| CS20N020 | 80 | 40 | 10 | 160 | 640 | 1280 | <10 | |
| CS20N025 | 80 | 40 | 20 | 160 | 640 | 1280 | 10 | |
| CS20N028 | 80 | 40 | 10 | 160 | 320 | 640 | <10 | |
| CS20N029 | 80 | 40 | 20 | 160 | 640 | 1280 | <10 | |
| CS20N031 | 80 | 40 | 10 | 160 | 320 | 640 | <10 | |
| CS20N036 | 80 | 40 | 20 | 160 | 640 | 1280 | 10 | |
| CS20N053 | 80 | 40 | 10 | 160 | 640 | 1280 | <10 | |
| CS20N056 | 80 | 40 | 20 | 160 | 640 | 1280 | <10 | |
| CS20N057 | 40 | 20 | 10 | 80 | 320 | 640 | <10 | |
| CS20N062 | 40 | 40 | 10 | 160 | 640 | 1280 | <10 | |
| CS20N068 | 40 | 20 | 10 | 80 | 640 | 1280 | <10 | |
| CS20N091 | 80 | 40 | 10 | 160 | 320 | 640 | <10 | |
| CS20N098 | 40 | 20 | 10 | 80 | 320 | 640 | <10 | |
| CS20N113 | 40 | 20 | 10 | 160 | 320 | 640 | <10 | |
| CS20N126 | 40 | 20 | 10 | 80 | 320 | 640 | <10 | |
| CS20N137 | 80 | 20 | 10 | 160 | 320 | 640 | <10 | |
| CS20N140 | 80 | 20 | 10 | 160 | 320 | 640 | <10 | |
| CS20N168 | 80 | 40 | 20 | 160 | 640 | 1280 | <10 | |
| CS20N174 | 80 | 40 | 10 | 160 | 320 | 640 | <10 | |
| CS20N210 | 80 | 40 | 20 | 160 | 320 | 640 | 10 | |
| CS20N212 | 80 | 40 | 20 | 160 | 640 | 1280 | <10 | |
| CS20N213 | 80 | 40 | 20 | 160 | 320 | 640 | <10 | |
| CS20N214 | 80 | 40 | 20 | 160 | 320 | 640 | <10 | |
| CS20N228 | 80 | 40 | 20 | 160 | 640 | 1280 | 10 | |
| CS20N274 | 80 | 40 | 10 | 160 | 320 | 640 | 10 | |
| CS20N294 | 80 | 40 | 10 | 80 | 320 | 640 | <10 | |
| CS20N349 | 40 | 20 | 10 | 80 | 320 | 640 | <10 | |
| CS20N351 | 80 | 40 | 10 | 160 | 640 | 1280 | <10 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, C.Y.; Segovia, K.; McElroy, J.A.; Li, T.; Guan, M.; Zhang, X.; Misra, S.; Hang, J.; Wan, X.-F. Cell-Adapted Mutations and Antigenic Diversity of Influenza B Viruses in Missouri, 2019–2020 Season. Viruses 2021, 13, 1896. https://doi.org/10.3390/v13101896
Tang CY, Segovia K, McElroy JA, Li T, Guan M, Zhang X, Misra S, Hang J, Wan X-F. Cell-Adapted Mutations and Antigenic Diversity of Influenza B Viruses in Missouri, 2019–2020 Season. Viruses. 2021; 13(10):1896. https://doi.org/10.3390/v13101896
Chicago/Turabian StyleTang, Cynthia Y., Karen Segovia, Jane A. McElroy, Tao Li, Minhui Guan, Xiaojian Zhang, Shamita Misra, Jun Hang, and Xiu-Feng Wan. 2021. "Cell-Adapted Mutations and Antigenic Diversity of Influenza B Viruses in Missouri, 2019–2020 Season" Viruses 13, no. 10: 1896. https://doi.org/10.3390/v13101896