 Samantha L. Tucker
Samantha L. Tucker Demba Sarr
Demba Sarr Balázs Rada*
Balázs Rada*- Department of Infectious Diseases, College of Veterinary Medicine, The University of Georgia, Athens, GA, United States
Cystic Fibrosis (CF) is a genetic disease that causes chronic and severe lung inflammation and infection associated with high rates of mortality. In CF, disrupted ion exchange in the epithelium results in excessive mucus production and reduced mucociliary clearance, leading to immune system exacerbation and chronic infections with pathogens such as P. aeruginosa and S. aureus. Constant immune stimulation leads to altered immune responses including T cell impairment and neutrophil dysfunction. Specifically, CF is considered a Th17-mediated disease, and it has been proposed that both P. aeruginosa and a subset of neutrophils known as granulocytic myeloid suppressor cells (gMDSCs) play a role in T cell suppression. The exact mechanisms behind these interactions are yet to be determined, but recent works demonstrate a role for arginase-1. It is also believed that P. aeruginosa drives gMDSC function as a means of immune evasion, leading to chronic infection. Herein, we review the current literature regarding immune suppression in CF by gMDSCs with an emphasis on T cell impairment and the role of P. aeruginosa in this dynamic interaction.
Neutrophil Dysfunction in CF
Cystic Fibrosis (CF) is an autosomal recessive disease caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene (1–3). CF is primarily found in the Caucasian population, with an estimated 70,000 individuals affected by the disease (1, 4). Disruption in CFTR function leads to ion dysregulation, abnormal pH, mucus build-up, chronic inflammation, and infection with pathogens such as Pseudomonas aeruginosa and Staphylococcus aureus. Symptoms in the lungs cause most of the morbidity and mortality in CF (4–13). Neutrophils are major drivers of chronic inflammation in CF airways (14–18). Neutrophils in general are inefficient at pathogen clearance in CF (19–24). In CF and other diseases, it is becoming more evident that different subpopulations of neutrophils exist that may be linked to varying forms of immune dysfunctions (25–30). An increasing body of work exists demonstrating the negative impact of neutrophils on lung disease outcome in CF. Excessive neutrophil recruitment to the lungs leads to increased levels of inflammatory cytokines such as IL-1β, IL-8, IL-17 and IL-6 (6, 31, 32). As neutrophils become activated, damaging granule components such as neutrophil elastase (NE) and metalloproteinase 9 (MMP9) are released into the extracellular space, resulting in perpetuated tissue injury and immune cell recruitment (5, 33, 34). NE has been described to inhibit the function of other cells found in the CF airways (epithelium, macrophages, dendritic cells) and represents a clinically highly relevant target for the pharmaceutical industry (35–39). CF sputum PMN counts, levels of ecDNA, myeloperoxidase (MPO), NE and PMN chemoattractants all correlate with CF lung disease severity (2–6). Phenotypic changes to neutrophils also occur upon entry into the CF airway environment including reduction in surface expression of the phagocytic markers CD16, CD14, and CD35, as well as increased surface expression of activation and degranulation markers CD66b and CD63 (40, 41). Additionally, changes in antigen presentation markers such as CD80, MHCII, and CD294 indicate that CF airway neutrophils potentially interact with T cells (40, 41).
Despite increased neutrophil recruitment to the CF airways, chronic infections with CF-related pathogens such as P. aeruginosa and Staphylococcus aureus suggest impairment of neutrophil-mediated killing of these pathogens (19, 21–23, 42). Exacerbated release of neutrophil extracellular traps (NETs) in CF airways (19, 43, 44), as well as increased NET formation in response to clinical isolates of P. aeruginosa from CF patients have been observed (17, 19, 20, 45–47). Another study demonstrated increased TLR5 surface expression on CF airway neutrophils compared to CF blood neutrophils and blood and airway neutrophils from healthy and non-CF bronchiectasis donors (48). This work further demonstrated that incubation of blood neutrophils in CF sputum supernatant increased TLR5 surface expression (48). It was previously shown that NETs represent a main mechanism of P. aeruginosa killing by neutrophils in in vitro suspension co-cultures (19). Mucoid P. aeruginosa was shown to be resistant to neutrophil-mediated killing (19, 46). Overall, these data suggest that antimicrobial effector functions of neutrophils are impaired in CF that could be due to enhanced immunosuppressive functions of the cells.
MDSCs
Immunosuppressive myeloid cells have been first described about three decades ago. While several names were proposed, in 2007 the term ‘Myeloid-derived suppressor cells (MDSCs)’ was coined to identify monocytes and neutrophils with powerful immunosuppressive features (49, 50). MDSCs have been mainly linked to pathological conditions in cancer, inflammation and autoimmune disease and their physiological roles have also been described (51). In general, two types of MDSCs have been distinguished: monocytic MDSCs (mMDSCs, M-MDSCs) and granulocytic/polymorphonuclear MDSCs (gMDSCs, also abbreviated as G-MDSCs or PMN-MDSCs) (51). There are several reviews that summarize current knowledge on MDSCs and their detailed role in diseases (51). The purpose of this review is to provide a brief summary and introduction to MDSCs and to specifically summarize the proposed roles of gMDSCs in CF only (Figure 1). Even though MDSCs have been studied for years, their origin and development remain largely unclear (51). A consensus among scientists exists related to the development of MDSCs from myeloid cells that are in an immature state (51). MDSCs are primarily defined by their immunosuppressive function and myeloid origin, and do not represent a well-defined, single cell subset (51). This is also reflected by the fact that cell surface markers specific to MDSCs that have been widely accepted by the scientific community have not been identified yet.
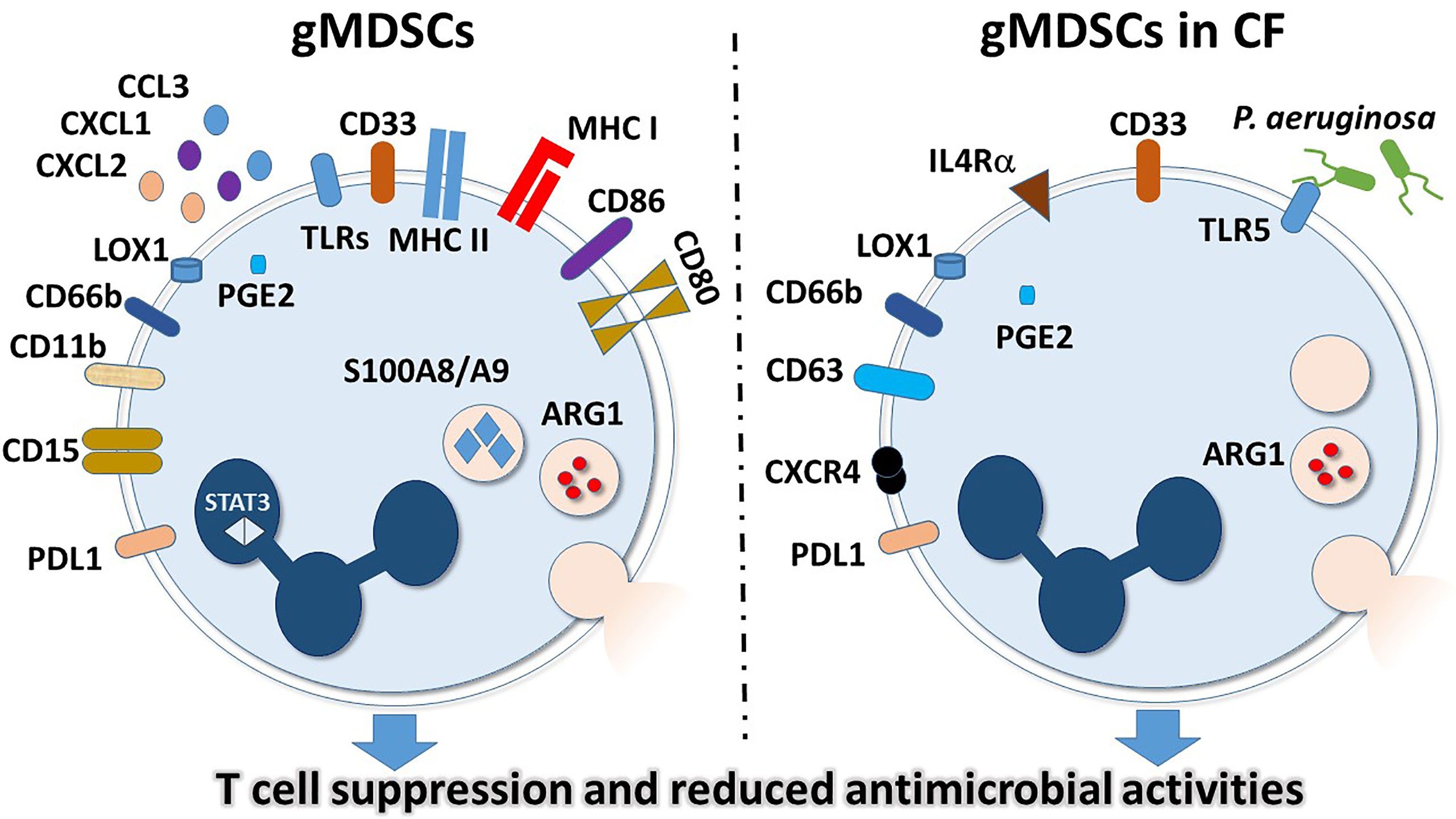
Figure 1 General and CF-specific features of gMDSCs. Surface markers and intracellular molecules are indicated that have higher expressions or activities in granulocytic MDSCs in general (left panel) or in gMDSCs in CF (right panel).
Neutrophils were originally thought to be terminally differentiated, proinflammatory cells, only responsible for and capable of pathogen elimination. However, it has recently become apparent that neutrophils represent a heterogeneous population that differ in maturity, density and inflammatory properties (25–28, 30, 52). The heterogeneity of neutrophils was first discovered in cancer patients, where a portion of neutrophils co-purified in the mononuclear cell fraction during peripheral blood cell isolation (53). In this study, it was determined that these lower density neutrophils (LDNs) were activated, less dense than normal neutrophils and capable of suppressing T cell signaling in a hydrogen peroxide-dependent manner (53). Because of the suppressor phenotype of these LDNs, the term granulocytic myeloid-derived suppressor cells (gMDSCs, also called PMN-MDSCs) was coined (53). gMDSCs describe a subset of myeloid cells expressing neutrophilic markers and are characterized by an immunosuppressive phenotype. This is in contrast to monocyte-derived MDSCs, which have similar functions, but stem from a different, monocytic lineage (51). Additional works demonstrating TGF-β-modulated polarization of protumor and anti-tumor neutrophils added to the clear presence of multiple neutrophil types (26). Specifically, blockage of TGF-β signaling resulted in increased cytotoxic, hypersegmented anti-tumor neutrophils (N1), whereas the presence of TGF-β resulted in less aggressive protumor neutrophils (N2) (26). Transcriptomic analyses in mice examined normal neutrophils from healthy animals, tumor-associated neutrophils (TANs) and splenic gMDSCs from cancer-positive animals distinguished the phenotypic differences of these cells (27). It was shown that while normal neutrophils and gMDSCs likely come from the same progenitors, they have very different mRNA profiles. Specifically, gMDSCs are primed for antigen presentation and highly express MHC class I and II as well as the co-stimulatory molecules CD80 and CD86 (27) (Figure 1). Enhanced antigen-presenting capacity is in line with data demonstrating gMDSC interactions with T cells (53). Additional changes in gMDSCs included increased expressions of TLRs and BCL-2-related apoptotic genes (27). Lastly, the expressions of neutrophil chemoattractants CXCL1, CXCL2 and CCL3 were markedly higher in gMDSCs compared to normal neutrophils (27). These studies highlight the phenotypic variability that occurs among neutrophils within an individual (Figure 1).
In the past decade, multiple studies have come out addressing additional differences between normal neutrophils and gMDSCs, and brought more questions than answers. For example, while gMDSCs were originally considered immunosuppressive LDNs and co-purified by density centrifugation with mononuclear cells in cancer patients, other LDNs have been found in autoimmune diseases such as systemic lupus erythematosus (SLE), are known to be hyper-inflammatory and cause vascular damage (25, 52, 54). It has recently been determined that the presence or absence of CD10 determines the maturity status of LDNs, and can distinguish between mature cells (CD10+) which have an immunosuppressive phenotype, and immature cells (CD10-) with an immune-stimulatory phenotype (28, 54). Maturation resulting in CD10 expression and immune suppression appears to be driven by G-CSF (28).
Numerous reviews exist describing the current literature available on the heterogeneity of neutrophils as well as gMDSCs and LDGs (29, 30, 51). The currently accepted characterization for gMDSCs isolated from human peripheral blood describes these cells as low-density neutrophils expressing CD11b, CD15, CD66b, LOX-1, and lacking CD14 (51, 53, 55). In mice, gMDSCs are defined as CD11b+ Ly6G+ while mMDCS are CD11b+ Ly6C+, the same way by which mature neutrophils and monocytes are determined. Additionally, gMDSCs suppress T cell proliferation as a functional marker, and have very high reactive oxygen species (ROS), ARG-1, PGE2, S100A8/A9, and STAT3 activities, as well as high levels of ER stress (51, 55, 56). Differences in signaling including strength, duration, and major pathway play pivotal roles in the abundance and function of gMDSCs within an individual. It is evident that chronic conditions such as cancer, pregnancy, obesity, or persistent infection lead to a sustained, low level immune response (52). This constant, weak stimulation results in the increased presence of gMDSCs that have reduced phagocytosis, increased ROS production, and are capable of suppressing T cells (17, 18, 51, 55, 57–59). Therefore, in comparison to normal neutrophils, gMDSCs seem to be reducing inflammation and cease the perpetual signaling that results from chronic immune stimulation (Figure 1).
gMDSCs in Cystic Fibrosis
It remains unclear in chronic diseases whether gMDSCs are only generated in the bone marrow alongside normal neutrophils, or if normal neutrophils can also develop into gMDSCs or gMDSC-like cells at the site of inflammation. One study demonstrated that ER stress leading to the upregulation of LOX-1 expression resulted in neutrophils with gene expression patterns and suppressive capabilities similar to gMDSCs (55). These data suggest that suppressive actions of neutrophils are possibly inducible; however, this has not been examined in CF. In the case of CF, reports of gMDSCs are conflicting with regards to phenotypic differences between normal neutrophils, gMDSCs from peripheral blood, and neutrophils isolated from the lungs (16–18).
Although the initial cause of CF is genetic, the symptoms of reduced ASL and excessive mucus production ultimately result in immune cell recruitment, tissue damage and perpetuated inflammation, which is further exacerbated by chronic bacterial and fungal infections (1, 13, 60, 61). As previously mentioned, neutrophils are abundantly present in CF airways, but fail to clear certain pathogens. This leads to the hypothesis that neutrophils are playing an alternate, immunosuppressive role in the CF airways. Given that P. aeruginosa induces T cell suppression as well as TLR5 expression in neutrophils, it was initially proposed that P. aeruginosa induces gMDSC production in CF as a means to evade the T cell immune response (16). To this end, it was demonstrated that gMDSCs, identified as CD33high/CD66bhigh/IL-4Rαinter/HLA-DRdim populations in the PBMC fraction were higher in CF patients compared to healthy controls (16). More importantly, gMDSCs in the PBMC fraction were higher in P. aeruginosa-positive individuals compared to P. aeruginosa-negative individuals (16). Additionally, while there was no correlation between blood gMDSCs and lung function of P. aeruginosa-negative individuals, the number of gMDSCs positively correlated with lung function of P. aeruginosa-positive individuals (16). It was further demonstrated that in vitro incubation of PBMCs with P. aeruginosa, or its flagellin alone induces gMDSCs that highly express TLR5 and CXCR4 in a CFTR-independent manner (16). Lastly, this report demonstrated that both CF-gMDSCs and in vitro P.aeruginosa-induced gMDSCs suppress CD4+ and CD8+ T cell proliferation, as well as IL-17 secretion (16). This study was the first to link gMDSCs to CF disease pathogenesis, and to suggest that in response to prolonged inflammation and infection, gMDSCs may be playing an anti-inflammatory role of reducing T cell proliferation, recruitment of other proinflammatory cells and tissue damage in response to P. aeruginosa infection (16) (Figure 1).
The report by Rieber et al. defined a function for gMDSCs circulating in the blood; however, to truly understand the role of these cells in CF, it is imperative to assess samples from the lungs and broncheoalveolar lavage fluid (BAL) (16). It has been demonstrated that gMDSCs can suppress T cells through the actions of Programmed Death Ligand 1 (PD-L1), arginase-1 (Arg-1) and ROS (53, 55, 57, 59, 62–66). PD-L1-mediated suppression, by interaction with PD-1, results in activated T cell exhaustion and blockade of secondary signals for activation (57, 59, 66). Arg-1 suppresses T cells by competitively binding arginine and generating L-ornithine (62). The lack of arginine prevents the expression of the ζ-chain of the T cell receptor (TCR) complex and therefore inhibits T cell function (62). To determine the mechanism of T cell suppression by CF-gMDSCs, a study was performed measuring PD-L1 and Arg-1 in both the blood and airways of CF patients (17). Here it was shown that mature airway neutrophils, defined as CD66b+/CD63+/CXCR4+/CD62Llo suppress T cell proliferation through the action of Arg-1, but not PD-L1 (17). Specifically, Arg-1 activity was shown to be higher in CF airway neutrophils compared to healthy controls. Additionally, incubation of PBMCs with CF airway supernatant resulted in reduced T cell proliferation that could be inhibited by a combination treatment with excess arginine and arginase inhibitor, but not by blockage of PD-L1 (17). Lastly, Arg-1 activity positively correlated with total airway neutrophils and negatively correlated with lung function (17). Interestingly, a more recent study determined that mMDSCs isolated from CF patients, characterized as CD14+ cells inhibited T cells in a PD-L1-dependent manner (67), suggesting that additional mechanisms exist for immune disruption in CF. Although this work clearly demonstrated the suppressive capabilities of CF airway neutrophils, it did not definitively conclude that these cells represent airway gMDSCs. That being said, the population isolated had many features of gMDSCs from peripheral blood, including CXCR4 expression, Arg-1 activity, and T cell suppression, suggesting the presence of gMDSCs or gMDSC-like cells in the CF airways (17) (Figure 1).
The data available on gMDSCs in CF airway disease suggest contributions from both host-driven responses as well as P. aeruginosa-mediated responses (16, 17, 40). To further investigate the impact of gMDSCs in the CF airway in regards to P. aeruginosa infection, animal studies using cftr-deficient mice were performed (18). The number and percent of gMDSCs, defined as CD11b+/Ly6Cinter/Ly6Ghigh cells as well as that of mMDSCs CD11b+/Ly6Chigh/Ly6G- were measured in the BAL, lungs, bone marrow, and spleens of cftr-deficient mice with or without P. aeruginosa infection (18). It was shown that P. aeruginosa infection recruits gMDSCs capable of T cell suppression to the lungs and BAL of cftr-deficient mice (18). In contrast, more gMDSCs were present in the bone marrow of uninfected mice, compared to infected animals (18). While a similar trend was noted for mMDSCs in the lung, they were present at a much lower percent compared to gMDSCs (18). To further understand the role of P. aeruginosa in gMDSCs’ suppressor activity, gMDSCs were isolated from the lung, spleen, and bone marrow of infected wild-type mice and co-cultured with T cells in vitro. This experiment demonstrated that gMDSCs from the lung and bone marrow were both capable of suppressing T cell proliferation (18). Lastly, this paper examined the role of cftr in gMDSC function, and showed a slight impairment of T cell suppression in cftr-/- gMDSCs; however, this impairment only occurred at very high gMDSC to T cell ratios, suggesting that cftr is only minimally involved in T cell suppression by gMDSCs (18, 19). Overall, this study demonstrates that gMDSCs are intrinsic to a CF mouse model, but that P. aeruginosa infection is also involved in gMDSC recruitment to the lungs and T cell suppression (18).
T Cell Function in Cystic Fibrosis
Several reports demonstrate altered T cell responses in CF, with a bias towards Th17 cell production and activity that has been linked to and could be mediated by gMDSCs (14, 15). Th17 cells, IL-17, and other Th17-associated cytokines have been shown to be increased in the BAL of patients with CF (15). The same study reported an association between high IL-17 levels in the BAL and a greater chance of developing P. aeruginosa infection within 2 years’ time (15). A negative correlation between lung function (FEV1%) and the number of Th17 cells in the peripheral blood has also been demonstrated in CF, suggesting that an increased Th17 response is associated with poorer disease outcome (68). Disruption of regulatory T cells (Tregs) has also been reported in CF (14). Specifically, the percent of Tregs compared to other cell populations was significantly lower in the peripheral blood and BAL of CF patients compared to healthy controls and non-CF bronchiectasis controls (14). It was also demonstrated that patients with chronic P. aeruginosa infection had even further reduced amounts of Tregs. These data were confirmed with cftr-/- mouse studies, showing decreased Treg numbers in the spleen and lung, as well as a further reduction in Tregs upon P. aeruginosa infection (14). This Treg disruption was not correlated with any other CF-associated pathogen (14). To further confirm the disruption of Tregs in CF, Hector et al. showed that both CFTR inhibitors, as well as incubation with clinically relevant P. aeruginosa reduced the percent of Tregs in the peripheral blood isolated from healthy individuals, and that Tregs isolated from CF patients were less suppressory than Tregs from healthy donors (14). This reduction in suppression was further enhanced in CF patients with chronic P. aeruginosa infection (14). Finally, this study analyzed Tregs and memory Tregs as a function of age and demonstrated a decline in CF Tregs with age that was increased by chronic P. aeruginosa infection, as well as a reduced capacity for generating memory Tregs in these individuals (14). Taken together, these studies demonstrate an impaired adaptive immune response in CF (Figure 1).
Conclusions
Immune system dysregulation is a driving force in CF disease progression and morbidity. Specifically, neutrophils in the lungs are inefficient killers and contribute to tissue damage, inflammation, and chronic infection. Additionally, gMDSCs or gMDSC-like neutrophils could mediate T cell suppression in CF. Suppression of T cells can result in systemic immune system disruption. This interaction between gMDSCs and T cells is further complicated by P. aeruginosa infection, which enhances the T cell suppressor phenotype of these neutrophils, and may enhance immune evasion by these bacteria (Figure 1). Although additional investigation is needed to fully elucidate how the dynamic relationship between P. aeruginosa, gMDSCs, and T cells impact disease exacerbation in CF; these interactions may serve as therapeutic targets for immune dysregulation. Future research into the impact of gMDSCs on T cells and other immune responses will help to determine the multifunctional capacity of neutrophils in CF as well as other chronic inflammatory and infectious diseases.
Author Contributions
ST conceptualized the idea, wrote and revised the manuscript. DS reviewed the manuscript. BR conceptualized the idea, obtained funding and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NIH grant R01HL136707 (to BR).
Author Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping. Science (1989) 245(4922):1059–65. doi: 10.1126/science.2772657
2. Yoshimura K, Nakamura H, Trapnell BC, Chu CS, Dalemans W, Pavirani A, et al. Expression of the Cystic Fibrosis Transmembrane Conductance Regulator Gene in Cells of non-Epithelial Origin. Nucleic Acids Res (1991) 19(19):5417–23. doi: 10.1093/nar/19.19.5417
3. Trapnell BC, Chu CS, Paakko PK, Banks TC, Yoshimura K, Ferrans VJ, et al. Expression of the Cystic Fibrosis Transmembrane Conductance Regulator Gene in the Respiratory Tract of Normal Individuals and Individuals With Cystic Fibrosis. Proc Natl Acad Sci USA (1991) 88(15):6565–9. doi: 10.1073/pnas.88.15.6565
4. Khan MA, Ali ZS, Sweezey N, Grasemann H, Palaniyar N. Progression of Cystic Fibrosis Lung Disease From Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes (Basel) (2019) 10(3). doi: 10.3390/genes10030183
5. Margaroli C, Garratt LW, Horati H, Dittrich AS, Rosenow T, Montgomery ST, et al. Elastase Exocytosis by Airway Neutrophils Is Associated With Early Lung Damage in Children With Cystic Fibrosis. Am J Respir Crit Care Med (2019) 199(7):873–81. doi: 10.1164/rccm.201803-0442OC
6. Kim JS, Okamoto K, Rubin BK. Pulmonary Function is Negatively Correlated With Sputum Inflammatory Markers and Cough Clearability in Subjects With Cystic Fibrosis But Not Those With Chronic Bronchitis. Chest (2006) 129(5):1148–54. doi: 10.1378/chest.129.5.1148
7. Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, et al. From CFTR Biology Toward Combinatorial Pharmacotherapy: Expanded Classification of Cystic Fibrosis Mutations. Mol Biol Cell (2016) 27(3):424–33. doi: 10.1091/mbc.e14-04-0935
8. Cholon DM, Esther CR Jr, Gentzsch M. Efficacy of Lumacaftor-Ivacaftor for the Treatment of Cystic Fibrosis Patients Homozygous for the F508del-CFTR Mutation. Expert Rev Precis Med Drug Dev (2016) 1(3):235–43. doi: 10.1080/23808993.2016.1175299
9. Condren ME, Bradshaw MD. Ivacaftor: A Novel Gene-Based Therapeutic Approach for Cystic Fibrosis. J Pediatr Pharmacol Ther (2013) 18(1):8–13. doi: 10.5863/1551-6776-18.1.8
10. Singh VK, Schwarzenberg SJ. Pancreatic Insufficiency in Cystic Fibrosis. J Cyst Fibros (2017) 16 Suppl 2:S70–8. doi: 10.1016/j.jcf.2017.06.011
11. Doring G, Flume P, Heijerman H, Elborn JS, Consensus Study G. Treatment of Lung Infection in Patients With Cystic Fibrosis: Current and Future Strategies. J Cyst Fibros (2012) 11(6):461–79. doi: 10.1016/j.jcf.2012.10.004
12. Davis PB. Cystic Fibrosis Since 1938. Am J Respir Crit Care Med (2006) 173(5):475–82. doi: 10.1164/rccm.200505-840OE
13. Cohen TS, Prince A. Cystic Fibrosis: A Mucosal Immunodeficiency Syndrome. Nat Med (2012) 18(4):509–19. doi: 10.1038/nm.2715
14. Hector A, Schafer H, Poschel S, Fischer A, Fritzsching B, Ralhan A, et al. Regulatory T-Cell Impairment in Cystic Fibrosis Patients With Chronic Pseudomonas Infection. Am J Respir Crit Care Med (2015) 191(8):914–23. doi: 10.1164/rccm.201407-1381OC
15. Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, et al. A Th17- and Th2-Skewed Cytokine Profile in Cystic Fibrosis Lungs Represents a Potential Risk Factor for Pseudomonas Aeruginosa Infection. Am J Respir Crit Care Med (2013) 187(6):621–9. doi: 10.1164/rccm.201206-1150OC
16. Rieber N, Brand A, Hector A, Graepler-Mainka U, Ost M, Schafer I, et al. Flagellin Induces Myeloid-Derived Suppressor Cells: Implications for Pseudomonas Aeruginosa Infection in Cystic Fibrosis Lung Disease. J Immunol (2013) 190(3):1276–84. doi: 10.4049/jimmunol.1202144
17. Ingersoll SA, Laval J, Forrest OA, Preininger M, Brown MR, Arafat D, et al. Mature Cystic Fibrosis Airway Neutrophils Suppress T Cell Function: Evidence for a Role of Arginase 1 But Not Programmed Death-Ligand 1. J Immunol (2015) 194(11):5520–8. doi: 10.4049/jimmunol.1500312
18. Oz HH, Zhou B, Voss P, Carevic M, Schroth C, Frey N, et al. Pseudomonas Aeruginosa Airway Infection Recruits and Modulates Neutrophilic Myeloid-Derived Suppressor Cells. Front Cell Infect Microbiol (2016) 6:167. doi: 10.3389/fcimb.2016.00167
19. Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, et al. Neutrophil Extracellular Trap (NET)-Mediated Killing of Pseudomonas Aeruginosa: Evidence of Acquired Resistance Within the CF Airway, Independent of CFTR. PloS One (2011) 6(9):e23637. doi: 10.1371/journal.pone.0023637
20. Yoo DG, Floyd M, Winn M, Moskowitz SM, Rada B. NET Formation Induced by Pseudomonas Aeruginosa Cystic Fibrosis Isolates Measured as Release of Myeloperoxidase-DNA and Neutrophil Elastase-DNA Complexes. Immunol Lett (2014) 160(2):186–94. doi: 10.1016/j.imlet.2014.03.003
21. Rada B. Interactions Between Neutrophils and Pseudomonas Aeruginosa in Cystic Fibrosis. Pathogens (2017) 6(1). doi: 10.3390/pathogens6010010
22. Watt AP, Courtney J, Moore J, Ennis M, Elborn JS. Neutrophil Cell Death, Activation and Bacterial Infection in Cystic Fibrosis. Thorax (2005) 60(8):659–64. doi: 10.1136/thx.2004.038240
23. Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, et al. The Role of Chloride Anion and CFTR in Killing of Pseudomonas Aeruginosa by Normal and CF Neutrophils. J Leukoc Biol (2008) 83(6):1345–53. doi: 10.1189/jlb.0907658
24. Fantone K, Tucker SL, Miller A, Yadav R, Bernardy EE, Fricker R, et al. Cystic Fibrosis Sputum Impairs the Ability of Neutrophils to Kill Staphylococcus Aureus. Pathogens (2021) 10(6). doi: 10.3390/pathogens10060703
25. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A Distinct Subset of Proinflammatory Neutrophils Isolated From Patients With Systemic Lupus Erythematosus Induces Vascular Damage and Synthesizes Type I IFNs. J Immunol (2010) 184(6):3284–97. doi: 10.4049/jimmunol.0902199
26. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: “N1” Versus “N2” TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
27. Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, et al. Transcriptomic Analysis Comparing Tumor-Associated Neutrophils With Granulocytic Myeloid-Derived Suppressor Cells and Normal Neutrophils. PloS One (2012) 7(2):e31524. doi: 10.1371/journal.pone.0031524
28. Marini O, Costa S, Bevilacqua D, Calzetti F, Tamassia N, Spina C, et al. Mature CD10(+) and Immature CD10(-) Neutrophils Present in G-CSF-Treated Donors Display Opposite Effects on T Cells. Blood (2017) 129(10):1343–56. doi: 10.1182/blood-2016-04-713206
29. Christoffersson G, Phillipson M. The Neutrophil: One Cell on Many Missions or Many Cells With Different Agendas? Cell Tissue Res (2018) 371(3):415–23. doi: 10.1007/s00441-017-2780-z
30. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of Neutrophils. Nat Rev Immunol (2019) 19(4):255–65. doi: 10.1038/s41577-019-0141-8
31. Stecenko AA, King G, Torii K, Breyer RM, Dworski R, Blackwell TS, et al. Dysregulated Cytokine Production in Human Cystic Fibrosis Bronchial Epithelial Cells. Inflammation (2001) 25(3):145–55. doi: 10.1023/A:1011080229374
32. Conese M, Copreni E, Di Gioia S, De Rinaldis P, Fumarulo R. Neutrophil Recruitment and Airway Epithelial Cell Involvement in Chronic Cystic Fibrosis Lung Disease. J Cyst Fibros (2003) 2(3):129–35. doi: 10.1016/S1569-1993(03)00063-8
33. Taylor PR, Bonfield TL, Chmiel JF, Pearlman E. Neutrophils From F508del Cystic Fibrosis Patients Produce IL-17A and Express IL-23 - Dependent IL-17rc. Clin Immunol (2016) 170:53–60. doi: 10.1016/j.clim.2016.03.016
34. Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell (2019) 176(1-2):113–.e15. doi: 10.1016/j.cell.2018.12.002
35. Le Gars M, Descamps D, Roussel D, Saussereau E, Guillot L, Ruffin M, et al. Neutrophil Elastase Degrades Cystic Fibrosis Transmembrane Conductance Regulator via Calpains and Disables Channel Function In Vitro and In Vivo. Am J Respir Crit Care Med (2013) 187(2):170–9. doi: 10.1164/rccm.201205-0875OC
36. Ma J, Kummarapurugu AB, Hawkridge A, Ghosh S, Zheng S, Voynow JA. Neutrophil Elastase-Regulated Macrophage Sheddome/Secretome and Phagocytic Failure. Am J Physiol Lung Cell Mol Physiol (2021). doi: 10.1152/ajplung.00499.2019
37. McKelvey MC, Weldon S, McAuley DF, Mall MA, Taggart CC. Targeting Proteases in Cystic Fibrosis Lung Disease. Paradigms, Progress, and Potential. Am J Respir Crit Care Med (2020) 201(2):141–7. doi: 10.1164/rccm.201906-1190PP
38. Roghanian A, Drost EM, MacNee W, Howie SE, Sallenave JM. Inflammatory Lung Secretions Inhibit Dendritic Cell Maturation and Function via Neutrophil Elastase. Am J Respir Crit Care Med (2006) 174(11):1189–98. doi: 10.1164/rccm.200605-632OC
39. Roghanian A, Sallenave JM. Neutrophil Elastase (NE) and NE Inhibitors: Canonical and Noncanonical Functions in Lung Chronic Inflammatory Diseases (Cystic Fibrosis and Chronic Obstructive Pulmonary Disease). J Aerosol Med Pulm Drug Deliv (2008) 21(1):125–44. doi: 10.1089/jamp.2007.0653
40. Forrest OA, Ingersoll SA, Preininger MK, Laval J, Limoli DH, Brown MR, et al. Frontline Science: Pathological Conditioning of Human Neutrophils Recruited to the Airway Milieu in Cystic Fibrosis. J Leukoc Biol (2018) 104(4):665–75. doi: 10.1002/JLB.5HI1117-454RR
41. Makam M, Diaz D, Laval J, Gernez Y, Conrad CK, Dunn CE, et al. Activation of Critical, Host-Induced, Metabolic and Stress Pathways Marks Neutrophil Entry Into Cystic Fibrosis Lungs. Proc Natl Acad Sci USA (2009) 106(14):5779–83. doi: 10.1073/pnas.0813410106
42. Sagel SD, Gibson RL, Emerson J, McNamara S, Burns JL, Wagener JS, et al. Impact of Pseudomonas and Staphylococcus Infection on Inflammation and Clinical Status in Young Children With Cystic Fibrosis. J Pediatr (2009) 154(2):183–8. doi: 10.1016/j.jpeds.2008.08.001
43. Dwyer M, Shan Q, D'Ortona S, Maurer R, Mitchell R, Olesen H, et al. Cystic Fibrosis Sputum DNA has NETosis Characteristics and Neutrophil Extracellular Trap Release Is Regulated by Macrophage Migration-Inhibitory Factor. J Innate Immun (2014) 6(6):765–79. doi: 10.1159/000363242
44. Marcos V, Zhou-Suckow Z, Onder Yildirim A, Bohla A, Hector A, Vitkov L, et al. Free DNA in Cystic Fibrosis Airway Fluids Correlates With Airflow Obstruction. Mediators Inflamm (2015) 2015:408935. doi: 10.1155/2015/408935
45. Yoo DG, Winn M, Pang L, Moskowitz SM, Malech HL, Leto TL, et al. Release of Cystic Fibrosis Airway Inflammatory Markers From Pseudomonas Aeruginosa-Stimulated Human Neutrophils Involves NADPH Oxidase-Dependent Extracellular DNA Trap Formation. J Immunol (2014) 192(10):4728–38. doi: 10.4049/jimmunol.1301589
46. Floyd M, Winn M, Cullen C, Sil P, Chassaing B, Yoo DG, et al. Swimming Motility Mediates the Formation of Neutrophil Extracellular Traps Induced by Flagellated Pseudomonas Aeruginosa. PloS Pathog (2016) 12(11):e1005987. doi: 10.1371/journal.ppat.1005987
47. Oz HH, Hartl D. Innate Immunity in Cystic Fibrosis: Novel Pieces of the Puzzle. J Innate Immun (2016) 8(6):529–30. doi: 10.1159/000448285
48. Koller B, Kappler M, Latzin P, Gaggar A, Schreiner M, Takyar S, et al. TLR Expression on Neutrophils at the Pulmonary Site of Infection: TLR1/TLR2-Mediated Up-Regulation of TLR5 Expression in Cystic Fibrosis Lung Disease. J Immunol (2008) 181(4):2753–63. doi: 10.4049/jimmunol.181.4.2753
49. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67(1):425. author reply 426. doi: 10.1158/0008-5472.CAN-06-3037
50. Krystal G, Sly L, Antignano F, Ho V, Ruschmann J, Hamilton M. Re: The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67(8):3986. doi: 10.1158/0008-5472.CAN-07-0211
51. Veglia F, Perego M, Gabrilovich D. Myeloid-Derived Suppressor Cells Coming of Age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
52. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (2011) 187(1):538–52. doi: 10.4049/jimmunol.1100450
53. Schmielau J, Finn OJ. Activated Granulocytes and Granulocyte-Derived Hydrogen Peroxide Are the Underlying Mechanism of Suppression of T-Cell Function in Advanced Cancer Patients. Cancer Res (2001) 61(12):4756–60.
54. Mistry P, Nakabo S, O'Neil L, Goel RR, Jiang K, Carmona-Rivera C, et al. Transcriptomic, Epigenetic, and Functional Analyses Implicate Neutrophil Diversity in the Pathogenesis of Systemic Lupus Erythematosus. Proc Natl Acad Sci USA (2019) 116(50):25222–8. doi: 10.1073/pnas.1908576116
55. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci Immunol (2016) 1(2). doi: 10.1126/sciimmunol.aaf8943
56. Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, et al. Polyunsaturated Fatty Acids Promote the Expansion of Myeloid-Derived Suppressor Cells by Activating the JAK/STAT3 Pathway. Eur J Immunol (2013) 43(11):2943–55. doi: 10.1002/eji.201343472
57. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers JW, et al. A Subset of Neutrophils in Human Systemic Inflammation Inhibits T Cell Responses Through Mac-1. J Clin Invest (2012) 122(1):327–36. doi: 10.1172/JCI57990
58. Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, et al. Neutrophils With Myeloid Derived Suppressor Function Deplete Arginine and Constrain T Cell Function in Septic Shock Patients. Crit Care (2014) 18(4):R163. doi: 10.1186/cc14003
59. Bowers NL, Helton ES, Huijbregts RP, Goepfert PA, Heath SL, Hel Z. Immune Suppression by Neutrophils in HIV-1 Infection: Role of PD-L1/PD-1 Pathway. PloS Pathog (2014) 10(3):e1003993. doi: 10.1371/journal.ppat.1003993
60. Coutinho CP, Dos Santos SC, Madeira A, Mira NP, Moreira AS, Sa-Correia I. Long-Term Colonization of the Cystic Fibrosis Lung by Burkholderia Cepacia Complex Bacteria: Epidemiology, Clonal Variation, and Genome-Wide Expression Alterations. Front Cell Infect Microbiol (2011) 1:12. doi: 10.3389/fcimb.2011.00012
61. Morgan WJ, Butler SM, Johnson CA, Colin AA, FitzSimmons SC, Geller DE, et al. Epidemiologic Study of Cystic Fibrosis: Design and Implementation of a Prospective, Multicenter, Observational Study of Patients With Cystic Fibrosis in the U.S. And Canada. Pediatr Pulmonol (1999) 28(4):231–41. doi: 10.1002/(SICI)1099-0496(199910)28:4<231::AID-PPUL1>3.0.CO;2-2
62. Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, et al. L-Arginine Modulates CD3zeta Expression and T Cell Function in Activated Human T Lymphocytes. Cell Immunol (2004) 232(1-2):21–31. doi: 10.1016/j.cellimm.2005.01.004
63. Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, et al. Suppression of T-Cell Functions by Human Granulocyte Arginase. Blood (2006) 108(5):1627–34. doi: 10.1182/blood-2006-11-010389
64. Rotondo R, Bertolotto M, Barisione G, Astigiano S, Mandruzzato S, Ottonello L, et al. Exocytosis of Azurophil and Arginase 1-Containing Granules by Activated Polymorphonuclear Neutrophils Is Required to Inhibit T Lymphocyte Proliferation. J Leukoc Biol (2011) 89(5):721–7. doi: 10.1189/jlb.1109737
65. Jacobsen LC, Theilgaard-Monch K, Christensen EI, Borregaard N. Arginase 1 Is Expressed in Myelocytes/Metamyelocytes and Localized in Gelatinase Granules of Human Neutrophils. Blood (2007) 109(7):3084–7. doi: 10.1182/blood-2006-06-032599
66. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed Death-1 Ligand 1 Interacts Specifically With the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity (2007) 27(1):111–22. doi: 10.1016/j.immuni.2007.05.016
67. Avendano-Ortiz J, Llanos-Gonzalez E, Toledano V, Del Campo R, Cubillos-Zapata C, Lozano-Rodriguez R, et al. Pseudomonas Aeruginosa Colonization Causes PD-L1 Overexpression on Monocytes, Impairing the Adaptive Immune Response in Patients With Cystic Fibrosis. J Cyst Fibros (2019) 18(5):630–5. doi: 10.1016/j.jcf.2018.11.002
Keywords: cystic fibrosis, myeloid-derived suppressor cell, neutrophil, immunosuppression, gMDSC
Citation: Tucker SL, Sarr D and Rada B (2021) Granulocytic Myeloid-Derived Suppressor Cells in Cystic Fibrosis. Front. Immunol. 12:745326. doi: 10.3389/fimmu.2021.745326
Received: 21 July 2021; Accepted: 24 August 2021;
Published: 21 September 2021.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Alessandro Rimessi, University of Ferrara, ItalyJean-Michel Sallenave, INSERM U1152 Physiopathologie et Epidémiologie des Maladies Respiratoires, France
Copyright © 2021 Tucker, Sarr and Rada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Balázs Rada, radab@uga.edu