
A new tyramine derivative, glycosamide A (1), together with two known amide alkaloids, N-benzoyltyramine methyl ether (2) and aurantiamide acetate (3), was isolated from the branches and leaves of Glycosmis craibii Tanaka. Their structures were elucidated through extensive 1D and 2D NMR and HR-ESI-MS spectroscopic analyses. All known compounds were isolated from the genus Glycosmis for the first time. Furthermore, the antiproliferative activities of all isolated alkaloids in vitro were evaluated. Compound 2 displayed weak antiproliferative activities against five human cancer cell lines, HL-60, SMMC-7721, A-549, MCF-7, and SW480.
Similar content being viewed by others
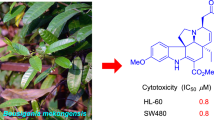
The genus Glycosmis, a member of the Rutaceae family, contains approximately 50 species distributed widely in subtropical and tropical regions of Asia [1]. In China, there are about 11 species along with two varieties that grow mainly in Southern China. Previous phytochemical investigations on the genus Glycosmis have led to the isolation of a wide variety of natural products containing acridones, quinolones, furoquinolines, sulfur-containing amides, macrocyclic nonapeptides, as well as carbazoles. Earlier pharmacological studies have shown that these compounds isolated from the genus Glycosmis display various pharmacological activities, including anti-inflammatory, antitumor, antibacterial, antimicrobial, and DYRK1A inhibitory, along with Th17 differentiation inhibitory activities [2,3,4,5,6,7,8,9,10,11,12]. Some plants of this genus were used in traditional/folk medicine to treat protracted diseases, soreness, numbness, fever, liver complaints, and other specific diseases [9]. Our preliminary experimental data revealed that the 95% EtOH extract of the branches and leaves of G. craibii showed remarkable antiproliferative activities, displaying IC50 values ranging from 15.4 to 46.8 μg/mL. In our ongoing search for bioactive natural products as potential antitumor agents from tropical medicinal plants in China [13,14,15], a systematic and in-depth chemical research on the stems of G. craibii was conducted, which led to the isolation and characterization of a new tyramine derivative, glycosamide A (1), together with two known compounds that were isolated from G. craibii for the first time. Their chemical structures were elucidated by comprehensive spectroscopic methods. In addition, all isolated compounds were evaluated for their antiproliferative activities against five human cancer cell lines in vitro. Herein, we described the isolation, structure elucidation, and antiproliferative activities of these compounds.

The air-dried and powdered branches and leaves of G. craibii were extracted by maceration using 95% ethanol. The combined extract solutions were then concentrated under reduced pressure to produce a crude extract, which was further separated by silica gel, ODS gel, and Sephadex LH-20 CC and semipreparative HPLC to yield a new tyramine derivative, glycosamide A (1), together with two known compounds (2 and 3). The new compound was isolated as a pair of inseparable conformers and obtained as a white amorphous powder. The ratio of conformer (1a) and (1b) was about 1:1 by 1H NMR analysis in DMSO-d6 as solvent. The 1H and 13C spectra of the new compound indicated either overlapped signals for some resonances or even two sets of resonances for the two conformers at room temperature. To simplify the structural analysis, we discussed the structure identification of the new compound based on the NMR data of conformer (1a) (Table 1). The 1H NMR of conformer (1a) displayed the presence of two coupled triplets of methylenes, a para-substituted benzene ring, a singlet methyl, and a terminal methyl. In combination with 13C NMR, HSQC, and HMBC, it was postulated that the new compound belongs to a long alkanoyl chain amide that contains a long alkanoyl moiety and a tyramine part [16]. Due to the presence of the N-methyl group, the rotation of the amide C–N bond was restricted, which resulted in two conformers: 50% (1a) s-cis and 50% (1b) s-trans [17].
Glycosamide A (1) was obtained as a white amorphous powder. The molecular formula of 1 was assigned as C25H43NO2 according to its HR-ESI-MS spectrum displaying the [M + Na]+ ion peak at m/z 412.31857 (calcd for C25H43NO2Na, 412.31860), suggesting an index of hydrogen deficiency of 5. The IR absorption bands in the IR spectrum of 1 suggested the existence of a hydroxy group (3400 cm–1) and an amide carbonyl group (1670 cm–1) along with benzene ring groups (1610, 1558, and 1460 cm–1). The 1H NMR spectrum of 1 exhibited a free hydroxy group at δ 9.20 (1H, s) exchangeable with D2O; a typical AA′BB′ system was observed at δ 6.66 (2H, d, J = 8.4 Hz, H-5′, 7′) and 6.98 (2H, d, J = 8.4 Hz, H-4′, 8′), and two coupled triplets of methylene protons were observed at δ 2.66 (2H, t, J = 7.2 Hz, H-2′) and 3.40 (2H, t, J = 7.2 Hz, H-1′) in the tyramine moiety.
Furthermore, in the 1H NMR spectrum, a terminal methyl at δ 0.85 (3H, t, J = 6.8 Hz, H-16) and methylenes at δ 2.21 (2H, t, J = 7.2 Hz, H-2), 1.45 (2H, m, H-3), and 1.23 (24H, m, H-4–15) were also observed. These data suggested the presence of a long alkyl chain linked to tyramine. The 13C NMR combined with DEPT data of 1 indicated the existence of 25 carbons, including seven sp2 carbons (six aromatic carbons and one amide-type carbonyl carbon) and two methyls along with 16 sp3 methylenes. The above spectral data suggested that the chemical structure of 1 was similar to that of N-[2-(4-hydroxyphenyl)ethyl]-hexadecanamide [16], except that an N-methyl group of tertiary amine was present in 1 instead of a secondary amine group, which was verified by the HMBC correlations of N-CH3 (δH 2.86, 3H, s) to C-1′ (δ 51.0) and C-1 (δ 171.7). The chemical structure of 1 was further confirmed through comprehensive analyses of its 2D NMR spectra, as displayed in Fig. 1. Accordingly, the chemical structure of 1 was named N-methyl-N-[2-(4-hydroxyphenyl)ethyl] hexadecanamide.

Selected 2D NMR correlations for glycosamide A (1).
In addition to the new tyramine derivative, glycosamide A (1), two known compounds were isolated and identified as N-benzoyltyramine methyl ether (2) [18] and aurantiamide acetate (3) [19] by comparison of their experimental data with that reported in the literature.
All isolated compounds were evaluated for their antiproliferative activities against five human cancer cell lines, HL-60, SMMC-7721, A-549, MCF-7, and SW480, according to the MTT method, using cisplatin as positive control. Unfortunately, only compound 2 showed slight antiproliferative effects against diverse human cancer cell lines, exhibiting IC50 values in the range of 23.85 ± 0.05 to 39.43 ± 0.17 μM (see Table 2).
Experimental
General Experimental Procedures. UV spectra of glycosamide A (1) were recorded on a Beckman DU 640 spectrophotometer. IR spectra of 1 were obtained on a Nicolet 6700 spectrophotometer. The NMR spectra of compounds 1–3 were recorded on an Avance III-400 MHz NMR spectrometer using TMS as internal standard, with chemical shifts recorded as δ values. HR-ESI-MS spectra of 1 were measured on a Q-TOF Ultima Global GAA076 LC mass spectrometer. Semipreparative HPLC was performed on a Shimadzu LC-16 system with a PAD detector using a Waters XBridge C18 column (250 × 10 mm, 5 μm). Silica gel (Qing Dao Hai Yang Chemical Group Co.; 100–200 mesh, 200–300 mesh), ODS gel (YMC; 50 μm), and Sephadex LH-20 gel (Amersham Biosciences; 40–70 μm) were used for column chromatography (CC). Precoated silica gel plates (F-254) were the products of Qingdao Haiyang Chemical Co., Ltd. and were employed for thin-layer chromatography.
Plant Material. The branches and leaves of G. craibii were gathered from Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Mengla County, Yunnan Province, People′s Republic China in August 2018. The plant specimens were identified by Jun-feng Wang (Lishui Forestry Research Institute). A voucher specimen (No. W180801) has been deposited at the Institute of Natural Medicine and Health Products, Taizhou University.
Extraction and Isolation. The air-dried branches and leaves of G. craibii (10 kg) were powdered and then extracted by 50 L of 95% ethanol at room temperature three times, each time for 5 days. The solvent was combined and condensed in vacuum to yield a crude extract. After suspension in water (10.0 L), the crude extract was extracted successively with petroleum ether (10.0 L × 3) and ethyl acetate (10.0 L × 3) to obtain the petroleum ether extract (69 g) and the EtOAc extract (90 g). The EtOAc extract (90 g) was subjected to silica gel CC and eluted with petroleum ether–EtOAc (100:0–0:100) to yield 10 fractions (Frs. 1–10). Fraction 3 (10 g) was further chromatographed over an RP-18 gel medium-pressure column chromatograph (CH3OH–H2O, 20:80–100:0) to give seven subfractions (Subfrs. 3A–3G). Subfraction 3E (0.8 g) was separated by semipreparative HPLC (XBridge C18 column, 250 × 10 mm, 5 μm, 90% CH3CN, 3.0 mL/min, tR 45.5 min) to obtain 1 (6.0 mg, 0.00006%). Subfraction 3F (0.4 g) was purified using Sephadex LH-20 gel CC and eluted with CH3OH, then separated by semipreparative HPLC (XBridge C18 column, 250 × 10 mm, 5 μm, 90% CH3OH, 3.0 mL/min, tR 23.0 and 52.5 min) to afford 3 (12 mg, 0.00012%) and 2 (3 mg, 0.00003%).
Glycosamide A (1), white amorphous powder. UV (MeOH, λmax, nm) (log ε): 200 (4.51), 222 (4.25), 287 (4.54), 320 (4.75). IR (KBr, νmax, cm–1): 3400, 2980, 2830, 2744, 1670, 1610, 1558, 1460, 860. HR-ESI-MS m/z 412.31857 [M + Na]+ (calcd for C25H43NO2Na+, 412.31860). 1H and 13C NMR data, see Table 1.
Cytotoxicity Bioassays. The following human tumor cell lines were used: HL-60, SMMC-7721, A-549, MCF-7, and SW480. All cells were cultured in RPMI-1640 or DMEM medium (HyClone, Logan, UT, USA), supplemented with 10% fetal bovine serum (HyClone) in 5% CO2 at 37°C. Cytotoxic activities were determined using the MTT method [20]. All compounds in DMSO were serially diluted with culture medium. The final concentrations of each of the compounds were 0.0625, 0.32, 1.6, 8, and 40 μM, and the experiments were performed in quadruplicate for each concentration. Cisplatin (Sigma, St. Louis, MO, USA) was used as positive control. After treatment, cell viability was measured and the cell growth curve plotted. The IC50 values were calculated by the Reed and Muench method [21].
References
T. M. Yasir, M. K. Tripathi, P. Singh, and R. Shrivastava, Nat. Prod. J., 9, 98 (2019).
M. A. Beniddir, E. L. Borgne, B. I. Iorga, N. Loaec, O. Lozach, L. Meijer, and M. Litaudon, J. Nat. Prod., 77, 1117 (2014).
Y. H. Choi, C. Seo, W. Jeong, J. E. Lee, J. Y. Lee, E. K. Ahn, and D. Lee, Bioorg. Chem., 87, 714 (2019).
Y. Chen, C. Tang, Y. Wu, S. Mo, S. Wang, G. Yang, and Z. Mei, Org. Biomol. Chem., 13, 6773 (2015).
N. M. Cuong, P. N. Khanh, P. T. Huyen, H. V. Duc, T. T. Huong, V. T. Ha, and F. Fusi, J. Nat. Prod., 77, 1586 (2014).
A. K. Chakravarty, T. Sarkar, K. Masuda, and K. Shiojima, Phytochemistry, 50, 1263 (1999).
K. Piboonprai, P. Khumkhrong, M. Khongkow, T. Yata, N. Ruangrungsi, C. Chansriniyom, and T. Iempridee, Biochem. Biophys. Res. Commun., 500, 866 (2018).
T. Sripisut, W. Phakhodee, T. Ritthiwigrom, S. Cheenpracha, U. Prawat, S. Deachathai, and S. Laphookhieo, Phytochem. Lett., 6, 337 (2013).
G. Z. Yang, Y. Wu, and Y. Chen, Helv. Chim. Acta, 95, 1449 (2012).
K. Yang, S. S. Guo, Z. F. Geng, C. X. You, W. J. Zhang, Y. P. Li, and Z. W. Deng, Ind. Crop. Prod., 74, 628 (2015).
K. L. Ji, Y. Y. Fan, H. H. Kuok, Q. F. Liu, T. Li, and J. M. Yue, CCS Chem., 2, 844 (2020).
J. S. Wang, Y. T. Zheng, T. Efferth, R. R. Wang, Y. M. Shen, and X. J. Hao, Phytochemistry, 66, 697 (2005).
S. Y. Wu, Y. H. Fu, G. Y. Chen, X. B. Li, Q. Zhou, C. R. Han, X. P. Song, X. J. Du, M. L. Xie, and G. G. Yao, Phytochem. Lett., 11, 236 (2015).
S. Y. Wu, Y. H. Fu, Q. Zhou, M. Bai, G. Y. Chen, S. Y. Zhao, C. R. Han, and X. P. Song, Molecules, 23, 472 (2018a).
S. Y. Wu, Y. H. Fu, Q. Zhou, M. Bai, G. Y. Chen, S. Y. Zhao, C. R. Han, and X. P. Song, Nat. Prod. Res., 32, 953 (2018b).
J. D. Wansi, S. O. Nwozo, L. M. Mbaze, K. P. Devkota, S. M. D. Moladje, Z. T. Fomum, and N. Sewald, Planta Med., 75, 517 (2009).
Z. F. Geng, K. Yang, Y. P. Li, S. S. Guo, C. X. You, W. J. Zhang, Z. Zhang, and S. S. Du, Nat. Prod. Res., 31, 791 (2017).
Y. Q. Du, H. Li, C. J. Liu, J. Z. Yang, J. Ma, D. Zhang, H. Sun, and D. M. Zhang, J. Asian Nat. Prod. Res., 17, 1048 (2015).
Z. T. Wang, Y. H. Lu, W. C. Ye, L. S. Xu, G. J. Xu, and Y. Z. Shu, J. Chin. Pharm. Sci., 8, 171 (1999).
T. Mosmann, J. Immunol. Methods, 65, 55 (1983).
L. J. Reed and H. Muench, Am. J. Hyg., 27, 493 (1938).
Acknowledgment
This project was supported by the National Natural Science Foundation of China (Nos. 31900297 and 31670363), the Science and Technology Planning Project of Guangdong Province (Nos. 2016A030307021, 2018A0303070005, and 2018A030307037), the Science and Technology Projects of Taizhou (No. 1901ny11), and the Special program for Natural Science Talents of Lingnan Normal University (No. ZL1903).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2021, pp. 764–767.
Rights and permissions
About this article

Cite this article
Wu, SY., Chen, ZM., Huang, GL. et al. A New Tyramine Derivative from the Branches and Leaves of Glycosmis craibii. Chem Nat Compd 57, 894–898 (2021). https://doi.org/10.1007/s10600-021-03505-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-021-03505-9