
Abstract
Following repeated opioid use, some dependent individuals experience persistent cognitive deficits that contribute to relapse of drug-taking behaviors, and one component of this response may be mediated by the endogenous dynorphin/kappa opioid system in neocortex. In C57BL/6 male mice, we find that acute morphine withdrawal evokes dynorphin release in the medial prefrontal cortex (PFC) and disrupts cognitive function by activation of local kappa opioid receptors (KORs). Immunohistochemical analyses using a phospho-KOR antibody confirmed that both withdrawal-induced and optically evoked dynorphin release activated KOR in PFC. Using a genetically encoded sensor based on inert KOR (kLight1.2a), we revealed the in vivo dynamics of endogenous dynorphin release in the PFC. Local activation of KOR in PFC produced multi-phasic disruptions of memory processing in an operant-delayed alternation behavioral task, which manifest as reductions in response number and accuracy during early and late phases of an operant session. Local pretreatment in PFC with the selective KOR antagonist norbinaltorphimine (norBNI) blocked the disruptive effect of systemic KOR activation during both early and late phases of the session. The early, but not late phase disruption was blocked by viral excision of PFC KORs, suggesting an anatomically dissociable contribution of pre- and postsynaptic KORs. Naloxone-precipitated withdrawal in morphine-dependent mice or optical stimulation of pdynCre neurons using Channelrhodopsin-2 disrupted delayed alternation performance, and the dynorphin-induced effect was blocked by local norBNI. Our findings describe a mechanism for control of cortical function during opioid dependence and suggest that KOR antagonism could promote abstinence.
Similar content being viewed by others


Introduction
Cognitive dysfunction is a common feature of substance use disorders and other psychiatric illnesses [1]. In these clinical syndromes, stress-induced changes in cortical function can exacerbate vulnerability to behavioral disruptions evident during substance use [2]. The neurochemical basis of these responses is not known, but stress activates multiple brain systems that lead to the release of neuropeptide transmitters, including endogenous dynorphin opioids that activate Gαi protein-coupled kappa opioid receptors (KORs) [3, 4]. Selective KOR agonists, like those in the hallucinogenic sage Salvia divinorum, are psychotomimetic in humans [5] and functional polymorphisms in the prodynorphin gene have been correlated with disrupted reversal learning in humans [6]; however, little is known about the role of the dynorphin/KOR system in physiological control of human cognition.
Disruptions in cognitive elements including impulse control and attention during protracted abstinence are hypothesized to drive drug-seeking behaviors [7]. These disruptions may occur as a result of neural adaptations to binge drug use or stressors associated with chronic drug exposure [8]. Periods of drug withdrawal activate stress-related neuropeptide systems, including corticotrophin releasing factor (CRF) and glucocorticoids [9]. Genetic knockout of the CRF2 receptor blocks morphine withdrawal-mediated increases in aversion, anhedonia, and prodynorphin mRNA in the striatum [10]. Increased striatal prodynorphin mRNA expression following chronic cocaine, nicotine, and alcohol use has been reported in rodents and humans [11,12,–13] and systemic KOR antagonism can normalize striatal function to decrease anhedonia-like behaviors in humans [14]. Binge cocaine or alcohol use is also associated with increased dynorphin/KOR mRNA and transcriptional markers in dorsolateral prefrontal cortex, orbitofrontal cortex [15], and other cortical regions [16] in humans, suggesting a significant role for dynorphin in cognitive dysregulation following chronic drug use [17].
Although the psychopathology of substance use disorders is complex, dynorphins and KOR are expressed in the mammalian cortex [18, 19] and analysis of their role in mouse models of cognition will likely provide useful insights for developing treatments for cognitive dysfunction. In the present study, we demonstrate that pharmacological treatment with a KOR agonist, optogenetic stimulation of PFC prodynorphin mRNA-expressing neurons, or precipitated morphine withdrawal can each significantly increase KOR activation as demonstrated by increased receptor phosphorylation in the PFC of mice. We then reveal the time course of dynorphin release during precipitated opioid withdrawal in the prefrontal cortex using a genetically encoded fluorescent sensor for endogenous dynorphin in the synaptic space. Cortical regulation of working memory can be readily measured in mice using a delayed alternation procedure [20] and in the present study, we assessed mouse performance in a food-reinforced operant-delayed alternation task. We find that pharmacological activation of KOR expressed in PFC as well as endogenous dynorphin released by either naloxone-precipitated morphine withdrawal or optical stimulation of PFC dynorphin neurons disrupted delayed alternation performance. Together, these studies demonstrate that local actions of the dynorphin/KOR system in the prefrontal cortex disrupt cognition.
Materials and methods
Subjects
Adult male C57BL/6 mice ranging from 2 to 10 months of age were used in these experiments. All experimental procedures were approved by the University of Washington Institutional Animal Use and Care Committee and were conducted in accordance with National Institutes of Health (NIH) “Principles of Laboratory Animal Care” (NIH Publication No. 86-23, revised 1985). Mice were group housed (2–5 mice per cage) and food restricted to approximately 90% of ad libitum body weight when included in the delayed alternation assay. Food restriction was necessary to motivate operant task response. Water was freely available at all times in their home cages. All testing was conducted during the light phase of the 12-h light/dark cycle. The daily food allotment was ~2–3 g standard Purina 5053 rodent diet chow per mouse, in addition to food pellets (Bio-Serv Dustless Precision Pellet, Rodent, Purified, 20 mg) consumed during the operant behavioral sessions. We have previously reported estrogen-regulated sex differences in intracellular signaling pathways that alter female responses to KOR agonists and antagonists [21,22,–23]. We observed that the KOR antagonist norbinaltorphimine dihydrochloride (norBNI) is not consistently long lasting in females, confounding the use of this and other KOR ligands in these studies. For these reasons, we chose to focus these experiments on male mice. Floxed KOR (KORlox) mice were generated by the Institut Clinique de la Souris, in which exon 1 of KOR was flanked by loxP sites [24] and prodynorphin Cre (PdynCre) mice were obtained from Richard Palmiter (University of Washington).
Drugs
Racemic (±) U50488 hydrochloride (U50488), norBNI, and morphine sulfate were provided by the National Institute of Drug Abuse Drug Supply Program (Bethesda, MD) and naloxone hydrochloride dihydrate was acquired from Sigma-Aldrich (St. Louis, MO, USA). U50,488 (1, 5, and 10 mg/kg), norBNI (10 mg/kg), morphine (10 mg/kg), or naloxone (1 and 10 mg/kg) were dissolved in saline and administered intraperitoneally (IP) in a volume of 10 mL/kg. norBNI (2.5 μg/μL) was dissolved in sterile artificial cerebrospinal fluid (aCSF) and intracranially microinjected. Dynorphin B (Tocris, Bristol, UK) was dissolved in ethanol.
Detailed methods are described in the Supplementary information section.
Statistical analysis
All data are presented as mean ± SEM. Our sample sizes for behavioral studies are based on those used in Abraham et al. [25] for KOR agonist actions on operant behaviors. Individual data points are shown when possible, and data distribution was assumed to be normal, but not formally tested. Mice were removed from analyses if no viral expression was found (four kLight-injected mice were excluded). We used two-tailed t-tests, one-way and two-way ANOVA (incorporating repeated measures (paired t-test or RM-ANOVA) where appropriate), and performed Sidak’s, Dunnet’s, or Fisher’s post-hoc tests, specified in text. Mixed effects ANOVA were used when individual mice did not respond during particular time bins to account for missing values. Operant behavior data were collected through a custom-built MED-PC program. Data were analyzed using Prism 9.0 (GraphPad Software; San Diego, California, USA) and custom-built MATLAB software (MathWorks Inc.; Natick, Massachusetts, USA).
Results
Stimuli leading to kappa opioid receptor activation in the prefrontal cortex
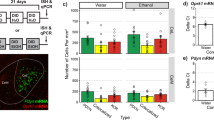
The dynorphin neuropeptides are expressed by neurons in the prefrontal cortex [26], but physiological and behavioral stimuli necessary to evoke dynorphin release in the cortex have not been identified. To address this question, we used a previously characterized phospho-selective antibody (KORp) [27] to detect agonist-activated KOR that has been phosphorylated by G protein Receptor Kinase 3 (GRK3) [28]. Prior KORp antibody characterization established that genetic deletion of either GRK3 [29] or prodynorphin [30] blocked stress-induced increases in KORp-immunoreactivity (KORp-IR). Kappa receptor phosphorylation at this site engages arrestin-dependent signaling, which has been associated with aversive and dysphoric qualities of KOR agonists in midbrain circuits [24]. We first confirmed that KOR agonist (U50,488, 10 mg/kg, i.p.) administration significantly increased KORp-IR in PFC (Fig. 1A) and this increase could be blocked by systemic pretreatment with the selective KOR antagonist, norBNI (10 mg/kg; Fig. 1B).

A Pharmacological validation of phospho-KOR antibody. Representative images (10×, 40× inset) show increased fluorescent signal from phospho-KOR (S369 site) immunoreactivity (KORp-IR) following KOR agonist (U50,488; 10 mg/kg) treatment, compared to saline. Dashed lines show boundaries of the prelimbic prefrontal cortex. Scale bar for 10× image shows 150 μm, scale bar for 40× image (inset) shows 30 μm. White arrows indicate KORp-IR+ cell. B Conditions for KOR activation in the prefrontal cortex. All groups were normalized to their corresponding control group. A one-way ANOVA showed an overall significant effect of treatment (F(9, 42) = 5.597, p < 0.0001). Fisher’s LSD post-hoc test was used for planned comparisons between groups that were run in the same cohorts and normalized to their own saline or no-stress controls. KOR agonist (10 mg/kg U50,488; n = 4) significantly increased KORp-IR compared to norBNI treatment alone (p = 0.0006). Pretreatment with norBNI (n = 4) blocked the increase in KORp-IR following U50,488 treatment (p = 0.0002). These groups were normalized to saline-treated controls (n = 4). Optogenetic stimulation (n = 4) of PFC dynorphin neurons (normalized to EYFP control; n = 2) significantly increased KORp IR, which was blocked by norBNI pretreatment prior to optogenetic stimulation (n = 3; p = 0.0468), demonstrating the presence of a local PFC dynorphin/kappa opioid receptor circuit. Repeated forced swim stress, a stressor known to cause dynorphin release in the brain, significantly increased (p = 0.0024) KORp IR (normalized to no-stress controls n = 5 dorsal raphe nucleus (DRN); n = 6 PFC) in the DRN (n = 6) compared to the prefrontal cortex (PFC; stress n = 6) in the same mice. This showed that swim stress did not cause the release of dynorphin in the prefrontal cortex. In contrast, repeated footshock (0.3 mA shock each min, for 15 min) increased KORp IR in the PFC compared to control mice (one sample t-test; p = 0.0036). Naloxone (1 mg/kg)-precipitated withdrawal (W/D) following chronic experimenter-administered morphine (10 mg/kg, 4 days twice per day, one morphine injection 2 h prior to naloxone on day 5) treatment (normalized to chronic saline treatment n = 8) also increased KORp IR in PFC (n = 9), which was blocked by pretreatment with norBNI (n = 5) when given 24 h prior to 1st morphine injection and 24 h prior to naloxone injection (p = 0.012). Error bars indicate SEM. *p < 0.05; **p < 0.01; ***p < 0.0001. C Representative image for ChR2 expression in PFC dynorphin neurons. Prodynorphin Cre (pdynCre) mice were injected in the PFC with Channelrhodopsin-2 (ChR2; AAV1-DIO-ChR2). Image shows ChR2 (cyan) expression in pdyn-Cre+ neurons in the PFC. 10× image; Scale bar shows 150 μm. D Representative image for ChR2 expression and KORp IR in PFC dynorphin neurons with saline pretreatment. Image shows 40× image of ChR2 (cyan) and KORp IR (magenta) in mice treated with saline prior to optical stimulation. For optical stimulation, mice were tethered to a fiberoptic patchcord and received 473 nm light (10 mW @ fiber tip) over 30 min (Duty cycle: 5 s on @ 20 Hz, 5 s off). Within 10 min after the end of optical stimulation, brains were perfused for tissue processing; 40× image; scale bar shows 20 μm. E Representative image for ChR2 and KORp IR in PFC dynorphin neurons with norBNI pretreatment. Pretreatment with norBNI (10 mg/kg; 24 h prior to stimulation) blocked ChR2-mediated increase in KORp IR; 40× image, scale bar shows 20 μm. F Representative images for morphine withdrawal (W/D) KORp IR. Saline treatment after repeated morphine treatment (left) did not produce withdrawal and did not increase KORp IR. Naloxone-precipitated morphine withdrawal increased KORp IR in the PFC during withdrawal, which was blocked by norBNI (10 mg/kg) pretreatment; 40× image; scale bars show 20 μm.
We determined that a subset of cortical neurons express dynorphins by injecting PdynCre mice [31] with AAV5-DIO-eYFP (Supplementary Fig. S1A). We hypothesized that these neurons were likely to release dynorphin locally within the PFC and tested this by optically stimulating the neuronal population and measuring KORp-IR in this region. PdynCre mice were injected in the PFC with AAV5-DIO-Channelrhodopsin-2 (ChR2; Fig. 1C) and >4 weeks later received a 30-min fiberoptic photostimulation period within the PFC (473 nm; 10 mW; 20 Hz; 5 s on/5 s off), previously shown to evoke dynorphin release in striatum [32]. Local photostimulation significantly increased KORp-IR in the PFC (Fig. 1B–D). This increase in KORp-IR was also blocked by pretreatment with systemic norBNI, indicating that the stimulation protocol evoked dynorphin release in the PFC (Fig. 1B, E).
Stress-induced release of the endogenous dynorphin opioid peptides is known to produce both analgesia [33] and aversion [34] in mice, and we next explored behavioral stimuli predicted to physiologically evoke dynorphin release in PFC. Repeated forced swim stress has been shown to stimulate dynorphin release in the dorsal raphe nucleus (DRN), ventral tegmental area, basolateral amygdala, and other brain regions [34, 35]. However, in the present study we found that 2 days of repeated swim stress did not significantly change KORp-IR in the PFC in C57BL/6 male mice, although we did confirm that KORp-IR was significantly increased in DRN in these same mice (Fig. 1B and Supplementary Fig. S1B). The anatomical selectivity of this stress effect was unexpected. In contrast, repeated footshock, which is known to produce intense cortical activation [36], significantly increased KORp-IR in the PFC (Fig. 1B).
Acute abstinence in persons with substance use disorder is profoundly dysphoric and animal models have demonstrated that enhanced dynorphin tone contributes to the aversive state and reinstatement risk during drug withdrawal [37, 38]. To assess whether dynorphin release contributes to the stress responses evident during opioid withdrawal, we made mice opioid-dependent by daily administration of morphine (10 mg/kg; twice per day for 4 days), then acutely precipitated morphine withdrawal with the opioid receptor antagonist naloxone (1 mg/kg) [39] 2 h following a single injection of 10 mg/kg morphine on day 5. Withdrawal behaviors were evoked by naloxone in morphine-dependent mice [40], and some of these behavioral responses (increased output of fecal boli) were significantly attenuated by norBNI (Supplementary Fig. S1C–E). We did not observe a significant increase in jumping behavior during withdrawal, suggesting a mild withdrawal response, as expected from our dosing procedure. Naloxone-precipitated morphine withdrawal significantly increased KORp-IR in the PFC (Fig. 1B, F). The increase in KORp-IR was blocked by norBNI pretreatment prior to morphine administration and withdrawal, demonstrating that dynorphin was released in the PFC during precipitated withdrawal.
A genetically encoded fluorescent sensor, kLight1.2a, reveals the dynamics of dynorphin release in vivo in the PFC during morphine withdrawal
Immunohistochemistry provides low temporal resolution for measuring the pattern of dynorphin release in the PFC. To achieve higher temporal resolution, we imaged dynorphin release using in vivo fiber photometry and an improved version of the ligand sensor kLight (kLight1.2a; Fig. 2A) [41]. We first confirmed that kLight1.2a had the sensitivity and selectivity required. kLight1.2a is based on an inert KOR and a circularly permuted green fluorescent protein (cpGFP) that couples ligand-induced conformational changes of KOR to the fluorescence changes of cpGFP. Neuro2A cells expressing AAV1-hSyn-kLight1.2a responded with a significant increase in fluorescence (ΔF/F) following treatment with the kappa agonists U50,488 (10 µM) or 10 µM dynorphin B (Fig. 2B and Supplementary Fig. S2A–E). In contrast, treatment with 10 µM morphine did not increase fluorescence. To determine in vivo sensitivity, AAV1-hSyn-kLight1.2a was injected into the PFC and after >4 weeks for expression (Fig. 2C, D), individual mice were injected with saline or U50,488 (1, 5, or 10 mg/kg) on separate days and kLight1.2a fluorescence was monitored by fiber photometry in the PFC as diagrammed (Fig. 2E). While saline injection did not affect fluorescence (ΔF/F) compared to the pre-injection 5-min baseline period, treatment with U50,488 (1–10 mg/kg) significantly increased fluorescence within 5 min following intraperitoneal administration (Fig. 2F and Supplementary Fig. S2E). When mice were pre-treated with a high dose of the short-acting, nonselective opioid antagonist naloxone (10 mg/kg) sufficient to block KOR [42] 30 min prior to a 10 mg/kg U50,488 injection, there was no significant increase in fluorescence compared to saline. A single dose of naloxone (1 mg/kg) did not affect kLight1.2a fluorescence alone but when administered simultaneously with a KOR agonist, decreased the magnitude of the agonist-induced signal (Supplementary Fig. S2F). Mice received naloxone rather than norBNI to avoid long-term inactivation of KORs. These experiments confirmed that kLight1.2a expressed in the PFC would respond to a systemic KOR agonist.

A kLight 1.2a diagram. kLight 1.2a is composed of an inert KOR protein fused to a circular green fluorescent protein that responds to conformational changes in KOR when bound to dynorphin or other KOR ligands. B KOR ligands increase kLight1.2a fluorescence. U50,488 (n = 6 cells) and dynorphin (n = 4) treatment significantly increased fluorescence (ΔF/F) during the 30-min imaging session starting at 5.75 min (U50,488) and 1.25 min (dynorphin B) following drug treatment. This increase persisted until the end of the imaging session. There was a significant interaction between drug treatment and time (F(180, 1380) = 5.930, p < 0.0001) and Dunnett’s post hoc showed significant (p < 0.05) differences between dynorphin and U50,488 compared to vehicle (n = 12). There was no significant effect of morphine treatment (n = 5) on kLight fluorescence. C kLight1.2a procedure. KORCre mice (n = 5) were unilaterally injected in the PFC with AAV-kLight1.2a and an optical fiber for photometry was implanted above the injection target region. D Representative images for kLight1.2a. kLight1.2a (green) expression was confirmed in mice after recordings. Fiber track for PFC implant visualized in 10× image (left), and 63× (right) image of expression shown on the right. Scale bar for 10× shows 100 μm and scale bar for 63× shows 20 μm. E Experimental timeline. kLight1.2a fluorescence was measured using fiber photometry (30 μW at fiber tip; 331 Hz; wavelength 470/405 nm). Following a 5-min baseline recording period, mice were injected with saline or U50,488 (1, 5, and 10 mg/kg) and allowed to freely explore a novel context. Naloxone (10 mg/kg) pretreatment occurred following a baseline period and 30 min later kLight1.2a response to 10 mg/kg U50,488 was recorded. For morphine withdrawal, mice underwent repeated morphine (10 mg/kg) injection procedure, and were tested for baseline fluorescence prior to naloxone (1 mg/kg) precipitated withdrawal. Each mouse received each condition on separate days. F kLight1.2a detects endogenous dynorphin release in the PFC during morphine withdrawal. Change in fluorescence (ΔF/F) over a 60-min period is shown (solid line). SEM. is shown above the mean with a filled area. A two-way ANOVA (drug × time) showed a significant interaction between treatment and time (F(3565, 14260) = 3.776, p < 0.0001). Dunnett’s post-hoc tests showed that U50,488 (10 mg/kg) injection significantly increased kLight fluorescence from 3.3 to 54.5 min compared to saline injection. Pretreatment with the nonselective opioid antagonist naloxone (10 mg/kg) blocked 10 mg/kg U50,488-mediated increases in kLight fluorescence. During the 60-min following 1 mg/kg naloxone administration to precipitate withdrawal in morphine-dependent mice, there was a significant increase in kLight fluorescence from 5.3 to 8.0, 9.0 to 13.0, 15.3 to 37.8, 39.0 to 41.5, and 43.5 to 44.8 min. This showed that kLight detected endogenous dynorphin release within the PFC of mice undergoing morphine withdrawal and that a 10 mg/kg dose of naloxone blocked U50488-mediated increases in kLight fluorescence. G Dose-dependent effects on kLight fluorescence. Area under peak value was compared between saline and all other conditions with a one-way ANOVA. There was a significant effect of treatment (F(5, 21378) = 561.6, p < 0.0001), and Dunnett’s post hoc showed that saline treatment was significantly different (p < 0.0001) compared to morphine withdrawal or U50,488 (1, 5, and 10 mg/kg). There was no difference between saline treatment and the naloxone/U50,488 group. Error bands above the mean indicate SEM. *p < 0.05.
To measure endogenous dynorphin release, we recorded kLight1.2a fluorescence in the PFC during naloxone-precipitated withdrawal in morphine-dependent mice. At a dose able to completely block mu but not KORs, naloxone (1 mg/kg) challenge significantly increased fluorescence in the PFC of morphine-treated mice (Fig. 2F). There was a significant increase in area under the curve between saline and U50,488 (1, 5, or 10 mg/kg) or during morphine withdrawal (Fig. 2G). These results confirmed the KORp imaging data and revealed that dynorphin release could be detected in the prefrontal cortex 5–45 min following the initiation of morphine withdrawal.
PFC KOR activation disrupts delayed alternation performance via pre- and postsynaptic mechanisms
We hypothesized that activation of the dynorphin/KOR system in the PFC would disrupt cognitive function in a working memory task. In an operant-delayed alternation task [20], mice were initially trained to press a retractable lever for a food pellet reward. An alternation contingency was then introduced, during which mice were required to make a response on one retractable lever, wait a specified delay for reintroduction of the levers, and then respond on the alternate lever for reinforcement (Fig. 3A). Mice were trained until reaching stable performance with a 10-s delay between lever presentations (>20 sessions). Raster plots of performance (Fig. 3B) show lever presses from a representative mouse early in training (Line 1) and following acquisition of the delayed alternation task (Line 2). After reaching stable performance in the 10-s delay contingency (Supplementary Fig. S3A), mice were stereotaxically injected bilaterally in the medial PFC with either aCSF or the long-lasting KOR antagonist norBNI (0.5 μg/0.2 μL aCSF) to locally inactivate KOR in PFC (Supplementary Fig. S3B, C). KOR inactivation following norBNI treatment persists for >2 weeks following drug clearance [43] by a molecular mechanism previously described [44]. To assess the role of postsynaptic KOR in PFC, we also injected AAV5-DIO-Cre in floxed KOR (KORlox/lox) mice [24] to selectively excise KOR in PFC neurons (PFC KORlox; Supplementary Fig. S2D) prior to training. Following recovery and re-stabilization of performance, mice were treated with either saline (IP) or U50,488 (5 mg/kg; IP) immediately before a delayed alternation session. In control mice receiving aCSF in the PFC (PFC aCSF), systemic KOR activation decreased the percentage of correct responses compared to saline treatment (Line 3; Fig. 3B). In mice having norBNI locally administered in the PFC (PFC norBNI), systemic KOR agonist administration produced no significant degradation in performance during early (0–15 min) or late phases (45–60 min) of the delayed alternation session (Line 4; Fig. 3B). In both the early phase and the late phase of the session, KOR activation disrupted performance in PFC of aCSF-injected mice by decreasing the percent of correct responses. There was also a KOR-mediated decrease in total responses during the early phase, but no significant difference in total response number during the late phase (Fig. 3C–F). PFC norBNI administration blocked the response suppressive effect of PFC KOR activation on early phase responding. KOR-mediated decreases in percent correct in the early phase were not evident in PFC KORlox mice, whereas disruptions in the late phase behaviors were still evident in mice having postsynaptic KOR deleted in PFC (Fig. 3C–F and Supplementary Fig. S3E). In contrast, local norBNI in PFC blocked disruptions during both phases. This pattern of behavioral disruptions suggests that KORs in postsynaptic PFC neurons are required for the early phase of cognitive disruptions and presynaptic PFC KORs mediate the later phase of cognitive disruption.

A Delayed alternation procedure. C57BL/6 male mice were food restricted and trained to complete lever alternations for reinforcement (detailed description in Methods). At the start of a trial, both levers were extended. Lever choice initiated a 10-s delay period during which both levers remained retracted and unavailable. When levers were reinserted, mice chose the alternate lever for food pellet reinforcement. Choosing the same lever as the previous response was recorded as an incorrect response and was not reinforced. After a 20 s intertrial interval, levers were reinserted to allow mice to start the next trial. The diagram of a hemi-coronal section outlines the PFC region (infralimbic and prelimbic cortex) targeted for injection; all placements were confirmed postmortem. B Raster plots of performance of a representative mouse during a 60-min trial session. Ticks (black) on the center line show trial initiating responses. Ticks above the center line show correct (reinforced) responses and ticks below show incorrect (nonreinforced) responses. Lever choice is shown by orange (left lever) or blue (right lever) ticks. Line 1 shows performance of a PFC aCSF mouse early in delayed alternation training (with a 2-s delay). Mice rarely alternated levers early in training, leading to a significant number of response errors. Raster line 2 shows improved performance in the same mouse after several training sessions, and saline injection (IP) prior to the delayed alternation session did not significantly alter performance in the task. Raster line 3 shows that treatment with the KOR agonist U50,488 (5 mg/kg, IP) in the same mouse decreased the percent of correct responses and decreased total response number during the delayed alternation session. Line 4 shows that bilateral local injection of the KOR antagonist (norBNI; 0.5 μg/0.2 μL) in the PFC blocked the disruptive effects of systemically administered U50,488 on both total response number and percent correct responses. Line 5 shows that U50,488 treatment decreased the correct responses in mice with postsynaptic KOR deletion (KORlox/lox mice injected with 0.2 μL AAV5-Cre-eYFP bilaterally in the PFC; PFC KORlox) during the late phase of the session, but not during the early phase of the session. C Postsynaptic KORs are required for early phase agonist-mediated decreases in correct response % in delayed alternation. After training for 3 weeks, aCSF (n = 12) or norBNI (n = 14) was bilaterally injected into PFC (3–5 days prior to testing), and the first 15 min of performance were compared between saline and KOR agonist (U50,488) treatment days. There was a significant interaction between group and agonist treatment F(2, 30) = 5.600, p = 0.0086. Sidak’s post-hoc test showed a significant difference between U50 and Saline treatment in the PFC aCSF mice (p = 0.0003), but not in PFC norBNI or PFC KORlox mice (n = 7). Gray borders and filled symbols indicate mice that received 30 min, rather than 60 min, training and testing sessions. D Systemic KOR agonist significantly decreases total responses in PFC aCSF mice. Total number of alternation responses was significantly decreased in PFC aCSF mice during the first 15 min of the session (Drug × Group Interaction: F(2, 30) = 8.582, p = 0.0011; Sidak’s post hoc: p = 0.002). There was a nonsignificant trend toward a decrease in response number in the PFC KORlox group (p = 0.0921). Gray borders and filled symbols indicate mice that received 30 min, rather than 60 min, training and testing sessions. E Postsynaptic KOR deletion does not block late phase KOR-mediated decreases in delayed alternation performance. KOR activation (PFC aCSF n = 7; norBNI n = 10; KORlox n = 7) significantly decreased percent correct in the delayed alternation task (F(2, 21) = 4.761, p = 0.0197) during the last 15 min of the session (late phase; 45–60 min) in PFC aCSF (Sidak’s p = 0.0171) and PFC KORlox (p = 0.0011) mice. F KOR activation did not affect total response number during the late phase of a delayed alternation session. There was no significant effect of drug on total response number following KOR activation. Dashed lines connect responses of individual mice receiving saline 1 day prior to the U50,488 trial session. Error bars indicate SEM. *p < 0.05, **p < 0.01; ***p < 0.0001.
Dynorphin release in the PFC disrupts delayed alternation performance
We tested whether opioid withdrawal-induced dynorphin release would alter delayed alternation performance. Mice were initially trained to stable performance in the task with a 10-s delay as described above. Following a baseline testing session, mice received 4 days of 10 mg/kg morphine twice daily. On the fifth day, mice were treated with morphine 2 h before the test session, then received naloxone immediately prior to entry into the operant chamber (Fig. 4A). The mild withdrawal procedure enabled operant behavioral measurements during the hour following naloxone treatment. There was not a significant effect of precipitated withdrawal on early phase task performance (Fig. 4B). However, during the later phase of the delayed alternation session (30–45 min) mice showed a significant impairment in performance. This impairment was not blocked by postsynaptic deletion of KOR in PFC (Fig. 4C and Supplementary Fig. S4A–F), but was blocked by local norBNI injection in the PFC.

A Experimental timeline. Mice received training in delayed alternation until reaching stable performance, then were intracranially injected with aCSF or norBNI. Floxed KOR mice were injected prior to training. Following recovery, mouse performance in a baseline delayed alternation session was assessed. Mice then received morphine (10 mg/kg) for 4 days, and on the fifth day received naloxone (1 mg/kg) prior to entry into the operant chamber. B There was no significant effect of morphine withdrawal on performance during the early phase of the task in the aCSF (n = 7), norBNI (n = 6), or floxed KOR (n = 7) groups. C Morphine withdrawal disrupts delayed alternation performance after naloxone treatment. In the 30–45 min window following naloxone treatment, there was a significant decrease in percent correct responding during a delayed alternation session in the aCSF group (n = 5), and the disruptive effect was not blocked by postsynaptic KOR deletion (n = 5). However, local norBNI pretreatment (n = 5) in the PFC blocked the disruptive effect of morphine withdrawal on delayed alternation performance (main effect of drug; F(2, 12) = 4.628; p = 0.0324); Sidak’s post hoc compared to baseline: PFC aCSF (p = 0.0211), PFC norBNI (p = 0.959), KORlox (p = 0.0191). D PFC pdynCre optical stimulation procedure. PdynCre mice were injected with ChR2 in the PFC and were trained in delayed alternation. On baseline and test days, mice were tethered to an optical patchcord for 30-min prior to a delayed alternation session and received either no stimulation (tether) or optical stimulation (stim) on separate days. E Optical stimulation of PFC pdynCre neurons does not disrupt early phase delayed alternation. Optical stimulation had no significant effect on percent correct in delayed alternation in pdynCre mice (n = 5) during the early phase of the test session. F Optical stimulation of PFC pdynCre neurons disrupts late phase delayed alternation performance. Compared to a tether session, photostimulation significantly decreased the percent of correct responses 45–60 min after stimulation (t8 = 2.33, p = 0.048). Error bars indicate SEM. *p < 0.05.
We then tested whether optical stimulation of PFC dynorphin neurons that increased KORp-IR also significantly disrupted delayed alternation performance (Fig. 4D–F). For a baseline day, mice were tethered to the optical patchcord for 30 min then performance in the task was determined. On the test day, mice received optical stimulation (30-min session as described for Fig. 1B–D) then were placed in the operant chamber. There was no difference between optically stimulated or tethered mice during the early phase of the session (Fig. 4E and Supplementary Fig. S4G, H). However, optical stimulation significantly decreased percent correct responding during the late phase of the task (Fig. 4F).
Discussion
The principal conclusion from this study is that the dynorphin/KOR system functions in the mouse medial prefrontal cortex to control cognitive performance in a working memory task. Both systemic KOR agonist treatment and evoked release of dynorphins activated KORs to reduce response accuracy. The specific contribution of postsynaptic KORs was tested by AAV-Cre-induced excision in floxed KOR mice, which showed that KOR activation in cortical neurons reduced response number and accuracy in the early phase of the operant task. Dynorphin release in the PFC elicited by morphine withdrawal or optogenetic stimulation of PFC pdynCre neurons also disrupted delayed alternation performance during the session. Our studies strengthen the preclinical basis for investigating KOR antagonists to decrease drug withdrawal-induced cognitive dysfunction.
We show that some aversive stimuli will cause dynorphin release in the prefrontal cortex. Repeated swim stress produced dynorphin release in the DRN, but not in the prefrontal cortex of the same mice. This suggests a regional specificity to the types of stressors that engage dynorphin release in different brain areas. Morphine withdrawal increased dynorphin release within cortical areas, and this increased dynorphin may contribute to reported disruptions in working memory in humans during abstinence from opioids [45]. Persistent deficits in executive function in opioid-dependent individuals could also contribute to disruptions in mood, stress reactivity, and reward processing. KOR antagonists may be useful for decreasing some of these symptoms and could promote long-term abstinence by decreasing the disruptive effects of withdrawal stress on behavior and cortical substrates. Cognitive dysfunction and altered cortical function are also common symptoms of schizophrenia, and some evidence suggests a modulatory role of the dynorphin/KOR system in this disorder. Elevated dynorphin concentrations in cerebrospinal fluid have been reported in individuals with schizophrenia and are associated with poorer treatment outcomes [46, 47]. In double-blind clinical trials, nonselective opioid antagonists reduced auditory hallucinations [48], and KOR antagonism was sufficient to decrease some psychotic symptoms [49]. While still preliminary, these findings suggest that selective KOR antagonists might also have therapeutic utility in the adjunctive treatment of psychosis [50].
Pharmacological KOR activation in the PFC is known to produce aversion [51] and anxiety [52] and KOR/dynorphin activation in other brain areas can also alter cognition by disrupting attention [53,54,–55] or behavioral inhibition [25, 56]. KOR activation has immediate and remote effects on behavior [57, 58]. and we observed multiple phases of response disruptions during delayed alternation test sessions. During the initial period following systemic KOR agonist treatment, we reported a decrease in the total responses in control mice in the delayed alternation task. This decrease in responding likely corresponds with previously reported [59] observations that systemic KOR activation decreases locomotion. Drug withdrawal also decreased responding in the task. However, the source of these decreases in responding was previously undetermined. Microinjection of a KOR agonist into the substantia nigra pars reticulata in young rats showed that KOR activation in this region disinhibited motor responding [60], contrary to the suppressive effects of systemic KOR injections. We chose to examine our data in bins across the session due to these complex locomotor effects and based on our previous studies showing that KOR activation produces perseverative responses during the later phase of an operant test session in a behavioral inhibition task [25]. Although KOR activation can significantly suppress responding during operant sessions [53,54,–55], these suppressive effects were not evident when KOR activation was blocked in the PFC. Our data indicate that there is a component of the behavioral response being mediated by the prefrontal cortex that contributes to the prolonged suppression of responding caused by systemic KOR activation. The underlying elements contributing to this suppression have not yet been established in rodents. Many pharmacological manipulations that produce aversive or hallucinogenic effects in humans often alter locomotor behavior in rodents, but directly measuring hallucinatory effects in mice is challenging. We speculate that the initial period of behavioral suppression may be related to the dissociative or hallucinogenic effects of KOR activation, and the late phase error responses may reflect a perseverative mode of responding, leading to decreased lever switching or suppressed ability to inhibit responding. KOR activation likely contributes to disruptions in behavior over relatively long periods of time by producing complex patterns of circuit inhibition and excitation through recruitment of particular intracellular signaling elements and engagement of associated homeostatic responses.
Based on the wide pre- and postsynaptic distribution of KORs and dynorphins in the PFC [18, 26, 51, 52], it is likely that KOR in the PFC has actions in many different circuits and this coordinated disruption of multiple nodes of activity could lead to the loss of cognitive control following stress. Single-cell gene expression studies in cortical regions in mice have suggested diverse clusters of cells that are enriched in KOR and prodynorphin expression [61]. KOR activation in presynaptic terminals in the PFC decreases the local release of dopamine from ventral tegmental area neurons [51] and glutamate from basolateral amygdala neurons [52]. We aimed to dissociate pre- and postsynaptic contributions of KOR activation in the prefrontal cortex and found that virally mediated excision of KORs from the PFC blocked disruptions during the early phase of the task. Although adenoviral strategies can lead to some retrograde transport that may delete KOR from select inputs into the prefrontal cortex, we expect that the bulk of the KOR deletion in our experiments is likely to occur in prefrontocortical cells. We observed that KOR agonist-mediated disruptions were blocked by postsynaptic KOR deletion; however, withdrawal-mediated disruptions in behavior were not. Together, these data suggest that presynaptic KOR effects contribute to dynorphin-mediated changes in behavior during opioid withdrawal. Further studies using retrograde viral approaches could specify the contribution of each KOR-containing cortical input to shifts in behavioral strategies during withdrawal.
Clinical trials with KOR antagonists have shown promising results for further development, but short-acting KOR antagonists will only be effective if used during periods of time in which dynorphin is released. The ligand sensor kLight1.2a enables the measurement of dynorphin release dynamics in the brain and can reveal the types of behavioral or environmental events that are associated with increased dynorphin activity. Opioid withdrawal and abstinence have previously been shown to drive changes in the dynorphin/KOR system [62], but direct evidence for this relationship relied on techniques with low temporal precision, such as immunohistochemistry. In humans, binge psychostimulant use has been associated with increased dynorphin release in the prefrontal cortex [16], and chronic opioid use is likely to produce similar increases in PFC dynorphin release. Using kLight1.2a, we demonstrated that naloxone-precipitated morphine withdrawal increased dynorphin release in the prefrontal cortex of mice; however, the cell populations involved in this effect are unknown. Our immunohistochemical studies show that dynorphin may be released from pdynCre PFC neurons following optogenetic stimulation and the same stimulation paradigm produced disruptions in delayed alternation behavior, suggesting that PFC dynorphin neuron activation could contribute to dysfunction in working memory. PdynCre neurons are likely to release multiple signaling molecules following optical stimulation, and further characterization of the subpopulations of dynorphin neurons in the PFC is required to determine the signaling pathways through which withdrawal-mediated effects might occur. In addition, stimulation of pdynCre PFC neurons may produce downstream activity in other dynorphin-containing afferents to the PFC. Our data strongly suggest that local dynorphin neurons in the PFC are likely to contribute to the dynorphin-dependent behaviors that we observe; however, developing a comprehensive atlas of dynorphin projections into the prefrontal cortex and determining physiological conditions leading to dynorphin release from each subpopulation of dynorphin neurons would help to further resolve the circuits involved in drug withdrawal.
Our characterizations of kLight1.2a demonstrate that we can detect both pharmacological and endogenously released KOR ligands using this sensor. However, the use of naloxone to precipitate withdrawal produces challenges in determining whether the magnitude of changes with endogenous dynorphin corresponds to the fluorescent signals from differing concentrations of U50,488. We found that a 1 mg/kg dose of naloxone blunted the U50,488 signal when the two compounds were administered simultaneously. This suggests that the peak magnitude of endogenous dynorphin release during 1 mg/kg naloxone-precipitated morphine withdrawal may be limited by naloxone treatment. However, there are likely to be differences in the temporal dynamics between endogenously release dynorphin and experiment-administered compounds that could influence the detection of KOR ligands. Overall, our characterizations demonstrate the high specificity and sensitivity of kLight1.2a in detecting KOR ligands in vitro and in vivo and parameters of kLight1.2a interactions with nonselective opioid receptor-targeted compounds.
In summary, our studies determined spatial and temporal aspects of stress-induced cognitive dysfunction. Increased dynorphin release in the prefrontal cortex during drug withdrawal disrupted cognition, suggesting that KOR antagonists could be useful for treating cognitive symptoms in substance use disorders. Our studies indicate that effective clinical translation of kappa therapeutics will require further analysis of the contributions of the KOR/dynorphin system to cognition in animal models and humans.
References
Millan MJ, Agid Y, Brüne M, Bullmore ET, Carter CS, Clayton NS, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012;11:141–68.
Arnsten AFT. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–22.
Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology. 2010;210:137–47.
Knoll AT, Carlezon WA. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73.
Maqueda AE, Valle M, Addy PH, Antonijoan RM, Puntes M, Coimbra J, et al. Salvinorin-A induces intense dissociative effects, blocking external sensory perception and modulating interoception and sense of body ownership in humans. Int J Neuropsychopharmacol. 2015;18:pyv065.
Votinov M, Pripfl J, Windischberger C, Moser E, Sailer U, Lamm C. A functional polymorphism in the prodynorphin gene affects cognitive flexibility and brain activation during reversal learning. Front Behav Neurosci. 2015;9:172.
Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–69.
Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82.
Chartoff EH, Carlezon WA. Drug withdrawal conceptualized as a stressor. Behav Pharmacol. 2014;25:473–92.
Ingallinesi M, Rouibi K, Le Moine C, Papaleo F, Contarino A. CRF2 receptor-deficiency eliminates opiate withdrawal distress without impairing stress coping. Mol Psychiatry. 2012;17:1283–94.
Hurd YL, Herkenham M. Molecular alterations in the neostriatum of human cocaine addicts. Synapse. 1993;13:357–69.
Isola R, Zhang H, Tejwani GA, Neff NH, Hadjiconstantinou M. Acute nicotine changes dynorphin and prodynorphin mRNA in the striatum. Psychopharmacology. 2009;201:507–16.
Lindholm S, Ploj K, Franck J, Nylander I. Repeated ethanol administration induces short- and long-term changes in enkephalin and dynorphin tissue concentrations in rat brain. Alcohol. 2000;22:165–71.
Krystal AD, Pizzagalli DA, Smoski M, Mathew SJ, Nurnberger J, Lisanby SH, et al. A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating κ-opioid antagonism as a treatment for anhedonia. Nat Med. 2020;26:760–8.
Bazov I, Kononenko O, Watanabe H, Kuntić V, Sarkisyan D, Taqi MM, et al. The endogenous opioid system in human alcoholics: molecular adaptations in brain areas involved in cognitive control of addiction. Addiction Biol. 2013;18:161–9.
Martinez D, Slifstein M, Matuskey D, Nabulsi N, Zheng M-Q, Lin S-F, et al. Kappa-opioid receptors, dynorphin, and cocaine addiction: a positron emission tomography study. Neuropsychopharmacology. 2019;44:1720–7.
Sirohi S, Bakalkin G, Walker BM. Alcohol-induced plasticity in the dynorphin/kappa-opioid receptor system. Front Mol Neurosci. 2012;5:95.
Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Autoradiographic differentiation of mu, delta, and kappa opioid receptors in the rat forebrain and midbrain. J Neurosci. 1987;7:2445–64.
Sohn J, Hioki H, Okamoto S, Kaneko T. Preprodynorphin-expressing neurons constitute a large subgroup of somatostatin-expressing GABAergic interneurons in the mouse neocortex. J Comp Neurol. 2014;522:1506–26.
Rossi MA, Hayrapetyan VY, Maimon B, Mak K, Je HS, Yin HH. Prefrontal cortical mechanisms underlying delayed alternation in mice. J Neurophysiol. 2012;108:1211–22.
Abraham AD, Schattauer SS, Reichard KL, Cohen JH, Fontaine HM, Song AJ, et al. Estrogen regulation of GRK2 inactivates kappa opioid receptor signaling mediating analgesia, but not aversion. J Neurosci. 2018;38:8031–43.
Chavkin C, Cohen JH, Land BB. Repeated administration of norbinaltorphimine produces cumulative kappa opioid receptor inactivation. Front Pharmacol. 2019;10:88.
Reichard KL, Newton KA, Rivera ZMG, Menezes PMS, de, Schattauer SS, Land BB, et al. Regulation of kappa opioid receptor inactivation depends on sex and cellular site of antagonist action. Mol Pharmacol. 2020;98:548–58.
Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, et al. Kappa opioid receptor-induced aversion requires p38 MAPK activation in VTA dopamine neurons. J Neurosci. 2015;35:12917–31.
Abraham AD, Fontaine HM, Song AJ, Andrews MM, Baird MA, Kieffer BL, et al. Opioid receptor activation in dopamine neurons disrupts behavioral inhibition. Neuropsychopharmacology. 2018;43:362–72.
Khachaturian H, Watson SJ, Lewis ME, Coy D, Goldstein A, Akil H. Dynorphin immunocytochemistry in the rat central nervous system. Peptides. 1982;3:941–54.
Lemos JC, Roth CA, Chavkin C. Signaling events initiated by kappa opioid receptor activation: quantification and immunocolocalization using phospho-selective KOR, p38 MAPK, and KIR 3.1 antibodies. Methods Mol Biol. 2011;717:197–219.
Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–9.
McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, et al. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J Biol Chem. 2004;279:1810–8.
Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, et al. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–84.
Krashes MJ, Shah BP, Madara JC, Olson DP, Strochlic DE, Garfield AS, et al. A novel excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature. 2014;507:238–42.
Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, et al. Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron. 2015;87:1063–77.
Akil H, Young E, Walker JM, Watson SJ. The many possible roles of opioids and related peptides in stress-induced analgesia. Ann N Y Acad Sci. 1986;467:140–53.
Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin κ-opioid system. J Neurosci. 2008;28:407–14.
Lemos JC, Roth CA, Messinger DI, Gill HK, Phillips PEM, Chavkin C. Repeated stress dysregulates κ-opioid receptor signaling in the dorsal raphe through a p38α MAPK-dependent mechanism. J Neurosci. 2012;32:12325–36.
Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 2012;76:804–12.
Koob GF. Neurobiology of opioid addiction: opponent process, hyperkatifeia, and negative reinforcement. Biol Psychiatry. 2020;87:44–53.
Zan G-Y, Wang Q, Wang Y-J, Liu Y, Hang A, Shu X-H, et al. Antagonism of κ opioid receptor in the nucleus accumbens prevents the depressive-like behaviors following prolonged morphine abstinence. Behavioural Brain Res. 2015;291:334–41.
Kest B, Palmese CA, Hopkins E, Adler M, Juni A. Assessment of acute and chronic morphine dependence in male and female mice. Pharmacol Biochem Behav. 2001;70:149–56.
Kelsey JE, Verhaak AMS, Schierberl KC. The kappa-opioid receptor antagonist, nor-binaltorphimine (nor-BNI), decreases morphine withdrawal and the consequent conditioned place aversion in rats. Behavioural Brain Res. 2015;283:16–21.
Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong W-H, et al. Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science. 2018;360:eaat4422.
Douglas AJ, Clarke G, MacMillan SJ, Bull PM, Neumann I, Way SA, et al. Effects of the kappa-opioid agonist U50,488 on parturition in rats. Br J Pharmacol. 1993;109:251–8.
Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharm Exp Ther. 1992;260:1237–43.
Schattauer SS, Land BB, Reichard KL, Abraham AD, Burgeno LM, Kuhar JR, et al. Peroxiredoxin 6 mediates Gαi protein-coupled receptor inactivation by cJun kinase. Nat Commun. 2017;8:743.
Rapeli P, Kivisaari R, Autti T, Kähkönen S, Puuskari V, Jokela O, et al. Cognitive function during early abstinence from opioid dependence: a comparison to age, gender, and verbal intelligence matched controls. BMC Psychiatry. 2006;6:9.
Heikkilä L, Rimón R, Ternius L. Dynorphin A and substance P in the cerebrospinal fluid of schizophrenic patients. Psychiatry Res. 1990;34:229–36.
Lindström LH. Clinical and biological markers for outcome in schizophrenia: a review of a longitudinal follow-up study in Uppsala schizophrenia research project. Neuropsychopharmacology. 1996;14:23S–26S.
Pickar D, Vartanian F, Bunney WE, Maier HP, Gastpar MT, Prakash R, et al. Short-term naloxone administration in schizophrenic and manic patients: a World Health Organization Collaborative Study. Arch Gen Psychiatry. 1982;39:313–9.
Schmauss C, Yassouridis A, Emrich HM. Antipsychotic effect of buprenorphine in schizophrenia. Am J Psychiatry. 1987;144:1340–2.
Clark SD, Abi-Dargham A. The role of dynorphin and the kappa opioid receptor in the symptomatology of schizophrenia: a review of the evidence. Biol Psychiatry. 2019;86:502–11.
Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz-Kuszak KN, Bäckman CM, et al. Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology. 2013;38:1770–9.
Tejeda HA, Hanks AN, Scott L, Mejias-Aponte C, Hughes ZA, O’Donnell P. Prefrontal cortical kappa opioid receptors attenuate responses to amygdala inputs. Neuropsychopharmacology. 2015;40:2856–64.
Nemeth CL, Paine TA, Rittiner JE, Béguin C, Carroll FI, Roth BL, et al. Role of kappa-opioid receptors in the effects of salvinorin A and ketamine on attention in rats. Psychopharmacol (Berl). 2010;210:263–74.
Paine TA, Tomasiewicz HC, Zhang K, Carlezon WA. Sensitivity of the five-choice serial reaction time task to the effects of various psychotropic drugs in Sprague-Dawley rats. Biol Psychiatry. 2007;62:687–93.
Shannon HE, Eberle EL, Mitch CH, McKinzie DL, Statnick MA. Effects of kappa opioid receptor agonists on attention as assessed by a 5-choice serial reaction time task in rats. Neuropharmacology. 2007;53:930–41.
Walker BM, Kissler JL. Dissociable effects of kappa-opioid receptor activation on impulsive phenotypes in wistar rats. Neuropsychopharmacology. 2013;38:2278–85.
Ehrich JM, Phillips PEM, Chavkin C. Kappa opioid receptor activation potentiates the cocaine-induced increase in evoked dopamine release recorded in vivo in the mouse nucleus accumbens. Neuropsychopharmacology. 2014;39:3036–48.
Chartoff EH, Ebner SR, Sparrow A, Potter D, Baker PM, Ragozzino ME, et al. Relative timing between kappa opioid receptor activation and cocaine determines the impact on reward and dopamine release. Neuropsychopharmacology. 2016;41:989–1002.
Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, et al. Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria. Sci Signal. 2016;9:ra117.
Collins RL, Zavala AR, Nazarian A, McDougall SA. kappa-Opioid receptors in the substantia nigra pars reticulata mediate the U-50,488-induced locomotor activity of preweanling rats. Brain Res Dev Brain Res. 2000;119:97–103.
Smith SJ, Sümbül U, Graybuck LT, Collman F, Seshamani S, Gala R, et al. Single-cell transcriptomic evidence for dense intracortical neuropeptide networks. ELife. 2019;8:e47889.
Lalanne L, Ayranci G, Filliol D, Gavériaux-Ruff C, Befort K, Kieffer BL, et al. Kappa opioid receptor antagonism and chronic antidepressant treatment have beneficial activities on social interactions and grooming deficits during heroin abstinence. Addiction Biol. 2017;22:1010–21.
Acknowledgements
We thank Mackenzie M. Andrews, Zeena M. G. Rivera, and Juliana Chase for assistance with data collection and data analysis for this manuscript.
Funding
Funding sources: P50-MH106428 (CC), P30-DA048736 (CC), R01-DA030074 (CC), R21-MH108839 (BBL), and T32-DA07278 (CC and ADA).
Author information
Authors and Affiliations
Contributions
ADA, SMC, and SSS conducted the experiments, ADA and CC wrote the manuscript, BAW generated software for data extraction and analysis, GOM, KM, and SSS generated reagents for experiments, ADA, BBL, and CC designed the experiments, and CC and LT secured funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Abraham, A.D., Casello, S.M., Schattauer, S.S. et al. Release of endogenous dynorphin opioids in the prefrontal cortex disrupts cognition. Neuropsychopharmacol. 46, 2330–2339 (2021). https://doi.org/10.1038/s41386-021-01168-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-021-01168-2
This article is cited by
-
Dynorphin / kappa-opioid receptor regulation of excitation-inhibition balance toggles afferent control of prefrontal cortical circuits in a pathway-specific manner
Molecular Psychiatry (2023)
-
The claustrum-prelimbic cortex circuit through dynorphin/κ-opioid receptor signaling underlies depression-like behaviors associated with social stress etiology
Nature Communications (2023)
-
The dynorphin/kappa opioid receptor mediates adverse immunological and behavioral outcomes induced by repetitive blast trauma
Journal of Neuroinflammation (2022)
-
Pushing the frontiers: tools for monitoring neurotransmitters and neuromodulators
Nature Reviews Neuroscience (2022)
