
Patterns of Coevolutionary Adaptations across Time and Space in Mouse Gammaretroviruses and Three Restrictive Host Factors

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sources of Mouse DNAs and RNAs
2.2. Primers, PCR, Cloning and Sequencing
2.3. Identification of Variants in Fv1 and Slc7a1(CAT1) Genes in M. m. castaneus
2.4. Identification of the Subspecies Origin of Fv1
2.5. Phylogenetic and Positive Selection Analyses
3. Results and Discussion
3.1. E-MLVs and Their CAT1 Receptor
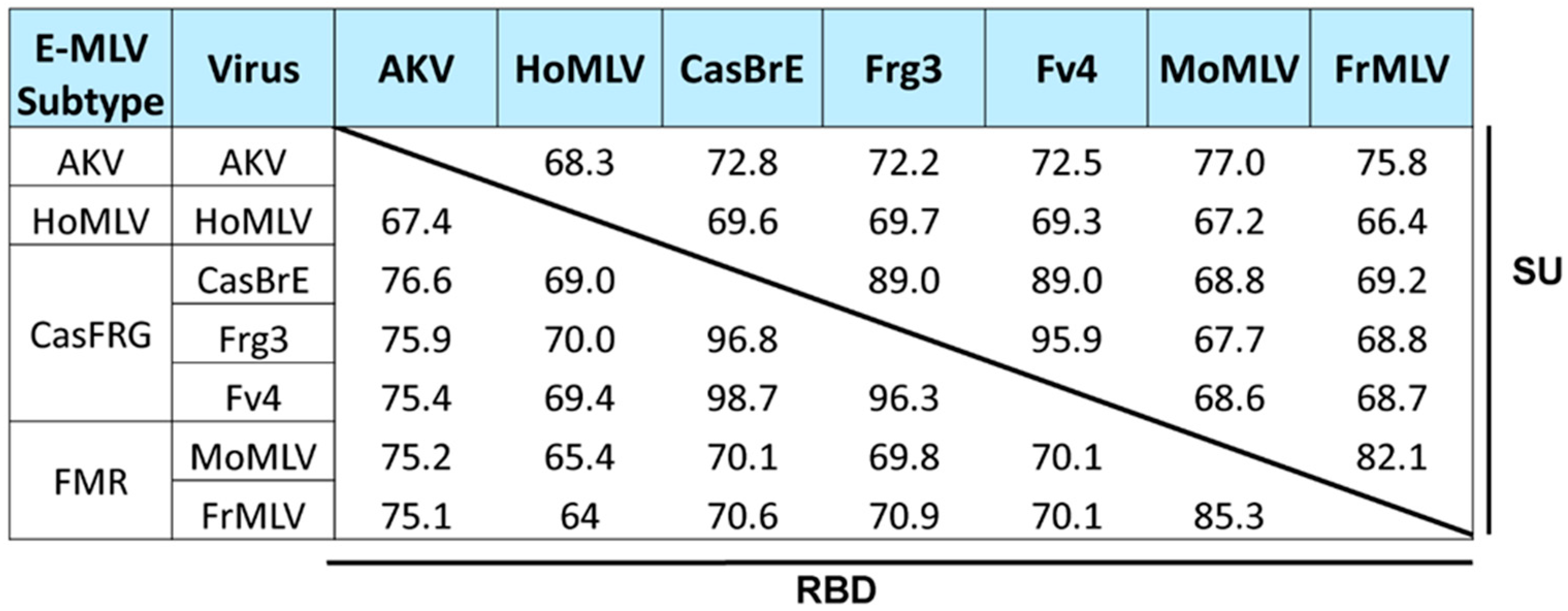
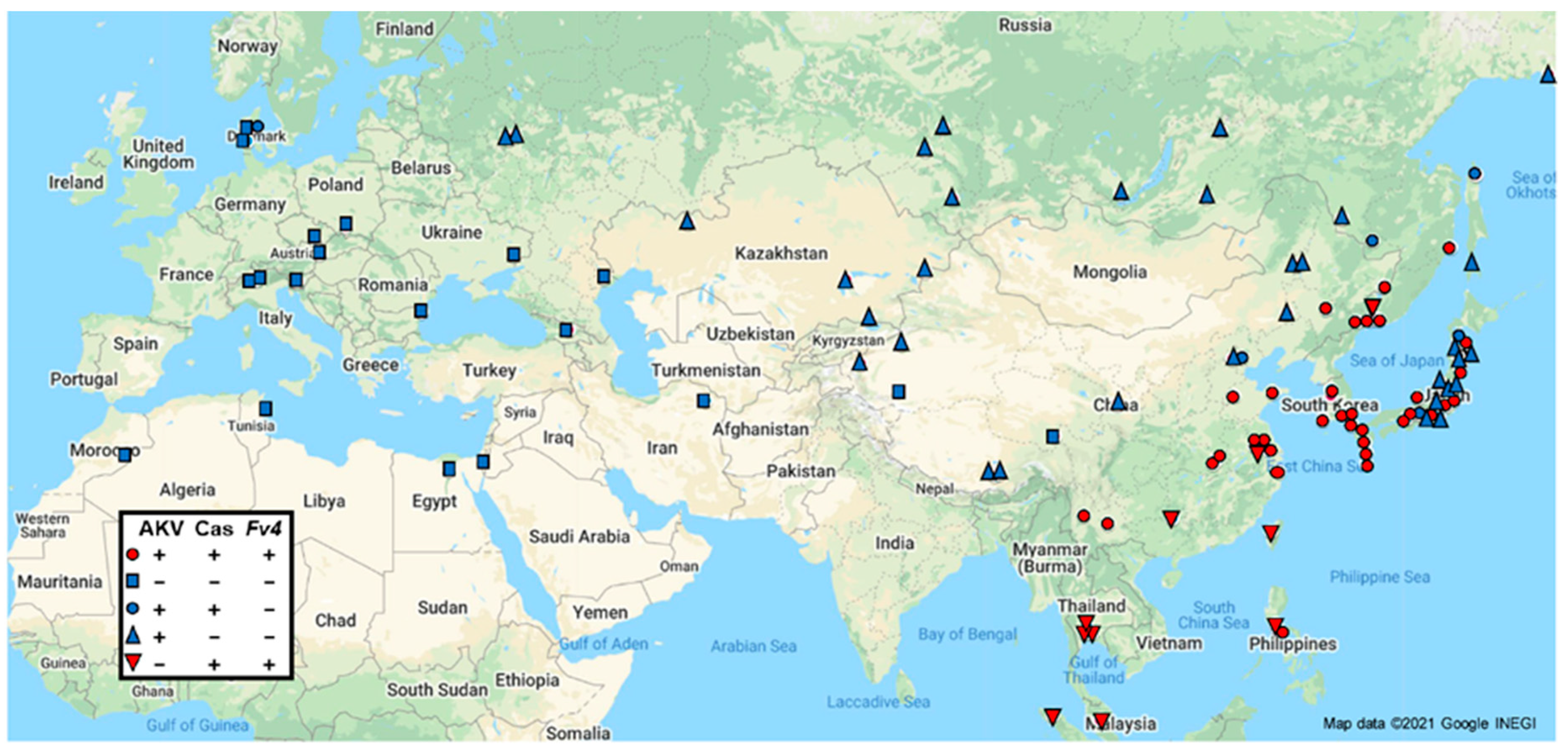
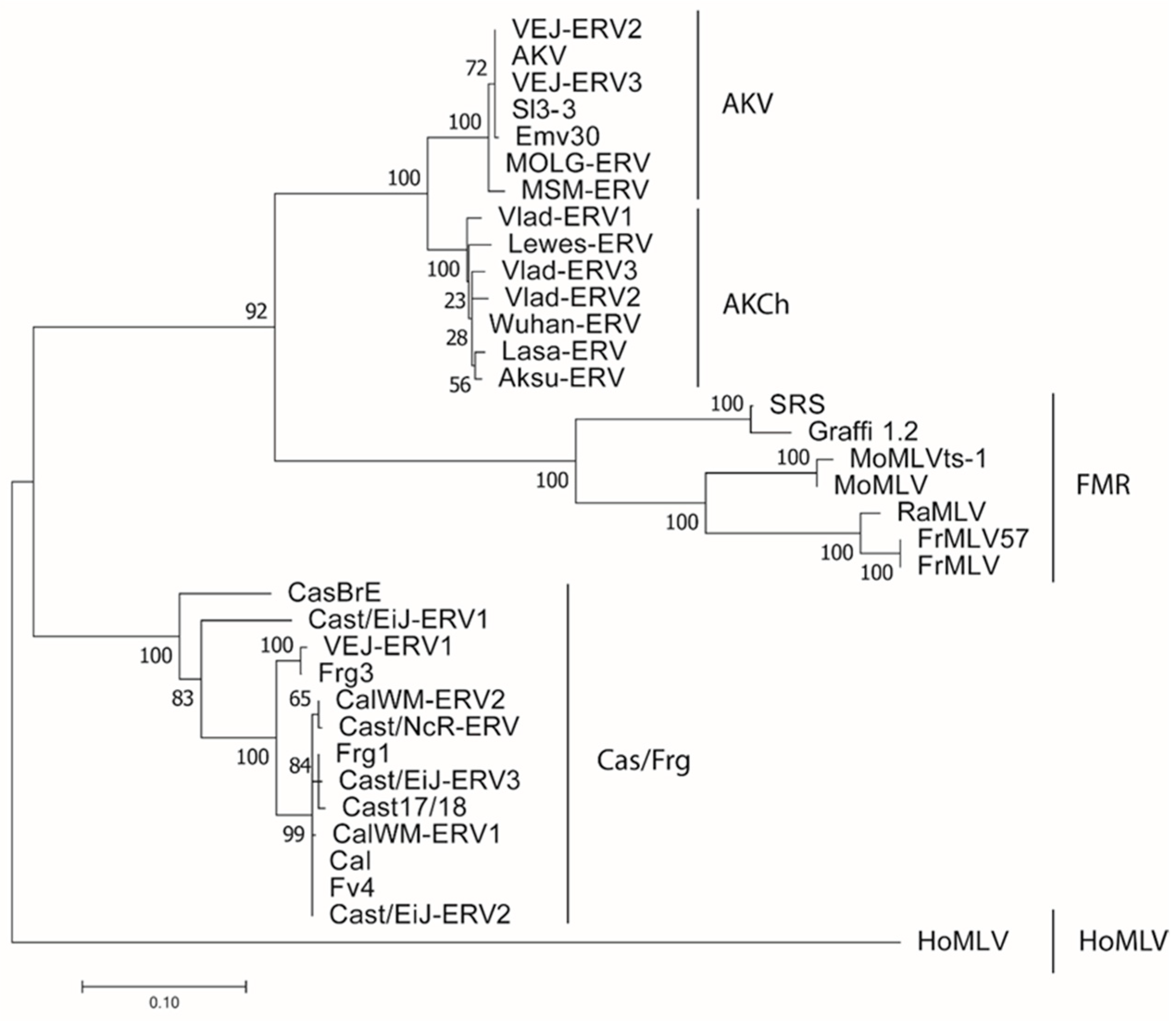
3.1.1. E-MLVs
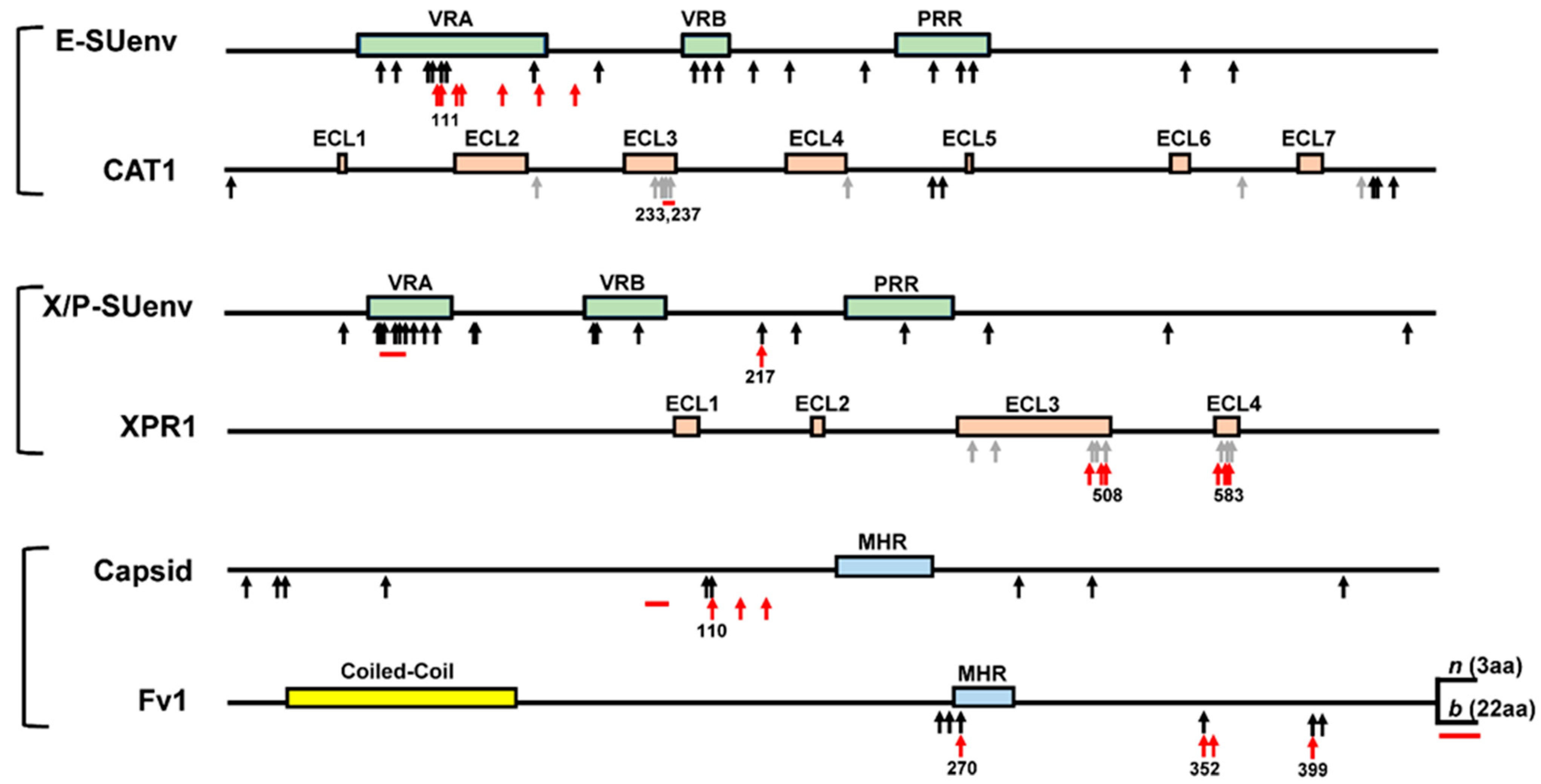
3.1.2. CAT1 Receptor
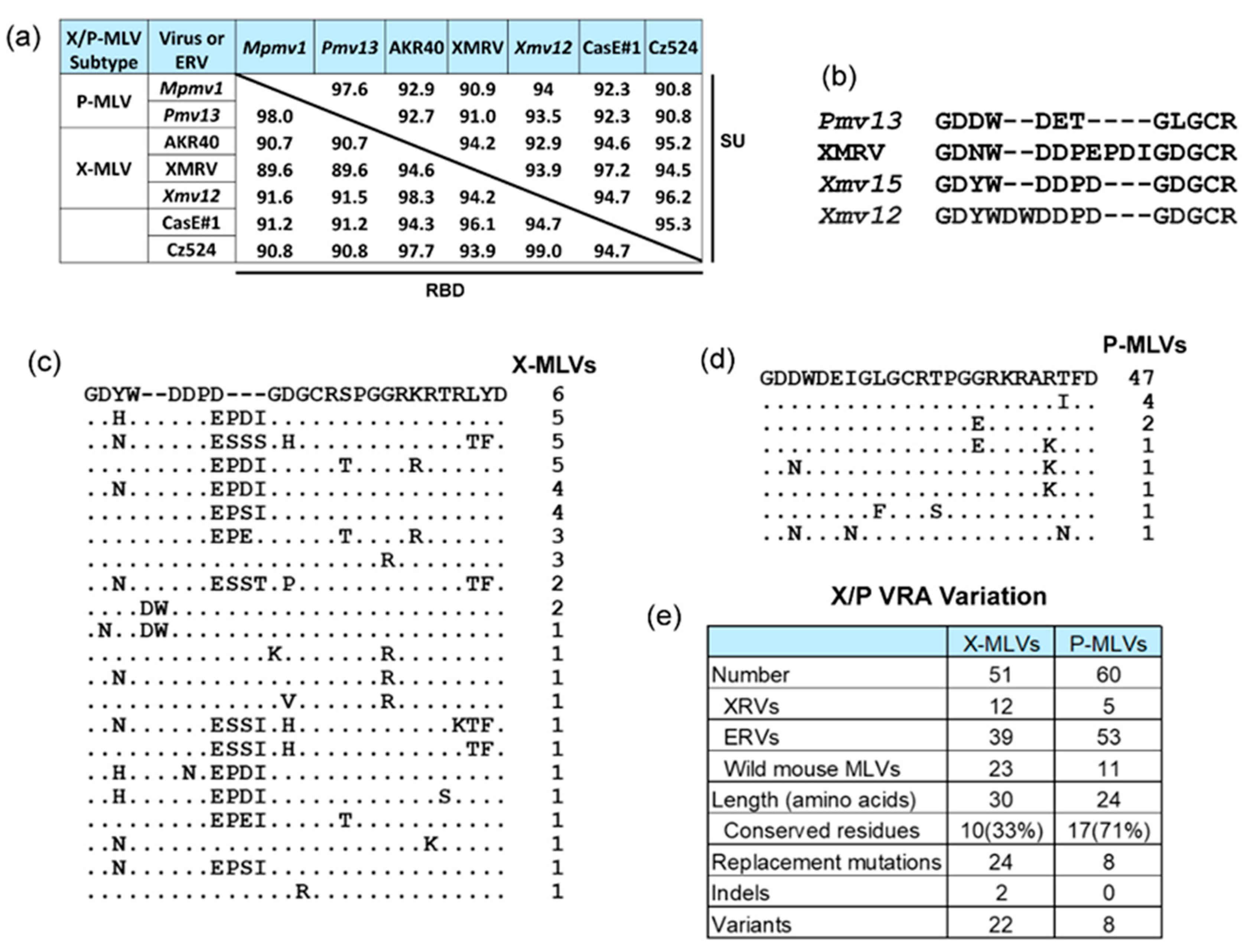
3.2. X/P-MLVs and Their XPR1 Receptor
3.2.1. XPR1
3.2.2. X/P-MLVs
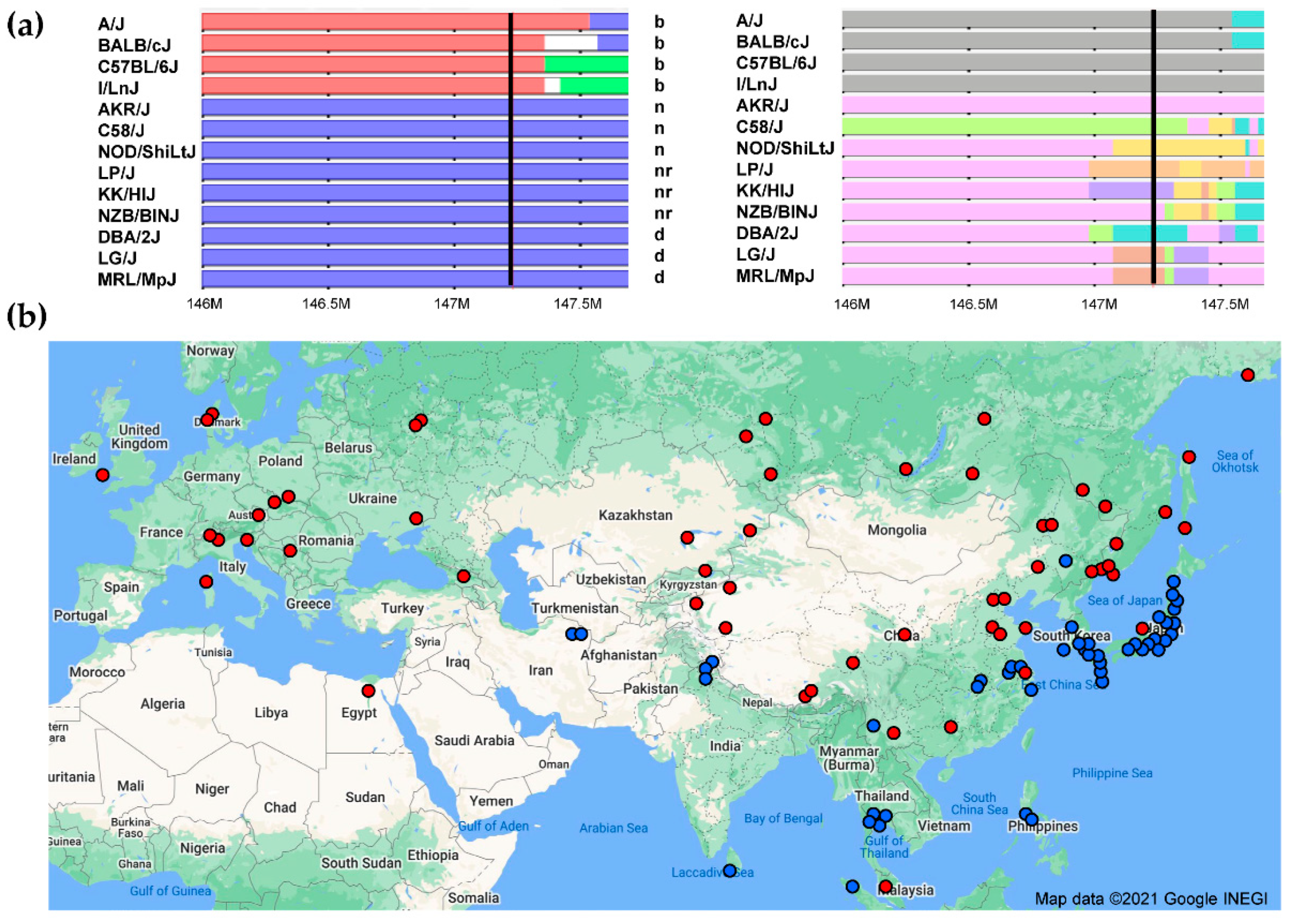
3.3. Fv1 and Its Capsid Target
3.3.1. Fv1
3.3.2. Viral Capsid Target of Fv1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jenkins, N.A.; Copeland, N.G.; Taylor, B.A.; Lee, B.K. Organization, distribution, and stability of endogenous ecotropic murine leukemia virus DNA sequences in chromosomes of Mus musculus. J. Virol. 1982, 43, 26–36. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, R.R.; Khan, A.S.; Hoggan, M.D.; Hartley, J.W.; Martin, M.A.; Repaske, R. Specific hybridization probes demonstrate fewer xenotropic than mink cell focus-forming murine leukemia virus env-related sequences in DNAs from inbred laboratory mice. J. Virol. 1986, 58, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Frankel, W.N.; Stoye, J.P.; Taylor, B.A.; Coffin, J.M. A linkage map of endogenous murine leukemia proviruses. Genetics 1990, 124, 221–236. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.R.; Didion, J.P.; Buus, R.J.; Bell, T.A.; Welsh, C.E.; Bonhomme, F.; Yu, A.H.; Nachman, M.W.; Pialek, J.; et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat. Genet. 2011, 43, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Kozak, C.A.; O’Neill, R.R. Diverse wild mouse origins of xenotropic, mink cell focus-forming, and two types of ecotropic proviral genes. J. Virol. 1987, 61, 3082–3088. [Google Scholar] [CrossRef] [Green Version]
- Cucchi, T.; Papayianni, K.; Cersoy, S.; Aznar-Cormano, L.; Zazzo, A.; Debruyne, R.; Berthon, R.; Balasescu, A.; Simmons, A.; Valla, F.; et al. Tracking the Near Eastern origins and European dispersal of the western house mouse. Sci. Rep. 2020, 10, 8276. [Google Scholar] [CrossRef]
- Macholan, M.; Munclinger, P.; Sugerkova, M.; Dufkova, P.; Bimova, B.; Bozikova, E.; Zima, J.; Pialek, J. Genetic analysis of autosomal and X-linked markers across a mouse hybrid zone. Evolution 2007, 61, 746–771. [Google Scholar] [CrossRef] [PubMed]
- Boso, G.; Kozak, C.A. Retroviral Restriction Factors and Their Viral Targets: Restriction Strategies and Evolutionary Adaptations. Microorganisms 2020, 8, 1965. [Google Scholar] [CrossRef] [PubMed]
- Van Valen, L. A new evolutionary law. Evol. Theory 1973, 1, 1–30. [Google Scholar]
- Yan, Y.; Buckler-White, A.; Wollenberg, K.; Kozak, C.A. Origin, antiviral function and evidence for positive selection of the gammaretrovirus restriction gene Fv1 in the genus Mus. Proc. Natl. Acad. Sci. USA 2009, 106, 3259–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Liu, Q.; Wollenberg, K.; Martin, C.; Buckler-White, A.; Kozak, C.A. Evolution of functional and sequence variants of the mammalian XPR1 receptor for mouse xenotropic gammaretroviruses and the human-derived XMRV. J. Virol. 2010, 84, 11970–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.N. The Geographic Mosaic of Coevolution; University of Chicago Press: Chicago, IL, USA, 2005. [Google Scholar]
- Jern, P.; Stoye, J.P.; Coffin, J.M. Role of APOBEC3 in genetic diversity among endogenous murine leukemia viruses. PLoS Genet. 2007, 3, e183. [Google Scholar] [CrossRef] [Green Version]
- Hasenkamp, N.; Solomon, T.; Tautz, D. Selective sweeps versus introgression–population genetic dynamics of the murine leukemia virus receptor Xpr1 in wild populations of the house mouse (Mus musculus). BMC Evol. Biol. 2015, 15, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamunusinghe, D.; Naghashfar, Z.; Buckler-White, A.; Plishka, R.; Baliji, S.; Liu, Q.; Kassner, J.; Oler, A.; Hartley, J.; Kozak, C.A. Sequence diversity, intersubgroup relationships, and origins of the mouse leukemia gammaretroviruses of laboratory and wild mice. J. Virol. 2016, 90, 4186–4198. [Google Scholar] [CrossRef] [Green Version]
- Halligan, D.L.; Kousathanas, A.; Ness, R.W.; Harr, B.; Eory, L.; Keane, T.M.; Adams, D.J.; Keightley, P.D. Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genet. 2013, 9, e1003995. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 11, 111011–111033. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.R.; de Villena, F.P.; McMillan, L. Comparative analysis and visualization of multiple collinear genomes. BMC Bioinform. 2012, 13 (Suppl. 3), S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nuc. Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Alamgir, A.S.; Owens, N.; Lavignon, M.; Malik, F.; Evans, L.H. Precise identification of endogenous proviruses of NFS/N mice participating in recombination with Moloney ecotropic murine leukemia virus (MuLV) to generate polytropic MuLVs. J. Virol. 2005, 79, 4664–4671. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battini, J.L.; Heard, J.M.; Danos, O. Receptor choice determinants in the envelope glycoproteins of amphotropic, xenotropic, and polytropic murine leukemia viruses. J. Virol. 1992, 66, 1468–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Laigret, F.; Martin, M.A.; Repaske, R. Characterization of a molecularly cloned retroviral sequence associated with Fv-4 resistance. J. Virol. 1985, 55, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Steffen, D.L.; Bird, S.; Weinberg, R.A. Evidence for the Asiatic origin of endogenous AKR-type murine leukemia proviruses. J. Virol. 1980, 35, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Inaguma, Y.; Miyashita, N.; Moriwaki, K.; Huai, W.C.; Jin, M.L.; He, X.Q.; Ikeda, H. Acquisition of two endogenous ecotropic murine leukemia viruses in distinct Asian wild mouse populations. J. Virol. 1991, 65, 1796–1802. [Google Scholar] [CrossRef] [Green Version]
- Voytek, P.; Kozak, C.A. Nucleotide sequence and mode of transmission of the wild mouse ecotropic virus, HoMuLV. Virology 1989, 173, 58–67. [Google Scholar] [CrossRef]
- Skorski, M.; Bamunusinghe, D.; Liu, Q.; Shaffer, E.; Kozak, C.A. Distribution of endogenous gammaretroviruses and variants of the Fv1 restriction gene in individual mouse strains and strain subgroups. PLoS ONE 2019, 14, e0219576. [Google Scholar] [CrossRef] [Green Version]
- Yonekawa, H.; Moriwaki, K.; Gotoh, O.; Miyashita, N.; Matsushima, Y.; Shi, L.; Cho, W.; Zhen, X.; Tagashira, Y. Hybrid origin of Japanese mice “Mus musculus molossinus”: Evidence from restriction analysis of mitochondrial DNA. Mol. Biol. Evol. 1988, 5, 63–78. [Google Scholar] [PubMed]
- Gardner, M.B. Type C viruses of wild mice: Characterization and natural history of amphotropic, ecotropic, and xenotropic MuLV. Curr. Top. Microbiol. Immunol. 1978, 79, 215–259. [Google Scholar] [PubMed]
- Rasheed, S.; Gardner, M.B. Resistance to fibroblasts and hematopoietic cells to ecotropic murine leukemia virus infection; an Akvr-1r gene effect. Int. J. Cancer 1983, 31, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Boursot, P.; Auffray, J.C.; Britton-Davidian, J.; Bonhomme, F. The evolution of house mice. Ann. Rev. Ecol. Syst. 1993, 24, 119–152. [Google Scholar] [CrossRef]
- Cloyd, M.W.; Hartley, J.W.; Rowe, W.P. Lymphomagenicity of recombinant mink cell focus-inducing murine leukemia viruses. J. Exp. Med. 1980, 151, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Oldstone, M.B.; Jensen, F.; Elder, J.; Dixon, F.J.; Lampert, P.W. Pathogenesis of the slow disease of the central nervous system associated with wild mouse virus. III. Role of input virus and MCF recombinants in disease. Virology 1983, 128, 154–165. [Google Scholar] [CrossRef]
- Kozak, C.A. Naturally occurring polymorphisms of the mouse gammaretrovirus receptors CAT-1 and XPR1 alter virus tropism and pathogenicity. Adv. Virol. 2011, 975801. [Google Scholar] [CrossRef] [Green Version]
- Davey, R.A.; Zuo, Y.; Cunningham, J.M. Identification of a receptor-binding pocket on the envelope protein of friend murine leukemia virus. J. Virol. 1999, 73, 3758–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.T.; Wu, T.; Kozak, C.A. Novel host range and cytopathic variant of ecotropic Friend murine leukemia virus. J. Virol. 2004, 78, 12189–12197. [Google Scholar] [CrossRef] [Green Version]
- Voytek, P.; Kozak, C. HoMuLV: A novel pathogenic ecotropic virus isolated from the European mouse, Mus hortulanus. Virology 1988, 165, 469–475. [Google Scholar] [CrossRef]
- Jung, Y.T.; Kozak, C.A. Generation of novel syncytium-inducing and host range variants of ecotropic moloney murine leukemia virus in Mus spicilegus. J. Virol. 2003, 77, 5065–5072. [Google Scholar] [CrossRef] [Green Version]
- Ferrarone, J.; Knoper, R.C.; Li, R.; Kozak, C.A. Second site mutation in the virus envelope expands the host range of a cytopathic variant of Moloney murine leukemia virus. Virology 2012, 433, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Park, B.H.; Matuschke, B.; Lavi, E.; Gaulton, G.N. A point mutation in the env gene of a murine leukemia virus induces syncytium formation and neurologic disease. J. Virol. 1994, 68, 7516–7524. [Google Scholar] [CrossRef] [Green Version]
- Masuda, M.; Hanson, C.A.; Hoffman, P.M.; Ruscetti, S.K. Analysis of the unique hamster cell tropism of ecotropic murine leukemia virus PVC-211. J. Virol. 1996, 70, 8534–8539. [Google Scholar] [CrossRef] [Green Version]
- Morse, H.C., III (Ed.) Introduction. In Origins of Inbred Mice; Academic Press: New York, NY, USA, 1978; pp. 1–31. [Google Scholar]
- Gross, L. “Spontaneous” leukemia developing in C3H mice following inoculation in infancy, with AK-leukemic extracts, or AK-embryos. Proc. Soc. Exp. Biol. Med. 1951, 76, 27–32. [Google Scholar] [CrossRef]
- Moloney, J.B. Biological studies on a lymphoid-leukemia virus extracted from sarcoma 37. I. Origin and introductory investigations. J. Natl. Cancer Inst. 1960, 24, 933–951. [Google Scholar] [PubMed]
- Craigie, J. Sarcoma 37 and ascites tumours. Ann. R. Coll. Surg. Engl. 1952, 11, 287–299. [Google Scholar]
- Friend, C. Cell-free transmission in adult Swiss mice of a disease having the character of a leukemia. J. Exp. Med. 1957, 105, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Henzy, J.E.; Johnson, W.E. Pushing the endogenous envelope. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albritton, L.M.; Tseng, L.; Scadden, D.; Cunningham, J.M. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell 1989, 57, 659–666. [Google Scholar] [CrossRef]
- Eiden, M.V.; Farrell, K.; Wilson, C.A. Glycosylation-dependent inactivation of the ecotropic murine leukemia-virus receptor. J. Virol. 1994, 68, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Tavoloni, N.; Rudenholz, A. Variable transduction efficiency of murine leukemia retroviral vector on mammalian cells: Role of cellular glycosylation. Virology 1997, 229, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Closs, E.I.; Graf, P.; Habermeier, A.; Cunningham, J.M.; Forstermann, U. Human cationic amino acid transporters hCAT-1, hCAT-2A, and hCAT-2B: Three related carriers with distinct transport properties. Biochemistry 1997, 36, 6462–6468. [Google Scholar] [CrossRef]
- Eiden, M.V.; Farrell, K.; Warsowe, J.; Mahan, L.C.; Wilson, C.A. Characterization of a naturally occurring ecotropic receptor that does not facilitate entry of all ecotropic murine retroviruses. J. Virol. 1993, 67, 4056–4061. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Kozak, C.A. Novel postentry resistance to AKV ecotropic mouse gammaretroviruses in the African pygmy mouse, Mus minutoides. J. Virol. 2008, 82, 6120–6129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, T.; Yoshimoto, E.; Meruelo, D. Identification of amino acid residues critical for infection with ecotropic murine leukemia retrovirus. J. Virol. 1993, 67, 1310–1314. [Google Scholar] [CrossRef] [Green Version]
- Albritton, L.M.; Kim, J.W.; Tseng, L.; Cunningham, J.M. Envelope-binding domain in the cationic amino acid transporter determines the host range of ecotropic murine retroviruses. J. Virol. 1993, 67, 2091–2096. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.M.; Gao, Y.; Sanders, D.A. Fv-4: Identification of the defect in Env and the mechanism of resistance to ecotropic murine leukemia virus. J. Virol. 2001, 75, 11244–11248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.A. Xenotropic viruses: Murine leukemia viruses associated with NIH Swiss, NZB, and other mouse strains. Science 1973, 182, 1151–1153. [Google Scholar] [CrossRef]
- Hartley, J.W.; Wolford, N.K.; Old, L.J.; Rowe, W.P. New class of murine leukemia-virus associated with development of spontaneous lymphomas. Proc. Natl. Acad. Sci. USA 1977, 74, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Kozak, C.A. The mouse “xenotropic” gammaretroviruses and their XPR1 receptor. Retrovirology 2010, 7, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, D.; Touhami, J.; Charnet, P.; Sitbon, M.; Battini, J.L. Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 2013, 3, 1866–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, C.A. Evolution of different antiviral strategies in wild mouse populations exposed to different gammaretroviruses. Curr. Opin. Virol. 2013, 3, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Marin, M.; Tailor, C.S.; Nouri, A.; Kozak, S.L.; Kabat, D. Polymorphisms of the cell surface receptor control mouse susceptibilities to xenotropic and polytropic leukemia viruses. J. Virol. 1999, 73, 9362–9368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Liu, Q.; Kozak, C.A. Six host range variants of the xenotropic/polytropic gammaretroviruses define determinants for entry in the XPR1 cell surface receptor. Retrovirology 2009, 6, 87. [Google Scholar] [CrossRef] [Green Version]
- Bamunusinghe, D.; Liu, Q.; Plishka, R.; Dolan, M.A.; Skorski, M.; Oler, A.J.; Yedavalli, V.R.K.; Buckler-White, A.; Hartley, J.W.; Kozak, C.A. Recombinant origins of pathogenic and nonpathogenic mouse gammaretroviruses with polytropic host range. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Lavignon, M.; Evans, L. A multistep process of leukemogenesis in Moloney murine leukemia virus-infected mice that is modulated by retroviral pseudotyping and interference. J. Virol. 1996, 70, 3852–3862. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.H.; Alamgir, A.S.; Owens, N.; Weber, N.; Virtaneva, K.; Barbian, K.; Babar, A.; Malik, F.; Rosenke, K. Mobilization of endogenous retroviruses in mice after infection with an exogenous retrovirus. J. Virol. 2009, 83, 2429–2435. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, S.K.; Oliff, A.I.; Linemeyer, D.L.; Lander, M.R.; Lowy, D.R. Genomes of murine leukemia viruses isolated from wild mice. J. Virol. 1981, 39, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Kozak, C.A.; Hartley, J.W.; Morse, H.C., 3rd. Laboratory and wild-derived mice with multiple loci for production of xenotropic murine leukemia virus. J. Virol. 1984, 51, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, S.; Duch, M.; Pedersen, F.S. Change of tropism of SL3-2 murine leukemia virus, using random mutational libraries. J. Virol. 2004, 78, 9343–9351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, J.W.; Yetter, R.A.; Morse, H.C. A mouse gene on chromosome 5 that restricts infectivity of mink cell focus-forming recombinant murine leukemia viruses. J. Exp. Med. 1983, 158, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.T.; Lyu, M.S.; Buckler-White, A.; Kozak, C.A. Characterization of a polytropic murine leukemia virus proviral sequence associated with the virus resistance gene Rmcf of DBA/2 mice. J. Virol. 2002, 76, 8218–8224. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Yan, Y.; Kozak, C.A. Rmcf2, a xenotropic provirus in the Asian mouse species Mus castaneus, blocks infection by polytropic mouse gammaretroviruses. J. Virol. 2005, 79, 9677–9684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lilly, F. Susceptibility to two strains of Friend leukemia virus in mice. Science 1967, 155, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.K.; Kiggans, J.O.; Yang, D.M.; Ou, C.Y.; Tennant, R.W.; Brown, A.; Bassin, R.H. Synthesis and circularization of N- and B-tropic retroviral DNA Fv-1 permissive and restrictive mouse cells. Proc. Natl. Acad. Sci. USA 1980, 77, 2994–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, M.W.; Colbeck, E.; Ellis, S.A.; Stoye, J.P. Evolution of the retroviral restriction gene Fv1: Inhibition of non-MLV retroviruses. PLoS Pathog. 2014, 10, e1003968. [Google Scholar] [CrossRef] [Green Version]
- Benit, L.; DeParseval, N.; Casella, J.F.; Callebaut, I.; Cordonnier, A.; Heidmann, T. Cloning of a new murine endogenous retrovirus, MuERV-L, with strong similarity to the human HERV-L element and with a gag coding sequence closely related to the Fv1 restriction gene. J. Virol. 1997, 71, 5652–5657. [Google Scholar] [CrossRef] [Green Version]
- Best, S.; LeTissier, P.; Towers, G.; Stoye, J.P. Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 1996, 382, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Boso, G.; Buckler-White, A.; Kozak, C.A. Ancient evolutionary origin and positive selection of the retroviral restriction factor Fv1 in Muroid Rodents. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Young, G.R.; Yap, M.W.; Michaux, J.R.; Steppan, S.J.; Stoye, J.P. Evolutionary journey of the retroviral restriction gene Fv1. Proc. Natl. Acad. Sci. USA 2018, 115, 10130–10135. [Google Scholar] [CrossRef] [Green Version]
- Hartley, J.W.; Rowe, W.P.; Huebner, R.J. Host-range restrictions of murine leukemia viruses in mouse embryo cell cultures. J. Virol. 1970, 5, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Kozak, C.A. Analysis of wild-derived mice for Fv-1 and Fv-2 murine leukemia virus restriction loci: A novel wild mouse Fv-1 allele responsible for lack of host range restriction. J. Virol. 1985, 55, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Kozak, C.A.; Chakraborti, A. Single amino acid changes in the murine leukemia virus capsid protein gene define the target of Fv1 resistance. Virology 1996, 225, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.N.; Bock, M.; Towers, G.; Stoye, J.P. Identification of the regions of Fv1 necessary for murine leukemia virus restriction. J. Virol. 2001, 75, 5182–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Bell, T.A.; Churchill, G.A.; Pardo-Manuel de Villena, F. On the subspecific origin of the laboratory mouse. Nat. Genet. 2007, 39, 1100–1107. [Google Scholar] [CrossRef]
- Qi, C.F.; Bonhomme, F.; Buckler-White, A.; Buckler, C.; Orth, A.; Lander, M.R.; Chattopadhyay, S.K.; Morse, H.C., 3rd. Molecular phylogeny of Fv1. Mamm. Genome 1998, 9, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Gautsch, J.W.; Elder, J.H.; Schindler, J.; Jensen, F.C.; Lerner, R.A. Structural markers on core protein p30 of murine leukemia virus: Functional correlation with Fv-1 tropism. Proc. Natl. Acad. Sci. USA 1978, 75, 4170–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, N.; Schindler, J.; Hynes, R. Six-NB-tropic murine leukemia viruses derived from a B-tropic virus of BALB/c have altered p30. J. Virol. 1977, 21, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimoto, A.; Hartley, J.W.; Rowe, W.P. Fv-1 restriction of xenotropic and amphotropic murine leukemia virus genomes phenotypically mixed with ecotropic virus. Virology 1979, 93, 215–225. [Google Scholar] [CrossRef]
- Jung, Y.T.; Kozak, C.A. A single amino acid change in the murine leukemia virus capsid gene responsible for the Fv1nr phenotype. J. Virol. 2000, 74, 5385–5387. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.; Bock, M.; Ellis, S.; LeTissier, P.; Bishop, K.N.; Yap, M.W.; Taylor, W.; Stoye, J.P. Retroviral capsid determinants of Fv1 NB and NR tropism. J. Virol. 2004, 78, 9592–9598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.; Risser, R. Genetic interactions in the spontaneous production of endogenous murine leukemia virus in low leukemic mouse strains. J. Exp. Med. 1982, 156, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Moll, B.; Hartley, J.W.; Rowe, W.P. Induction of B-tropic and N-tropic murine leukemia virus from B10.BR/SgLi mouse embryo cell lines by 5-iodo-2’-deoxyuridine. J. Natl. Cancer. Inst. 1979, 63, 213–217. [Google Scholar]
- Rosenke, K.; Lavignon, M.; Malik, F.; Kolokithas, A.; Hendrick, D.; Virtaneva, K.; Peterson, K.; Evans, L.H. Profound amplification of pathogenic murine polytropic retrovirus release from coinfected cells. J. Virol. 2012, 86, 7241–7248. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorphisms at Receptor Critical Sites | MLV or ERV | Phenotype | Reference |
|---|---|---|---|
| S76Δ,S77Δ | MoMLV | Reduces infection of M. dunni cells | [52] |
| S84A | F-S MLV | Induces syncytia in M. dunni, infects hamster cells | [51] |
| Cas/Frg E-ERVs | Unknown | ||
| S82F 1 | Mo-Spl574 | Induces syncytia in M. dunni, restricted in other mouse cells | [53] |
| S82F, E114G 1 | Spl574-E114G | Correction of host range restriction of Spl574 | [54] |
| W102G | TR1.3 (FrMLV) | Syncytia formation in SC-1 cells, neurologic disease | [55] |
| E116G, W129K | PVC-211 (FrMLV) | Enhanced ability to infect hamster cells | [56] |
| Fv1 Sites | ||||||
|---|---|---|---|---|---|---|
| Fv1 Type 1 | Subspecies, Name (Location) | 270 | 352 | 358 | 399 | C-Terminus 2 |
| n or n-like | domesticus, SK/Cam (UK) | S | K | V | n | |
| domesticus, CalWM (USA) | K | S | K | V | n | |
| musculus, PWK (Czech Republic) | K | S | K | V | n | |
| musculus, SKIVE (Denmark) | K | S | K | V | n | |
| nr or nr-like | musculus, (Novobirsk, Russia) | F | K | V | n | |
| musculus, CZII (Slovakia) | K | F | K | V | n | |
| spp., Aks (China) | K | F | K | V | n | |
| spp., Las (China) | K | F | K | V | n | |
| spp., (Vladivostok, Russia) | K | F | K | V | n | |
| domesticus, ZALENDE (Switzerland | F | K | V | n | ||
| domesticus, CLA (USA) | F | K | V | n | ||
| d-like | domesticus, PRAE (Morocco) | Q | S | K | V | n |
| b or b-like | molossinus, MOM (Japan) | K | S | E | R | b |
| bactrianus, (Iran) | S | K | V | b | ||
| castaneus, (Philippines) | S | K | V | b | ||
| castaneus, H12 (India) 5 | K/Q 3 | S | K | V | b/b2 3 | |
| castaneus, H30,34 (India) 5 | K/Q 3 | S | K | V | b3 | |
| castaneus, H15,27 (India) 5 | K/R 3 | S | K | V | b | |
| castaneus, H24,26,28,36 (India) 5 | K | S | K | V | b | |
| molossinus, MOLD (Japan) | K | S | K | R | b | |
| molossinus, MOLC (Japan) | K | F | K | R | b | |
| molossinus, MAE (Japan) | K | F | K | R | b | |
| molossinus, JF1/Ms (Japanese fancy mouse) | K | F | K | R | b | |
| molossinus, (Saitama, Japan) | F | K | R | b | ||
| spp., IAS3 (Korea) | F | K | R | b | ||
| spp., Wuh (China) | S | K | R | b | ||
| domesticus LEWES (USA) | Q | S | K | V | b3 | |
| castaneus, CAST/EiJ (Thailand) | Q | S | K | V | b4 | |
| castaneus, H14 (India) 5 | Q | - 4 | - 4 | - 4 | - 4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boso, G.; Lam, O.; Bamunusinghe, D.; Oler, A.J.; Wollenberg, K.; Liu, Q.; Shaffer, E.; Kozak, C.A. Patterns of Coevolutionary Adaptations across Time and Space in Mouse Gammaretroviruses and Three Restrictive Host Factors. Viruses 2021, 13, 1864. https://doi.org/10.3390/v13091864
Boso G, Lam O, Bamunusinghe D, Oler AJ, Wollenberg K, Liu Q, Shaffer E, Kozak CA. Patterns of Coevolutionary Adaptations across Time and Space in Mouse Gammaretroviruses and Three Restrictive Host Factors. Viruses. 2021; 13(9):1864. https://doi.org/10.3390/v13091864
Chicago/Turabian StyleBoso, Guney, Oscar Lam, Devinka Bamunusinghe, Andrew J. Oler, Kurt Wollenberg, Qingping Liu, Esther Shaffer, and Christine A. Kozak. 2021. "Patterns of Coevolutionary Adaptations across Time and Space in Mouse Gammaretroviruses and Three Restrictive Host Factors" Viruses 13, no. 9: 1864. https://doi.org/10.3390/v13091864