
Controlled Anchoring of (Phenylureido)sulfonamide-Based Receptor Moieties: An Impact of Binding Site Multiplication on Complexation Properties

 ,
,
Abstract
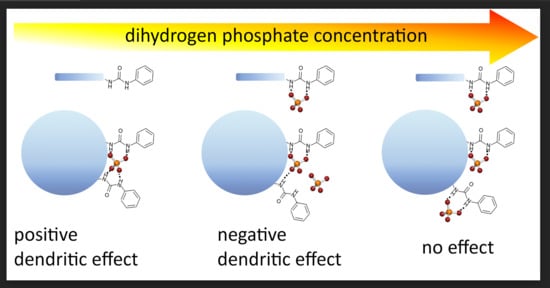
:
1. Introduction
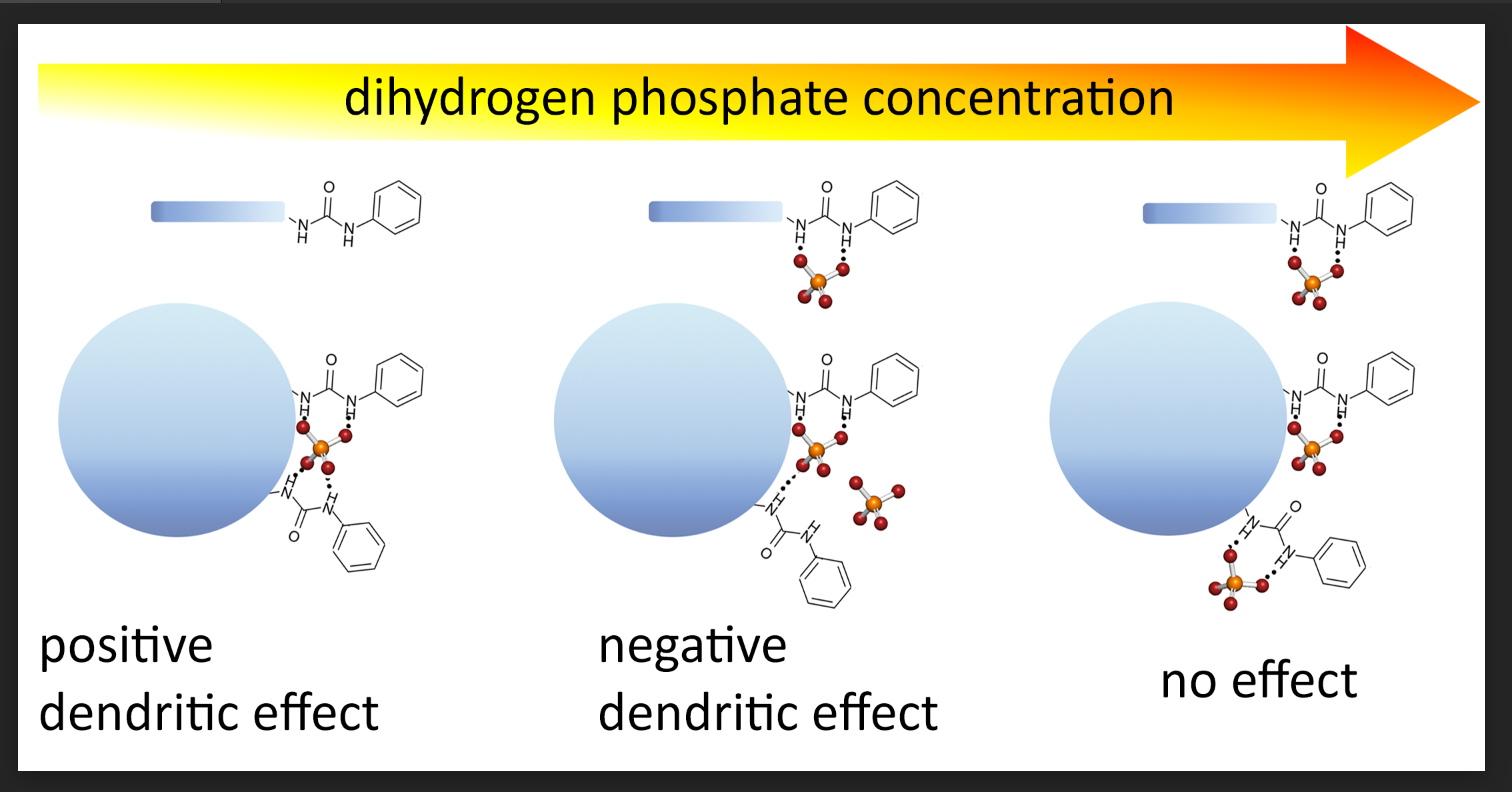
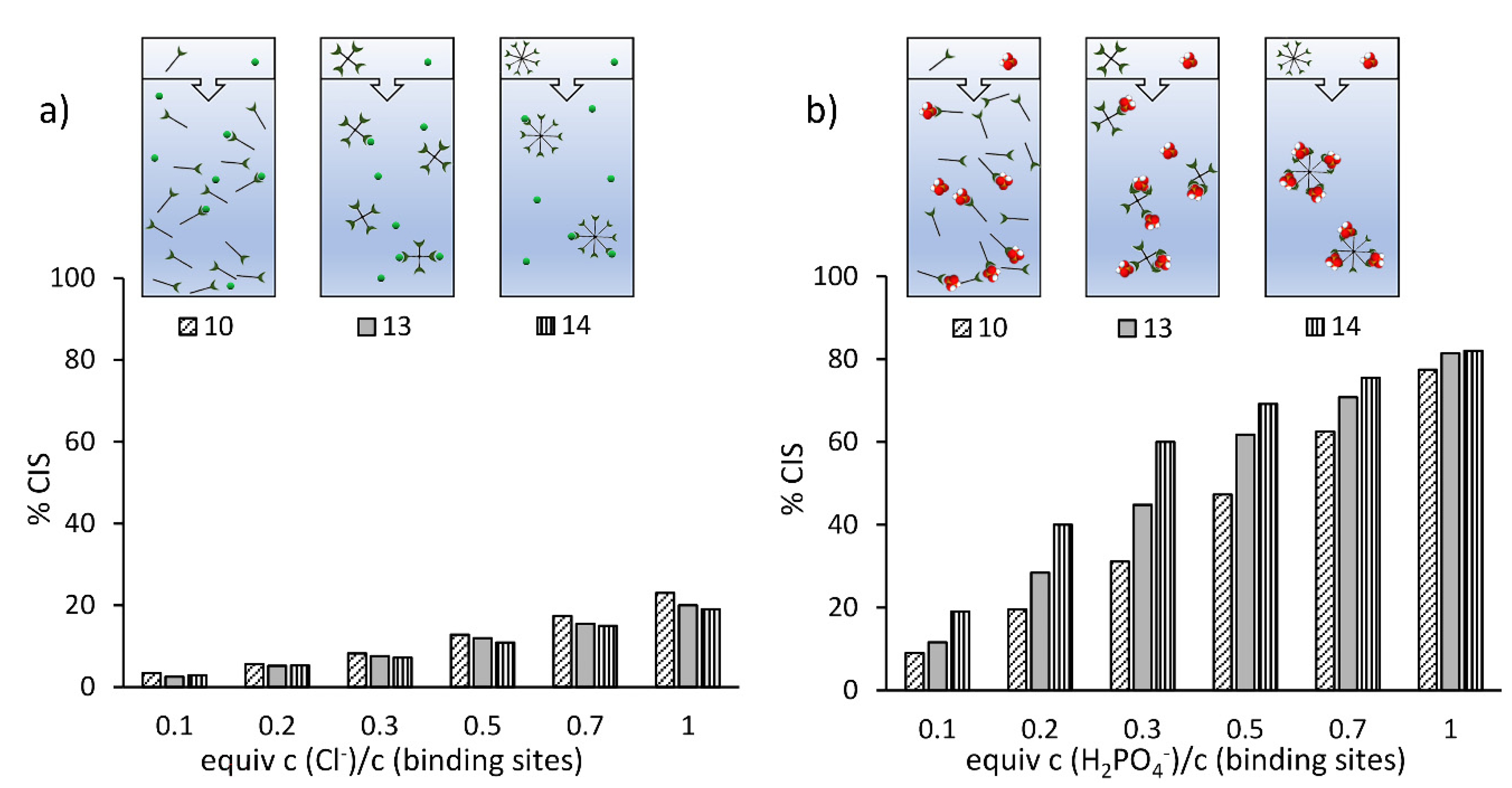
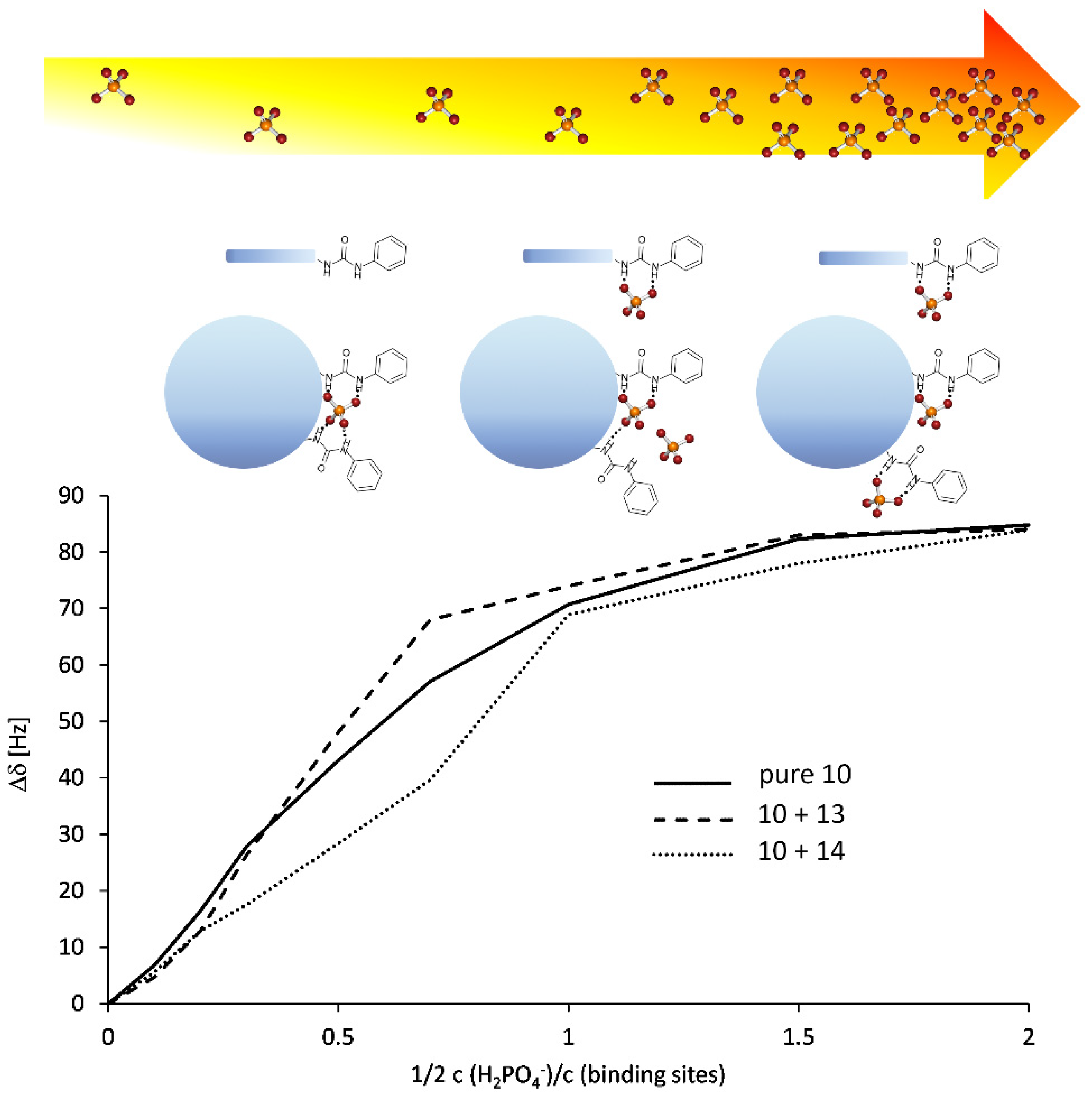
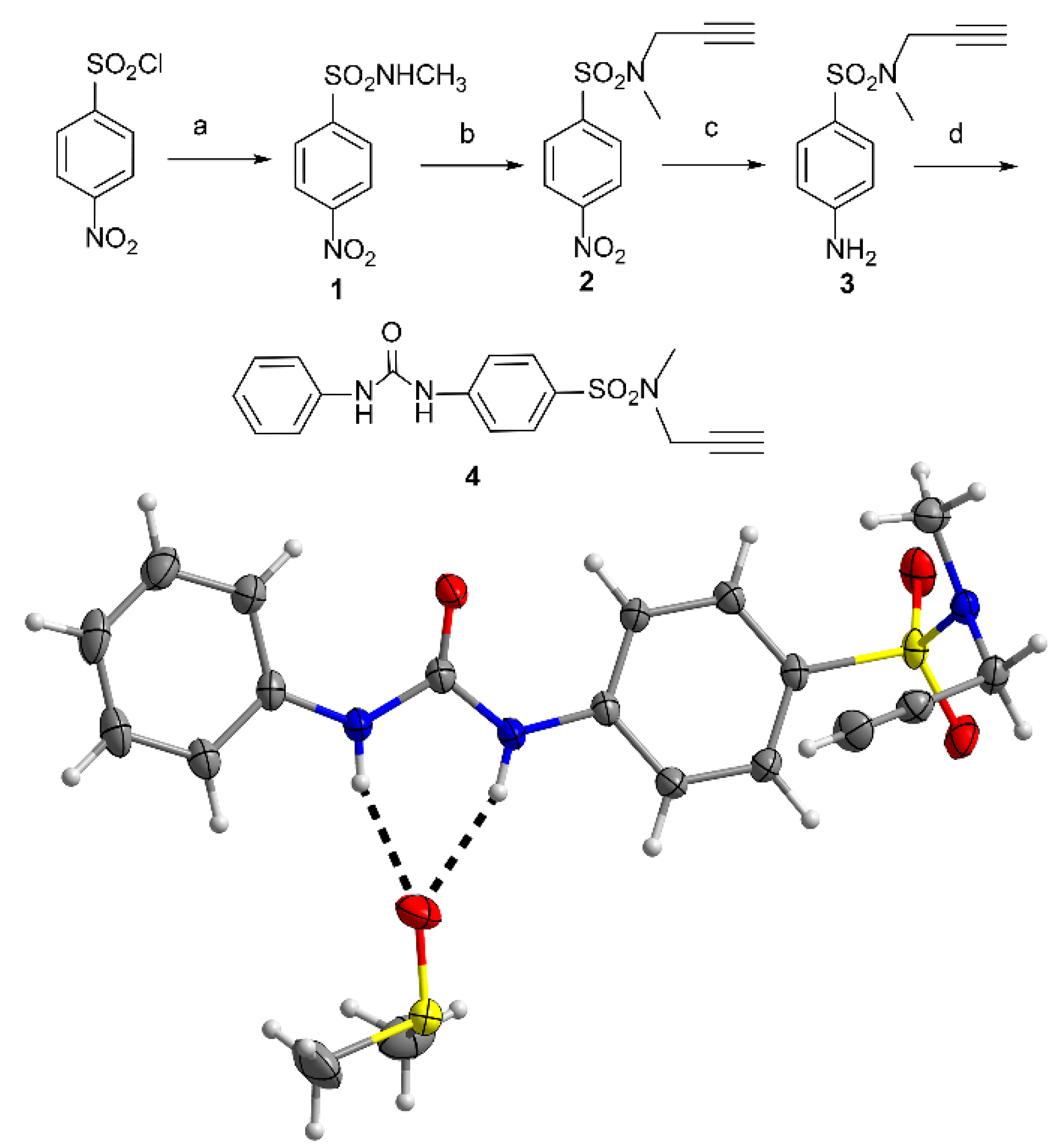
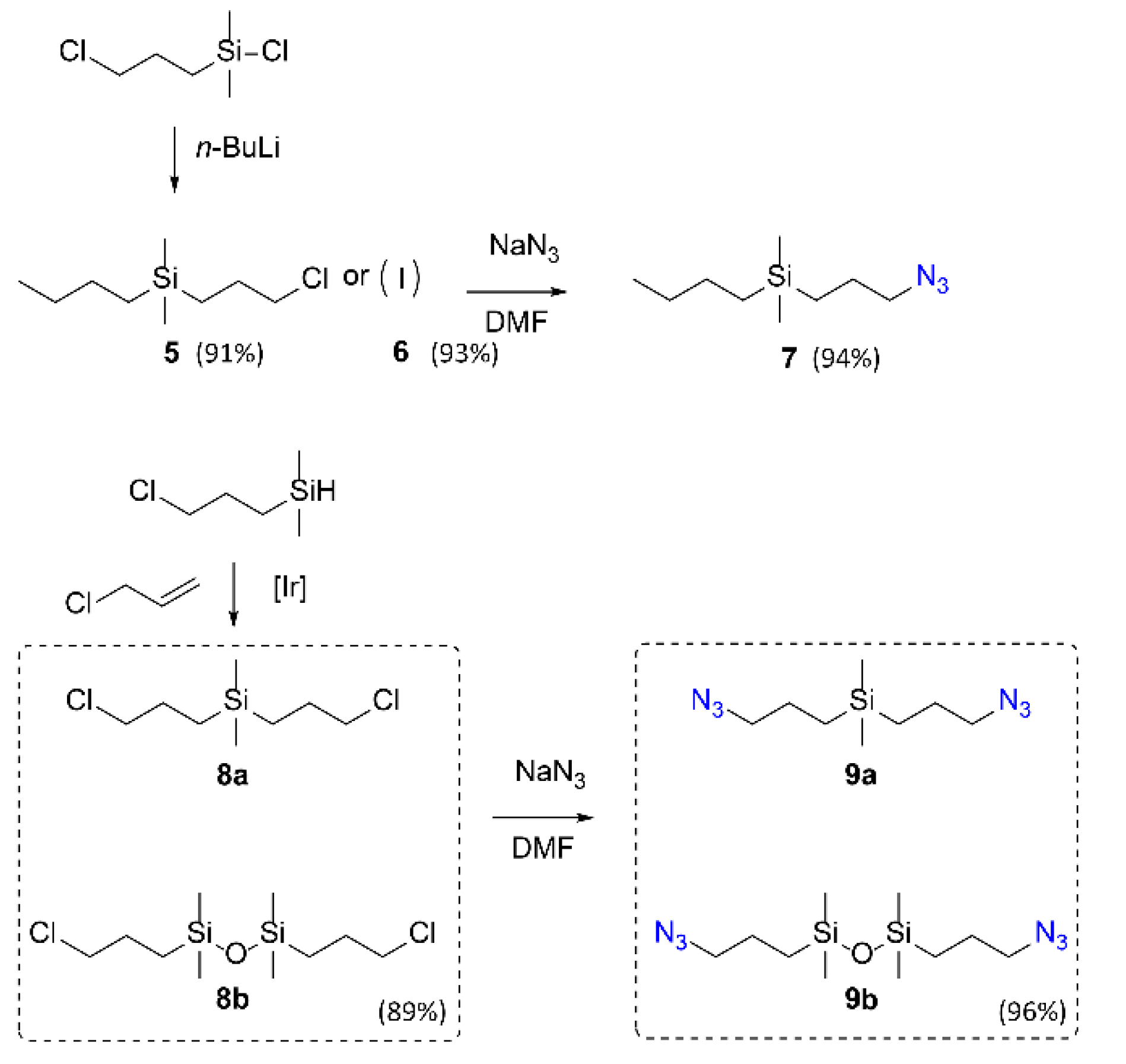
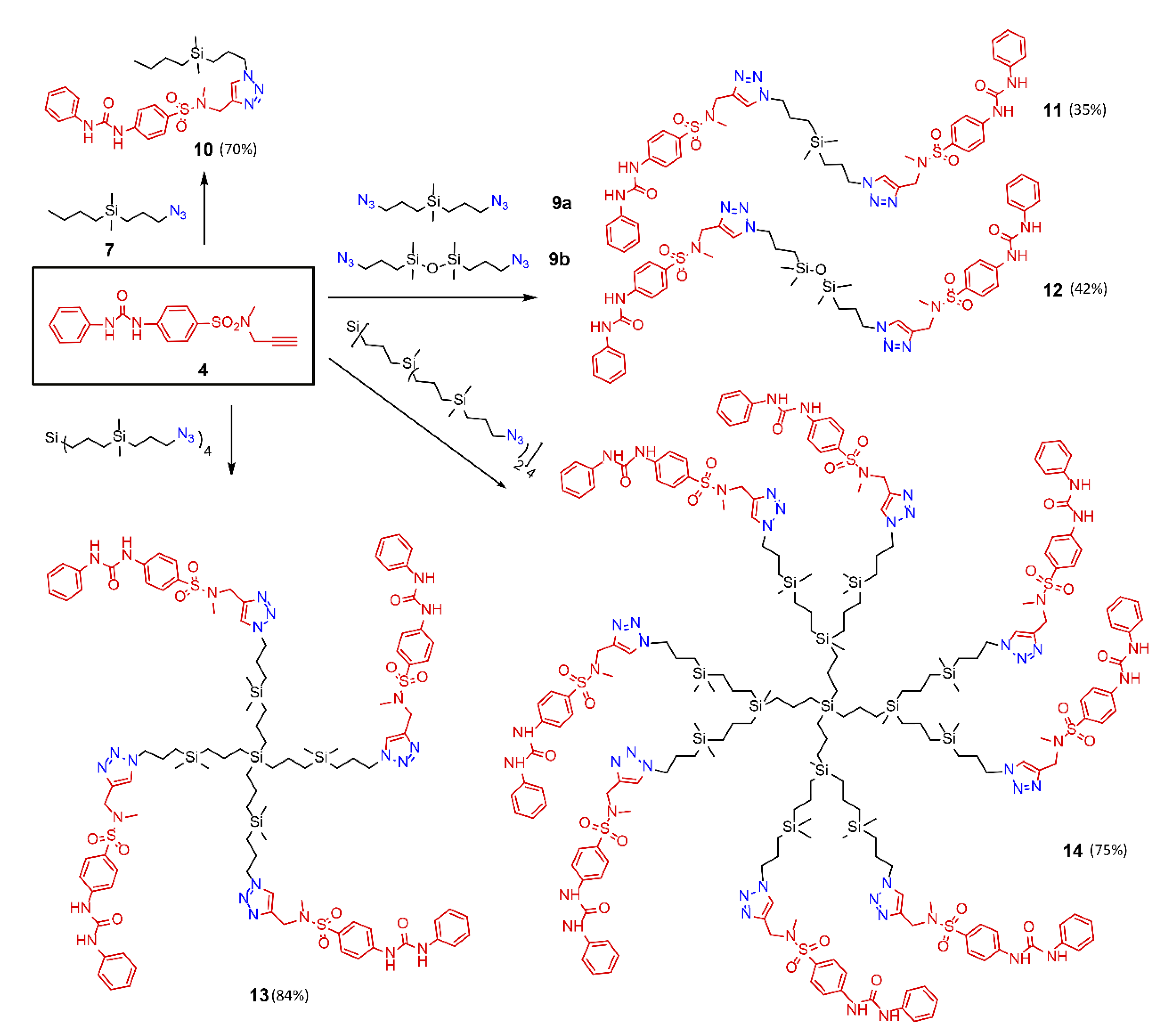
2. Results
3. Discussion
4. Materials and Methods
4.1. General
4.2. Determination of Complexation Constants
4.3. Synthesis
4.3.1. Synthesis of Precursors
4.3.2. Preparation of Receptors—A General Procedure
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Appendix A
References
- Tomalia, D.A. Dendrimers and Other Dendritic Polymers; Fréchet, J.M.J., Tomalia, D.A., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2001; Volume 1, ISBN 9780470845820. [Google Scholar]
- Newkome, G.R.; Moorefield, C.N.; Vögtle, F. Dendritic Molecules: Concepts, Syntheses, Perspectives; Wiley-VCH: Weinheim, Germany, 1996. [Google Scholar]
- Tomalia, D.A.; Christensen, J.B.; Boas, U. The dendritic effect. Dendrimers, Dendrons, and Dendritic Polym; Cambridge University Press: Cambridge, UK, 2012; pp. 276–292. [Google Scholar] [CrossRef]
- Caminade, A.M.; Ouali, A.; Laurent, R.; Turrin, C.O.; Majoral, J.P. The dendritic effect illustrated with phosphorus dendrimers. Chem. Soc. Rev. 2015, 44, 3890–3899. [Google Scholar] [CrossRef]
- Zaupa, G.; Scrimin, P.; Prins, L.J. Origin of the Dendritic Effect in Multivalent Enzyme-Like Catalysts. J. Am. Chem. Soc. 2008, 130, 5699–5709. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A. Dendritic effects: Dependency of dendritic nano-periodic property patterns on critical nanoscale design parameters (CNDPs). New J. Chem. 2012, 36, 264–281. [Google Scholar] [CrossRef]
- Chow, H.F.; Leung, C.F.; Wang, G.X.; Yang, Y.Y. Dendritic effects in functional dendrimer molecules. Comptes Rendus Chim. 2003, 6, 735–745. [Google Scholar] [CrossRef]
- Madaan, K.; Kumar, S.; Poonia, N.; Lather, V.; Pandita, D. Dendrimers in drug delivery and targeting: Drug-dendrimer interactions and toxicity issues. J. Pharm. Bioallied Sci. 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Gothwal, A.; Jain, K.; Kesharwani, P.; Gupta, U.; Chourasia, M.K.; Iyer, A.K. Dendrimer nanohybrid carrier systems: An expanding horizon for targeted drug and gene delivery. Drug Discov. Today 2017, 23, 300–314. [Google Scholar] [CrossRef]
- Bronstein, L.M. Magnetically Recoverable Catalysts with Dendritic Ligands for Enhanced Catalysis and Easy Separation. ChemCatChem 2015, 7, 1058–1060. [Google Scholar] [CrossRef]
- Caminade, A.M.; Laurent, R.; Chaudret, B.; Majoral, J.P. Phosphine-terminated dendrimers Synthesis and complexation properties. Coord. Chem. Rev. 1998, 178–180, 793–821. [Google Scholar] [CrossRef]
- Liang, L.; Ruiz, J.; Astruc, D. The efficient copper(I) (hexabenzyl)tren catalyst and dendritic analogues for green “click” reactions between azides and alkynes in organic solvent and in water: Positive dendritic effects and monometallic mechanism. Adv. Synth. Catal. 2011, 353, 3434–3450. [Google Scholar] [CrossRef]
- Jishkariani, D.; Lee, J.D.; Yun, H.; Paik, T.; Kikkawa, J.M.; Kagan, C.R.; Donnio, B.; Murray, C.B. The dendritic effect and magnetic permeability in dendron coated nickel and manganese zinc ferrite nanoparticles. Nanoscale 2017, 9, 13922–13928. [Google Scholar] [CrossRef]
- Ogasawara, S.; Ikeda, A.; Kikuchi, J.I. Positive dendritic effect in DNA/porphyrin composite photocurrent generators containing dendrimers as the stationary phase. Chem. Mater. 2006, 18, 5982–5987. [Google Scholar] [CrossRef]
- Hu, J.; Xu, T.; Cheng, Y. NMR insights into dendrimer-based host-guest systems. Chem. Rev. 2012, 112, 3856–3891. [Google Scholar] [CrossRef]
- Esipenko, N.A.; Koutnik, P.; Minami, T.; Mosca, L.; Lynch, V.M.; Zyryanov, G.V.; Anzenbacher, P. First supramolecular sensors for phosphonate anions. Chem. Sci. 2013, 4, 3617–3623. [Google Scholar] [CrossRef] [Green Version]
- Valerio, C.; Fillaut, J.L.; Ruiz, J.; Guittard, J.; Blais, J.C.; Astruc, D. The dendritic effect in molecular recognition: Ferrocene dendrimers and their use as supramolecular redox sensors for the recognition of small inorganic anions. J. Am. Chem. Soc. 1997, 119, 2588–2589. [Google Scholar] [CrossRef]
- Ornelas, C.; Ruiz, J.; Astruc, D. Dendritic and ion-pairing effects in oxo-anion recognition by giant alkylferrocenyl dendrimers. Organometallics 2009, 28, 4431–4437. [Google Scholar] [CrossRef]
- Boas, U.; Karlsson, A.J.; De Waal, B.F.M.; Meijer, E.W. Synthesis and properties of new thiourea-functionalized poly(propylene imine) dendrimers and their role as hosts for urea functionalized guests. J. Org. Chem. 2001, 66, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- Stephan, H.; Spies, H.; Johannsen, B.; Klein, L.; Vögtle, F. Lipophilic urea-functionalized dendrimers as efficient carriers for oxyanions. Chem. Commun. 1999, 1875–1876. [Google Scholar] [CrossRef]
- Cuřínová, P.; Winkler, M.; Krupková, A.; Císařová, I.; Budka, J.; Wun, C.N.; Blechta, V.; Malý, M.; Červenková Št’astná, L.; Sýkora, J.; et al. Transport of Anions across the Dialytic Membrane Induced by Complexation toward Dendritic Receptors. ACS Omega 2021, 6, 15514–15522. [Google Scholar] [CrossRef] [PubMed]
- Casado, C.M.; Cuadrado, I.; Alonso, B.; Morán, M.; Losada, J. Silicon-based ferrocenyl dendrimers as anion receptors in solution and immobilized onto electrode surfaces. J. Electroanal. Chem. 1999, 463, 87–92. [Google Scholar] [CrossRef]
- Villoslada, R.; Alonso, B.; Casado, C.M.; García-Armada, P.; Losada, J. Anion receptor electrochemical sensing properties of poly(propyleneimine) dendrimers with ferrocenylamidoalkyl terminal groups. Organometallics 2009, 28, 727–733. [Google Scholar] [CrossRef]
- Salvadori, K.; Šimková, L.; Císařová, I.; Sýkora, J.; Ludvík, J.; Cuřínová, P. Sulphonamidic Groups as Electron-Withdrawing Units in Ureido-Based Anion Receptors: Enhanced Anion Complexation versus Deprotonation. ChemPlusChem 2020, 85, 1401–1411. [Google Scholar] [CrossRef]
- Klejch, T.; Slavíček, J.; Hudeček, O.; Eigner, V.; Gutierrez, N.A.; Cuřínová, P.; Lhoták, P. Calix[4]arenes containing a ureido functionality on the lower rim as highly efficient receptors for anion recognition. New J. Chem. 2016, 40, 7935–7942. [Google Scholar] [CrossRef]
- Ramenda, T.; Steinbach, J.; Wuest, F. 4-[18F]Fluoro-N-methyl-N-(propyl-2-yn-1-yl)benzenesulfonamide ([18F]F-SA): A versatile building block for labeling of peptides, proteins and oligonucleotides with fluorine-18 via Cu(I)-mediated click chemistry. Amino Acids 2013, 44, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Lafay, J.; Latxague, L.; Lacroix, C.; Déléris, G. Synthesis of novel C-organosilicon derivatives, potential inhibitors of HIV reverse transcription. Phosphorus. Sulfur. Silicon Relat. Elem. 1995, 102, 155–168. [Google Scholar] [CrossRef]
- Strašák, T.; Malý, J.; Wróbel, D.; Malý, M.; Herma, R.; Čermák, J.; Müllerová, M.; Št′astná, L.Č.; Cuřínová, P. Phosphonium carbosilane dendrimers for biomedical applications-synthesis, characterization and cytotoxicity evaluation. RSC Adv. 2017, 7, 18724–18744. [Google Scholar] [CrossRef] [Green Version]
- Liegertová, M.; Wrobel, D.; Herma, R.; Müllerová, M.; Šťastná, L.Č.; Cuřínová, P.; Strašák, T.; Malý, M.; Čermák, J.; Smejkal, J.; et al. Evaluation of toxicological and teratogenic effects of carbosilane glucose glycodendrimers in zebrafish embryos and model rodent cell lines. Nanotoxicology 2018, 12, 797–818. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K. A Practical Guide for the Determination of Binding Constants. J. Incl. Phenom. Macrocycl. Chem. 2001, 39, 193–209. [Google Scholar] [CrossRef]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef]
- The Binding Constants Were Calculated Using the Bindfit Application Freely. Available online: http://supramolecular.org (accessed on 12 August 2021).
- Haav, K.; Kadam, S.A.; Toom, L.; Gale, P.A.; Busschaert, N.; Wenzel, M.; Hiscock, J.R.; Kirby, I.L.; Haljasorg, T.; Lõkov, M.; et al. Accurate Method to Quantify Binding in Supramolecular Chemistry. J. Org. Chem. 2013, 78, 7796–7808. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-L.; Roovers, J. Synthesis of Novel Carbosilane Dendritic Macromolecules. Macromolecules 1993, 26, 963–968. [Google Scholar] [CrossRef]
- Van Der Made, A.W.; Van Leeuwen, P.W.N.M.; De Wilde, J.C.; Brandes, R.A.C. Dendrimeric silanes. Adv. Mater. 1993, 5, 466–468. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP-a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anion | 1H-NMR KAss [M−1] (Error %) | UV-Vis KAss [M−1] (Error %) |
|---|---|---|
| H2PO4− | 5700 (36) | 6100 (20) |
| AcO− | 1300 (13) | 1730 (7) |
| BzO− | 2060 (14) | 2200 (6) |
| HSO4− | 190 (1.5) | - |
| Cl− | 70 (4) | - |
| Br− | 9 (0.8) | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvadori, K.; Krupková, A.; Šťastná, L.Č.; Müllerová, M.; Eigner, V.; Strašák, T.; Cuřínová, P. Controlled Anchoring of (Phenylureido)sulfonamide-Based Receptor Moieties: An Impact of Binding Site Multiplication on Complexation Properties. Molecules 2021, 26, 5670. https://doi.org/10.3390/molecules26185670
Salvadori K, Krupková A, Šťastná LČ, Müllerová M, Eigner V, Strašák T, Cuřínová P. Controlled Anchoring of (Phenylureido)sulfonamide-Based Receptor Moieties: An Impact of Binding Site Multiplication on Complexation Properties. Molecules. 2021; 26(18):5670. https://doi.org/10.3390/molecules26185670
Chicago/Turabian StyleSalvadori, Karolína, Alena Krupková, Lucie Červenková Šťastná, Monika Müllerová, Václav Eigner, Tomáš Strašák, and Petra Cuřínová. 2021. "Controlled Anchoring of (Phenylureido)sulfonamide-Based Receptor Moieties: An Impact of Binding Site Multiplication on Complexation Properties" Molecules 26, no. 18: 5670. https://doi.org/10.3390/molecules26185670