
Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia



Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Study Samples
2.3. Extraction of Double-Stranded RNA and cDNA Synthesis
2.4. DNA Library Preparation and Illumina® Sequencing
2.5. Genome Assembly
2.6. Identification of Genotype Constellations
2.7. Phylogenetic Analysis
2.8. Protein Modelling
3. Results
3.1. Genotyping Based on Whole Genome Constellations
3.2. Phylogenetic and Sequence Analysis
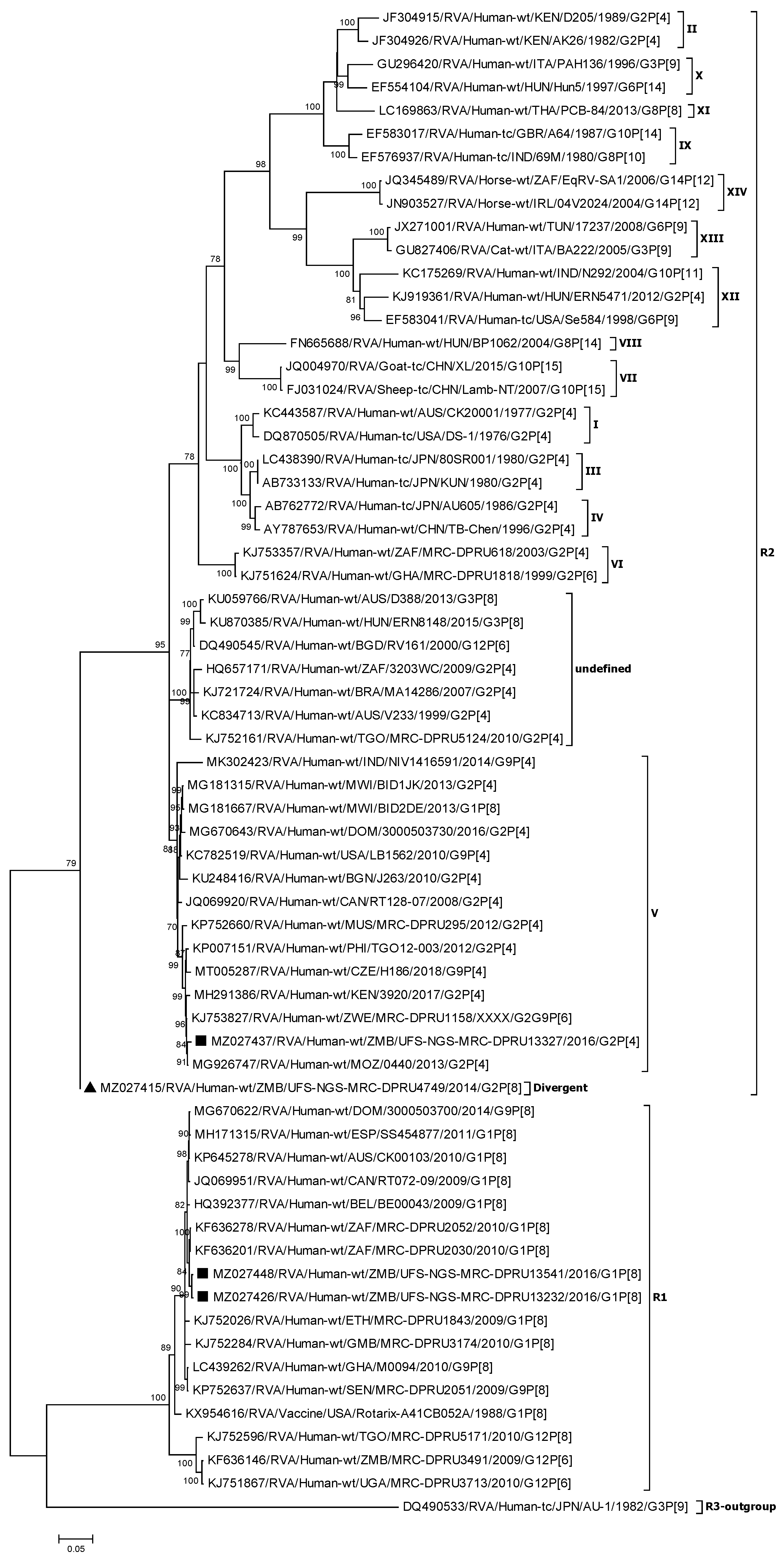
3.2.1. Phylogenetic Analysis of the VP7 Genes (G1 and G2)
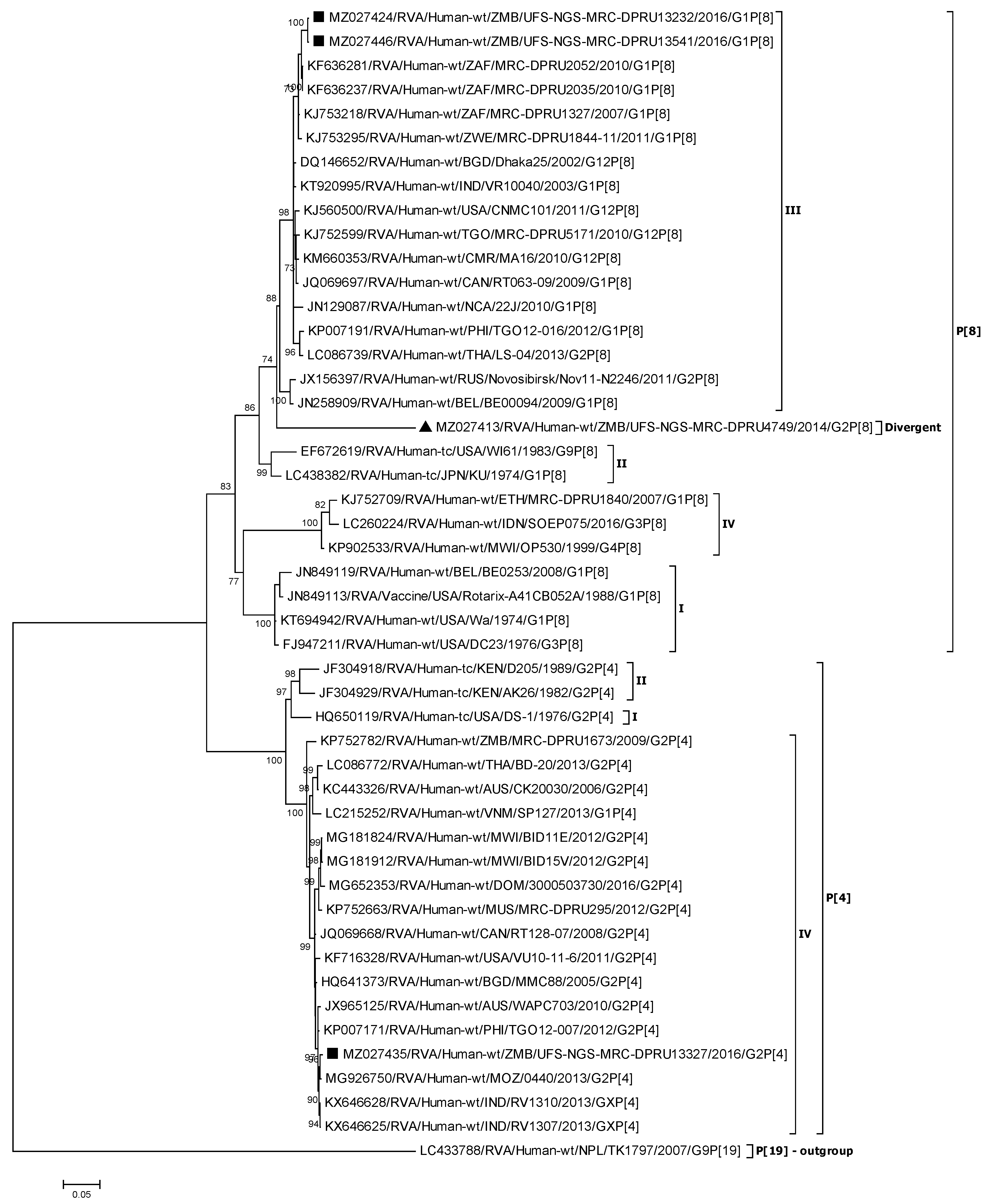
3.2.2. Phylogenetic Analysis of the VP4 Genes (P[4] and P[8])
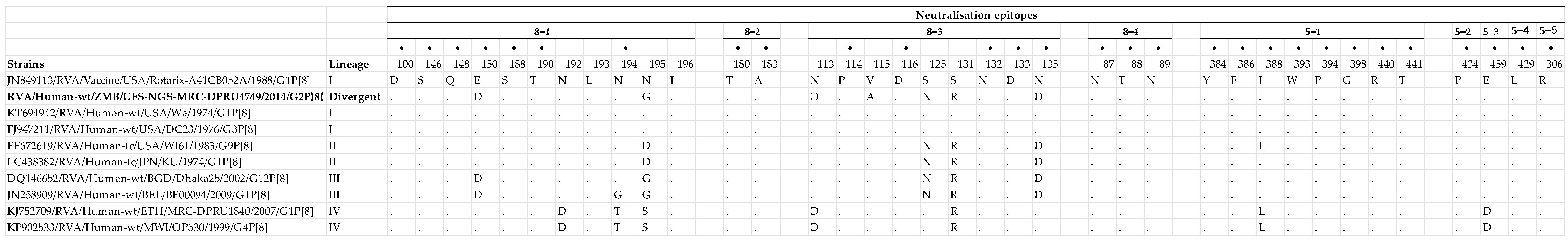
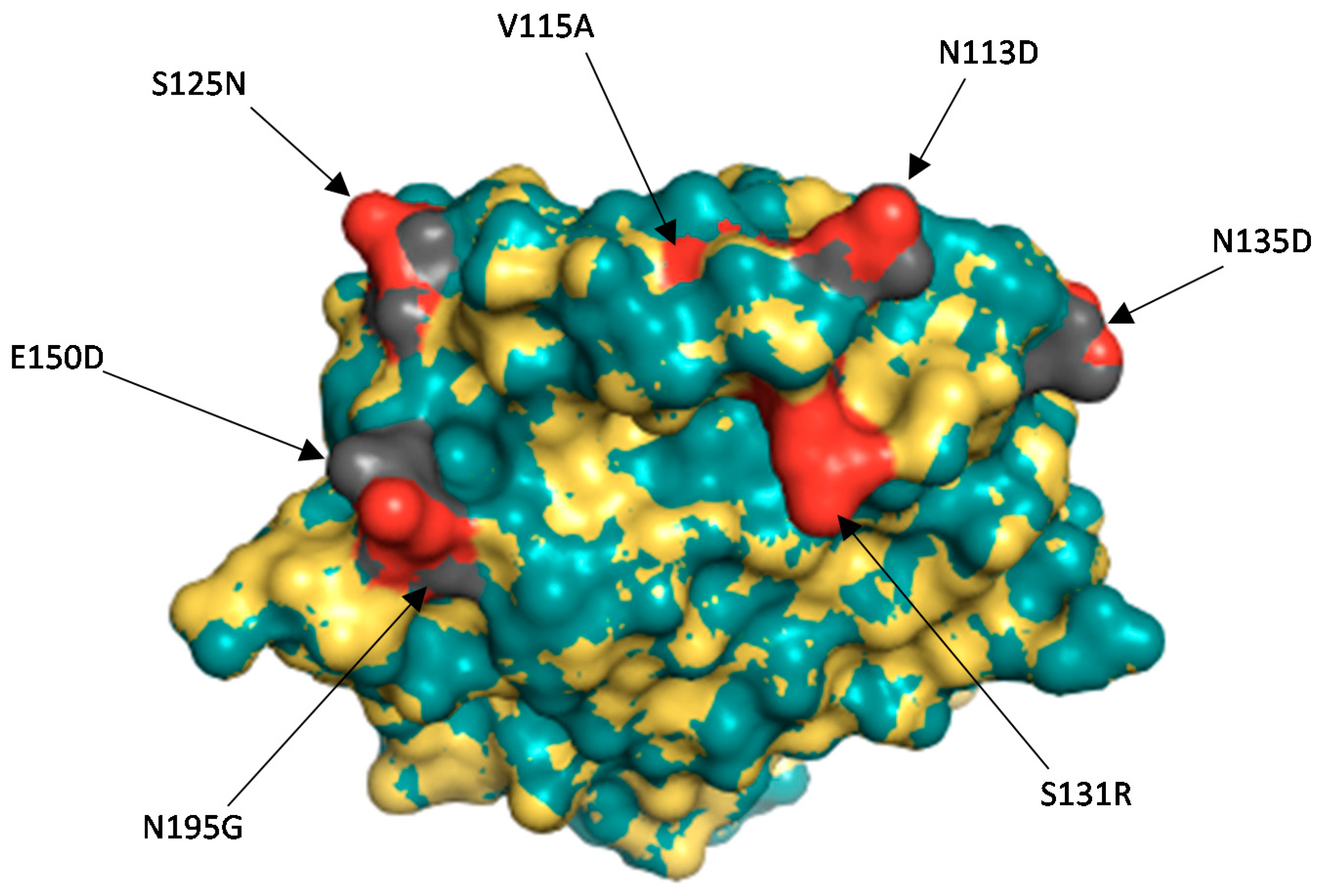
3.2.3. Comparison of the VP4 Antigenic Epitopes of Zambian G2P[8] to Rotarix®
3.2.4. Phylogenetic Analysis of the VP1 Gene
3.2.5. Phylogenetic Analysis of the VP6, VP2 and VP3 Genes
3.2.6. Phylogenetic Analysis of the NSP1-NSP5 Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus vaccination and the global burden of rotavirus diarrhoea among children younger than 5 years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Rotavirus vaccines WHO position paper: January 2013—Recommendations. Wkly. Epidemiol. Rec. 2013, 5, 49–64. [Google Scholar] [CrossRef]
- International Vaccine Access Centre, John Hopkins Bloomberg School of Public Health. Map. VIEW-hub. Available online: https://view-hub.org/ (accessed on 15 April 2021).
- Chilengi, R.; Rudd, C.; Bolton, C.; Guffey, B.; Masumbu, P.K.; Stringer, J. Successes, challenges and lessons learned in accelerating introduction of rotavirus immunisation in Zambia. World J. Vaccines 2015, 5, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Mpabalwani, E.M.; Simwaka, C.J.; Mwenda, J.M.; Mubanga, C.P.; Monze, M.; Matapo, B.; Parashar, U.D.; Tate, J.E. Impact of rotavirus vaccination on diarrhoeal hospitalisations in children aged <5 Years in Lusaka, Zambia. Clin. Infect. Dis. 2016, 62, S183–S187. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Vaccine-Preventable Diseases: Monitoring System. 2020 Global Summary. 2021. Available online: https://apps.who.int/immunization_monitoring/globalsummary (accessed on 15 April 2021).
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Hoshino, Y.; Kapikian, A.Z. Rotavirus Serotypes: Classification and Importance in Epidemiology, Immunity, and Vaccine Development. J. Health Popul. Nutr. 2000, 18, 5–14. Available online: https://www.jstor.org/stable/23499057 (accessed on 8 April 2021). [PubMed]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Kobayashi, N. Whole-genomic analysis of rotavirus strains: Current status and future prospects. Future Microbiol. 2011, 6, 1049–1065. [Google Scholar] [CrossRef]
- Donker, N.C.; Kirkwood, C.D. Selection and evolutionary analysis in the nonstructural protein NSP2 of rotavirus A. Infect. Genet. Evol. 2012, 12, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Hoxie, I.; Dennehy, J.J. Intragenic recombination influences rotavirus diversity and evolution. Virus Evol. 2020, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, C.D. Genetic and Antigenic Diversity of Human Rotaviruses: Potential Impact on Vaccination Programs. J. Infect. Dis. 2010, 202, S43–S48. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Heylen, E.; Zeller, M.; Rahman, M.; Lemey, P.; Van Ranst, M. Phylodynamic analyses of rotavirus genotypes G9 and G12 underscore their potential for swift global spread. Mol. Biol. Evol. 2010, 27, 431–2436. [Google Scholar] [CrossRef] [Green Version]
- Ramig, R.F. Genetics of the rotaviruses. Annu. Rev. Microbiol. 1997, 51, 225–255. [Google Scholar] [CrossRef]
- Banerjee, A.; Lo, M.; Indwar, P.; Deb, A.K.; Das, S.; Manna, B.; Dutta, S.; Bhadra, U.K.; Bhattacharya, M.; Okamoto, K.; et al. Upsurge and spread of G3 rotaviruses in Eastern India (2014–2016): Full genome analyses reveals heterogeneity within Wa-like genomic constellation. Infect. Genet. Evol. 2018, 63, 158–174. [Google Scholar] [CrossRef]
- Doro, R.; Farkas, S.L.; Martella, V.; Banyai, K. Zoonotic transmission of rotavirus: Surveillance and control. Expert Rev. Anti. Infect. Ther. 2015, 13, 1337–1350. [Google Scholar] [CrossRef]
- Ghosh, S.; Shintani, T.; Urushibara, N.; Taniguchi, K.; Kobayashi, N. Whole-genomic analysis of a human G1P[9] rotavirus strain reveals intergenogroup reassortment events. J. Gen. Virol. 2012, 93, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Nyaga, M.M.; Stucker, K.M.; Esona, M.D.; Jere, K.C.; Mwinyi, B.; Shonhai, A.; Tsolenyanu, E.; Mulindwa, A.; Chibumbya, J.N.; Adolfine, H.; et al. Whole-genome analyses of DS-1-like human G2P[4] and G8P[4] rotavirus strains from Eastern, Western and Southern Africa. Virus Genes 2014, 49, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Seheri, L.M.; Magagula, N.B.; Peenze, I.; Rakau, K.; Ndadza, A.; Mwenda, J.M.; Weldegebriel, G.; Steele, A.D.; Mphahlele, M.J. Rotavirus strain diversity in Eastern and Southern African countries before and after vaccine introduction. Vaccine 2018, 36, 7222–7230. [Google Scholar] [CrossRef] [PubMed]
- Heiman, E.M.; McDonald, S.M.; Barro, M.; Taraporewala, Z.F.; Bar-Magen, T.; Patton, J.T. Group A human rotavirus genomics: Evidence that gene constellations are influenced by viral protein interactions. J. Virol. 2008, 82, 11106–11116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary Dynamics of Human Rotaviruses: Balancing Reassortment with Preferred Genome Constellations. PLoS Pathog. 2009, 5, e1000634. [Google Scholar] [CrossRef]
- Ward, R.L.; Nakagomi, O.; Knowlton, D.R.; McNeal, M.M.; Nakagomi, T.; Clemens, J.D.; Sack, D.A.; Schiff, G.M. Evidence for natural reassortants of human rotaviruses belonging to different genogroups. J. Virol. 1990, 64, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Cowley, D.; Donato, C.M.; Roczo-Farkas, S.; Kirkwood, C.D. Emergence of a novel equine-like G3P[8] intergenogroup reassortant rotavirus strain associated with gastroenteritis in Australian children. J. Gen. Virol. 2016, 97, 403–410. [Google Scholar] [CrossRef]
- Heylen, E.; Likele, B.B.; Zeller, M.; Stevens, S.; De Coster, S.; Conceição-Neto, N.; Van Geet, C.; Jacobs, J.; Ngbonda, D.; Van Ranst, M.; et al. Rotavirus surveillance in Kisangani, the Democratic Republic of the Congo, reveals a high number of unusual genotypes and gene segments of animal origin in non-vaccinated symptomatic children. PLoS ONE 2014, 9, e100953. [Google Scholar] [CrossRef] [Green Version]
- Hoa-Tran, T.N.; Nakagomi, T.; Vu, H.M.; Nguyen, T.T.T.; Takemura, T.; Hasebe, F.; Dao, A.T.H.; Anh, P.H.Q.; Nguyen, A.T.; Dang, A.D.; et al. Detection of three independently-generated DS-1-like G9P[8] reassortant rotavirus A strains during the G9P[8] dominance in Vietnam, 2016–2018. Infect. Genet. Evol. 2020, 80, 104194. [Google Scholar] [CrossRef] [PubMed]
- Jere, K.C.; Chaguza, C.; Bar-zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B. Emergence of double- and triple-gene reassortant G1P[8] rotaviruses possessing a DS-1-like backbone after rotavirus vaccine introduction in Malawi. J. Virol. 2018, 92, e01246-17. [Google Scholar] [CrossRef] [Green Version]
- Katz, E.M.; Esona, M.D.; Betrapally, N.S.; De La Cruz De Leon, L.A.; Neira, Y.R.; Rey, G.J.; Bowen, M.D. Whole-gene analysis of inter-genogroup reassortant rotaviruses from the Dominican Republic: Emergence of equine-like G3 strains and evidence of their reassortment with locally-circulating strains. Virology 2019, 534, 114–131. [Google Scholar] [CrossRef]
- Komoto, S.; Tacharoenmuang, R.; Guntapong, R.; Ide, T.; Tsuji, T.; Yoshikawa, T.; Tharmaphornpilas, P.; Sangkitporn, S.; Taniguchi, K. Reassortment of Human and Animal Rotavirus Gene Segments in Emerging DS-1-Like G1P[8] Rotavirus Strains. PLoS ONE 2016, 11, e0148416. [Google Scholar] [CrossRef]
- Luchs, A.; da Costa, A.C.; Cilli, A.; Komninakis, S.C.V.; Carmona, R.D.C.C.; Morillo, S.G.; Sabino, E.C.; Timenetsky, M.D.C.S.T. First Detection of DS-1-like G1P[8] Double-gene Reassortant Rotavirus Strains on The American Continent, Brazil, 2013. Sci. Rep. 2019, 9, 2210. [Google Scholar] [CrossRef] [PubMed]
- Maringa, W.M.; Mwangi, P.N.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Peenze, I.; Esona, M.D.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Molecular Characterisation of a Rare Reassortant Porcine-Like G5P[6] Rotavirus Strain Detected in an Unvaccinated Child in Kasama, Zambia. Pathogens 2020, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Nyaga, M.M.; Tan, Y.; Seheri, M.L.; Halpin, R.A.; Akopov, A.; Stucker, K.M.; Fedorova, N.B.; Shrivastava, S.; Duncan Steele, A.; Mwenda, J.M.; et al. Whole-genome sequencing and analyses identify high genetic heterogeneity, diversity and endemicity of rotavirus genotype P[6] strains circulating in Africa. Infect. Genet. Evol. 2018, 63, 79–88. [Google Scholar] [CrossRef]
- Nyaga, M.M.; Jere, K.C.; Esona, M.D.; Seheri, M.L.; Stucker, K.M.; Halpin, R.A.; Akopov, A.; Stockwell, T.B.; Peenze, I.; Diop, A.; et al. Whole genome detection of rotavirus mixed infections in human, porcine and bovine samples co-infected with various rotavirus strains collected from sub-Saharan Africa. Infect. Genet. Evol. 2015, 31, 321–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simwaka, J.C.; Mpabalwani, E.M.; Seheri, M.; Peenze, I.; Monze, M.; Matapo, B.; Parashar, U.D.; Mufunda, J.; Mphahlele, J.M.; Tate, J.E.; et al. Diversity of rotavirus strains circulating in children under five years of age who presented with acute gastroenteritis before and after rotavirus vaccine introduction, University Teaching Hospital, Lusaka, Zambia, 2008–2015. Vaccine 2018, 36, 7243–7247. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; van Dijk, A.A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, P.; Conklin, D. NanoDrop Microvolume Quantitation of Nucleic Acids. J. Vis. Exp. 2010, 45, e2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organisation and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Centre for Biotechnology Information. Nucleic Acids 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schaffer, A.A.; Brister, J.R. Virus Variation Resource-improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence Limits on Phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Agbemabiese, C.A.; Nakagomi, T.; Damanka, S.A.; Dennis, F.E.; Lartey, B.L.; Armah, G.E.; Nakagomi, O. Sub-genotype phylogeny of the non-G, non-P genes of genotype 2 Rotavirus A strains. PLoS ONE 2019, 14, e0217422. [Google Scholar] [CrossRef] [PubMed]
- Aida, S.; Nahar, S.; Paul, S.K.; Hossain, M.A.; Kabir, M.R.; Sarkar, S.R.; Ahmed, S.; Ghosh, S.; Urushibara, N.; Kawaguchiya, M.; et al. Whole genomic analysis of G2P[4] human rotaviruses in Mymensingh, north-central Bangladesh. Heliyon 2016, 2, e00168. [Google Scholar] [CrossRef] [Green Version]
- Arista, S.; Giammanco, G.M.; De Grazia, S.; Ramirez, S.; Biundo, C.L.; Colomba, C.; Cascio, A.; Martella, V. Heterogeneity and temporal dynamics of evolution of G1 human rotaviruses in a settled population. J. Virol. 2006, 80, 10724–10733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, Y.H.; Nakagomi, T.; Agbemabiese, C.A.; Nakagomi, O. Changes in the distribution of lineage constellations of G2P[4] Rotavirus A strains detected in Japan over 32 years (1980-2011). Infect. Genet. Evol. 2015, 34, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Doan, Y.H.; Nakagomi, T.; Nakagomi, O. Repeated circulation over 6 years of intergenogroup mono-reassortant G2P[4] rotavirus strains with genotype N1 of the NSP2 gene. Infect. Genet. Evol. 2012, 12, 1202–1212. [Google Scholar] [CrossRef]
- Gouvea, V.; Lima, R.C.; Linhares, R.E.; Clark, H.F.; Nosawa, C.M.; Santos, N. Identification of two lineages (WA-like and F45-like) within the major rotavirus genotype P[8]. Virus Res. 1999, 59, 141–147. [Google Scholar] [CrossRef]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic analyses reveal differences in the VP7 and VP4 antigenic epitopes between human rotaviruses circulating in Belgium and rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunliffe, N.A.; Gondwe, J.S.; Graham, S.M.; Thindwa, B.D.; Dove, W.; Broadhead, R.L.; Molyneux, M.E.; Hart, C.A. Rotavirus strain diversity in Blantyre, Malawi, from 1997 to 1999. J. Clin. Microbiol. 2001, 39, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, S.; Kobayashi, N.; Paul, S.K.; Alam, M.M.; Chawla-Sarkar, M.; Krishnan, T. Characterisation of full-length VP4 genes of OP354-like P[8] human rotavirus strains detected in Bangladesh representing a novel P[8] subtype. Arch. Virol. 2009, 154, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Heylen, E.; Damanka, S.; Pietsch, C.; Donato, C.; Tamura, T.; Kulkarni, R.; Arora, R.; Cunliffe, N.; Maunula, L.; et al. Emerging OP354-like P[8] rotaviruses have rapidly dispersed from Asia to other continents. Mol. Biol. Evol. 2015, 32, 2060–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giammanco, G.M.; Bonura, F.; Zeller, M.; Heylen, E.; Van Ranst, M.; Martella, V.; Bányai, K.; Matthijnssens, J.; De Grazia, S. Evolution of DS-1-like human G2P[4] rotaviruses assessed by complete genome analysis. J. Gen. Virol. 2014, 95, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Reslan, L.; El-Husseini, M.; Raoof, H.; Finianos, M.; Guo, C.; Thakkar, R.; Inati, A.; Dbaibo, G.; Lipkin, W.; et al. Full genome characterisation of human G3P[6] and G3P[9] rotavirus strains in Lebanon. Infect. Genet. Evol. 2020, 78, 104133. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, C.; Liebert, U.G. Molecular characterisation of different equine-like G3 rotavirus strains from Germany. Infect. Genet. Evol. 2018, 57, 46–50. [Google Scholar] [CrossRef]
- Rasebotsa, S.; Uwimana, J.; Mogotsi, M.T.; Rakau, K.; Magagula, N.B.; Seheri, M.L.; Mwenda, J.M.; Mphahlele, M.J.; Sabiu, S.; Mihigo, R.; et al. Whole-genome analyses identifies multiple reassortant rotavirus strains in Rwanda post-vaccine introduction. Viruses 2021, 13, 95. [Google Scholar] [CrossRef]
- Thanh, H.D.; Tran, V.T.; Lim, I.; Kim, W. Emergence of human G2P[4] rotaviruses in the post-vaccination era in South Korea: Footprints of multiple interspecies reassortment events. Sci. Rep. 2018, 8, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Dormitzer, P.R.; Nason, E.B.; Prasad, B.V.V.; Harrison, S.C. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 2004, 430, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.Y.; Estes, M.K. Proteolytic enhancement of rotavirus infectivity: Biologic mechanisms. Virology 1980, 101, 432–439. [Google Scholar] [CrossRef]
- Denisova, E.; Dowling, W.; LaMonica, R.; Shaw, R.; Scarlata, S.; Ruggeri, F.; Mackow, E.R. Rotavirus capsid protein VP5* permeabilises membranes. J. Virol. 1999, 73, 3147–3153. [Google Scholar] [CrossRef] [Green Version]
- Dowling, W.; Denisova, E.; LaMonica, R.; Mackow, E.R. Selective membrane permeabilisation by the rotavirus Vp5* protein is abrogated by mutations in an internal hydrophobic domain. J. Virol. 2000, 74, 6368–6376. [Google Scholar] [CrossRef] [Green Version]
- Fiore, L.; Greenberg, H.B.; Mackow, E.R. The VP8 fragment of VP4 is the rhesus rotavirus hemagglutinin. Virology 1991, 181, 553–563. [Google Scholar] [CrossRef]
- Ruggeri, F.M.; Greenberg, H.B. Antibodies to the trypsin cleavage peptide VP8 neutralise rotavirus by inhibiting binding of virions to target cells in culture. J. Virol. 1991, 65, 2211–2219. [Google Scholar] [CrossRef] [Green Version]
- Trask, S.D.; McDonald, S.M.; Patton, J.T. Structural insights into the coupling of virion assembly and rotavirus replication. Nat. Rev. Microbiol. 2012, 10, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Rota Council. Available Rotavirus Vaccine Products—Rotavirus Vaccines Prequalified by WHO. 2020. Available online: https://preventrotavirus.org (accessed on 16 April 2021).
- Betts, M.J.; Russel, R.B. Amino acid properties and consequences of substitutions. In Bioinformatics for Geneticists; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2007; pp. 311–342. [Google Scholar] [CrossRef]
- Garnier, J.; Osguthorpe, D.J.; Robson, B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978, 120, 97–120. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID and Year | Hospital | The Child’s Place of Residence | Sex | Age | Presenting Illness Symptoms | Dehydration Status and Treatment Administered | Vaccination Status | Outcome of Illness |
|---|---|---|---|---|---|---|---|---|
| UFS-NGS-MRC-DPRU4749/2014 | ACDH Ndola | Chifubu | Female | 5 months | Diarrhoea for 4 days (4 episodes in 24 h), no vomiting, temperature of 39 °C | Moderate dehydration, treated with ORS | Not vaccinated | Alive |
| UFS-NGS-MRC-DPRU13232/2016 | ACDH Ndola | Kawama | Male | 7 months | Diarrhoea for 3 days (6 episodes in 24 h), vomiting for 2 days (4 episodes in 24 h), temperature of 38.2 °C | Severe dehydration, treated with IV fluids | Vaccinated (1 dose) | Alive |
| UFS-NGS-MRC-DPRU13541/2016 | ACDH Ndola | Mwange A | Male | 8 months | Diarrhoea for 3 days (8 episodes in 24 h), vomiting for 3 days (3 episodes in 24 h), no fever | Severe dehydration, treated with IV fluids | Not vaccinated | Alive |
| UFS-NGS-MRC-DPRU13327/2016 | UTH Lusaka | Kapata | Male | 20 months | Diarrhoea for 1 day (3 episodes in 24 h), vomiting for 3 days, no fever | No dehydration, treated with ORS | Vaccinated (2 doses) | Alive |
| Strain | VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UFS-NGS-MRC-DPRU13232 | Genotype | G1 | P[8] | I1 | R1 | C1 | M1 | A1 | N2 | T1 | E1 | H1 |
| Contig length | 1062 | 2359 | 1356 | 3301 | 2717 | 2591 | 1567 | 1059 | 1074 | 750 | 644 | |
| Reads mapped to contig | 21,238 | 4523 | 14,997 | 87,349 | 52,209 | 52,222 | 26,784 | 53,976 | 30,125 | 25,306 | 21,366 | |
| UFS-NGS-MRC-DPRU13541 | Genotype | G1 | P[8] | I1 | R1 | C1 | M1 | A1 | N2 | T1 | E1 | H1 |
| Contig length | 1063 | 2359 | 1352 | 3301 | 2729 | 2591 | 1567 | 1059 | 1074 | 750 | 663 | |
| Reads mapped to contig | 33,485 | 10,936 | 62,838 | 108,961 | 79,014 | 134,489 | 80,109 | 33,007 | 36,184 | 34,457 | 12,638 | |
| UFS-NGS-MRC-DPRU4749 | Genotype | G2 | P[8] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| Contig length | 1062 | 2360 | 1356 | 3302 | 2684 | 2591 | 1569 | 1059 | 1066 | 750 | 815 | |
| Reads mapped to contig | 1445 | 4513 | 2302 | 6738 | 4388 | 5214 | 2315 | 1063 | 1268 | 916 | 471 | |
| UFS-NGS-MRC-DPRU13327 | Genotype | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N1 | T2 | E2 | H2 |
| Contig length | 1062 | 2359 | 1354 | 3298 | 2684 | 2591 | 1566 | 1059 | 1066 | 751 | 798 | |
| Reads mapped to contig | 24,446 | 51,762 | 23,311 | 67,839 | 53,795 | 60,905 | 25,147 | 11,048 | 20,338 | 13,618 | 13,618 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maringa, W.M.; Simwaka, J.; Mwangi, P.N.; Mpabalwani, E.M.; Mwenda, J.M.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia. Viruses 2021, 13, 1872. https://doi.org/10.3390/v13091872
Maringa WM, Simwaka J, Mwangi PN, Mpabalwani EM, Mwenda JM, Mphahlele MJ, Seheri ML, Nyaga MM. Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia. Viruses. 2021; 13(9):1872. https://doi.org/10.3390/v13091872
Chicago/Turabian StyleMaringa, Wairimu M., Julia Simwaka, Peter N. Mwangi, Evans M. Mpabalwani, Jason M. Mwenda, M. Jeffrey Mphahlele, Mapaseka L. Seheri, and Martin M. Nyaga. 2021. "Whole Genome Analysis of Human Rotaviruses Reveals Single Gene Reassortant Rotavirus Strains in Zambia" Viruses 13, no. 9: 1872. https://doi.org/10.3390/v13091872