
Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors


College of Pharmacy, Umm Al-Qura University, Makkah 21955, Saudi Arabia
Molecules 2021, 26(18), 5678; https://doi.org/10.3390/molecules26185678
Submission received: 26 August 2021
/
Revised: 11 September 2021
/
Accepted: 14 September 2021
/
Published: 18 September 2021
(This article belongs to the Special Issue Kinase Inhibitors 2021)
Abstract
:Aurora-A kinase, a key mitosis regulator, is expressed in a cell cycle-dependent manner and has an essential role in maintaining chromosomal stability and the normal progression of the cell through mitosis. Aurora-A kinase is overexpressed in many malignant solid tumors, such as breast, ovarian, colon, and pancreatic cancers. Thus, inhibiting Aurora-A kinase activity is a promising approach for cancer treatment. Here, new triazole derivatives were designed as bioisosteric analogues of the known inhibitor JNJ-7706621. The new compounds showed interesting inhibitory activity against Aurora-A kinase, as attested by IC50s in the low to submicromolar range.
1. Introduction
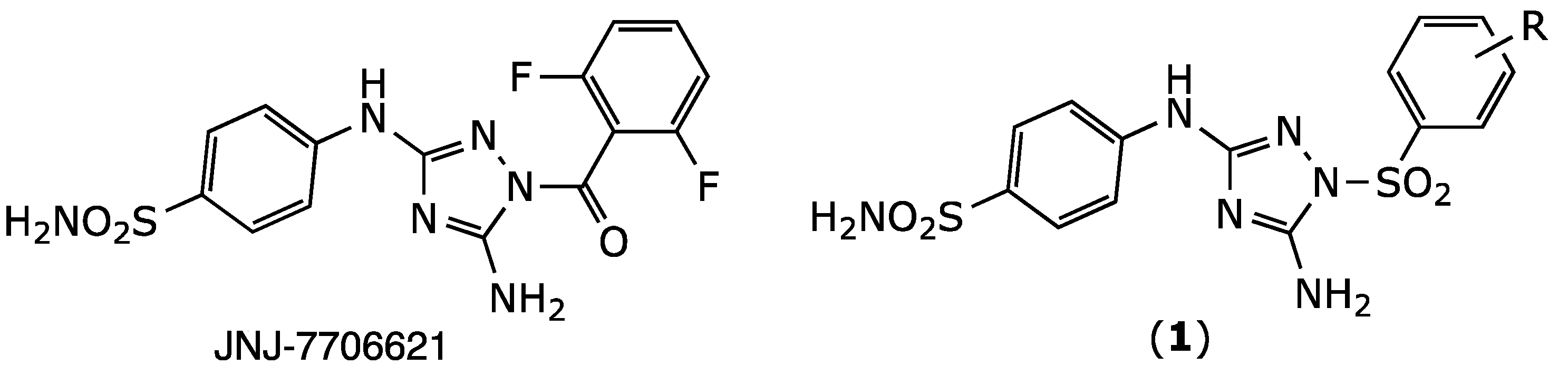
Most types of cancers are characterized by genomic instability, which can range from subtle DNA sequence changes to gene amplification, chromosome translocations, and alterations in chromosome numbers [1,2]. Chromosome changes are generally referred to by the general term chromosomal instability. Chromosome stability depends on Aurora-A kinase, a member of the serine/threonine kinase family and a key mitosis regulator. Aurora-A kinase is expressed in a cell cycle-dependent manner and has an essential role in maintaining chromosomal stability and the normal progression of the cell through mitosis [3]. Aurora-A kinase is overexpressed in many malignant solid tumors, such as breast, ovarian, colon, and pancreatic cancers [4]. For this reason, inhibiting Aurora-A kinase activity is a promising approach for cancer treatment [5]. One compound, JNJ-7706621 (Figure 1), is an inhibitor of aurora kinases and of cyclin-dependent kinases [6]. JNJ-7706621 has shown potent antiproliferative activity in various cancerous cell lines and was several folds less potent at inhibiting normal cell growth [7]. Furthermore, it significantly reduced the tumor size in an A375 melanoma human tumor xenograft model [7]. Recently, NJ-7706621 promoted the reversal of resistance to CD37-targeted radioimmunotherapy in DLBC lymphoma cell lines [8]. In the present study, we report the synthesis of JNJ-7706621 analogues (1). These derivatives were designed using an isosteric approach where the amide bond of JNJ-7706621 is replaced by a sulfonamide function (Figure 1). The sulfonamide group was chosen in this study as it is considered highly druggable thanks to its improved stability to hydrolysis and hydrogen bonding potential [9].
2. Results and Discussion
2.1. Chemistry
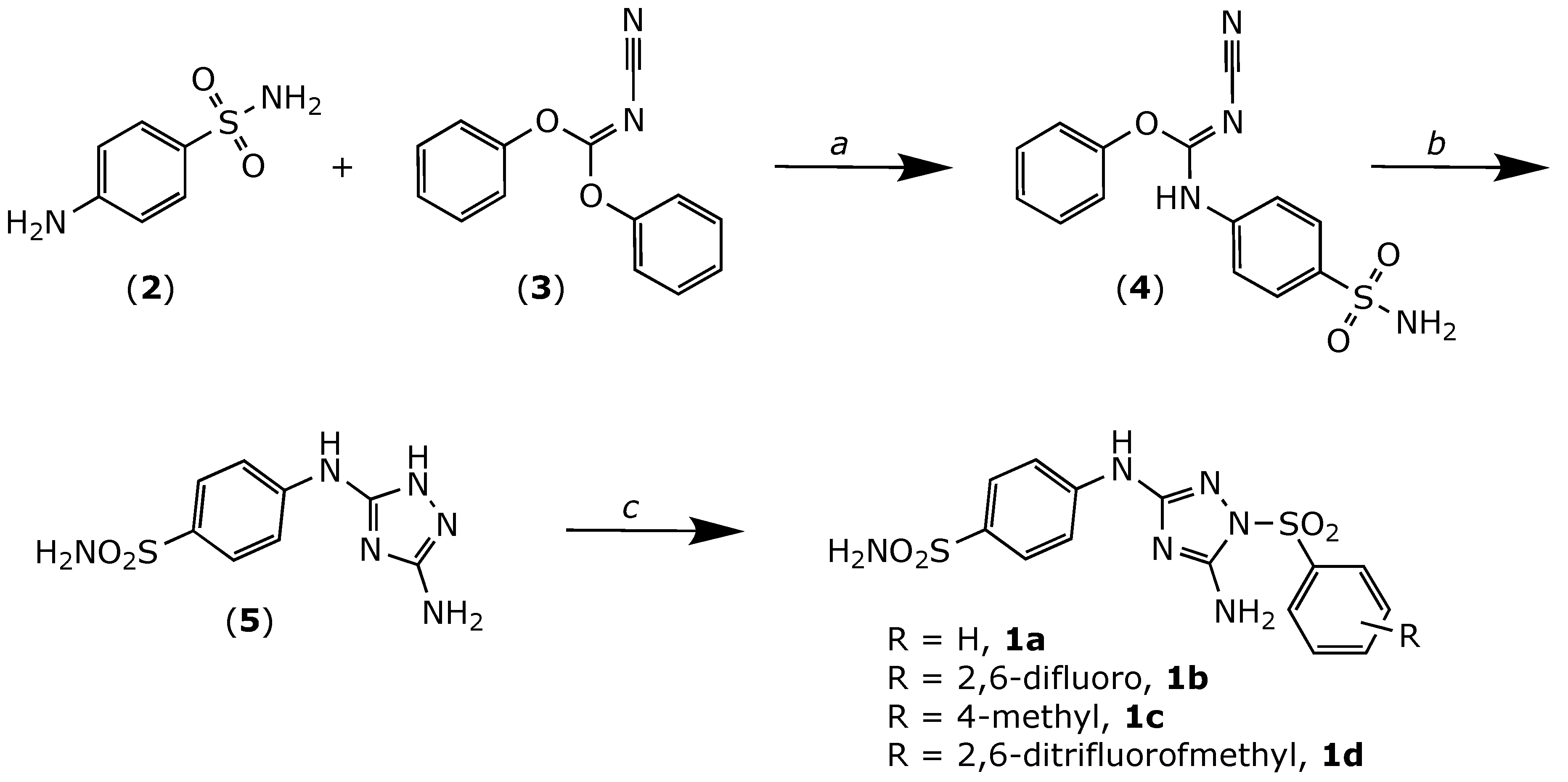
The compounds (1a–d) were obtained as shown in Figure 2. Briefly, the known triazole derivative (5) was synthesized as previously described [10], with slight modifications. First, 4-aminobenzensulfonamide (2) was heated in a sealed tube with diphenyl cyanocarbonimidate (3) at 90 °C to give intermediate (4). The latter was then cyclized by treatment with hydrazine to yield derivative 1H [1,2,4] triazole-3,5-diamine (5). Finally, the desired analogues were obtained by nucleophilic substitution of the corresponding phenylsulfonyl chlorides by the triazole derivative (5) using pyridine as a solvent.
2.2. Biological Evaluation
The inhibition of Aurora-A kinase activity by the designed compounds (Table 1) was evaluated by a method that involved the chelation-enhanced fluorescence mechanism (ChEF-based assay, PhosphoSens®) [11]. Compounds 1a–c inhibited the kinase activity of Aurora-A kinase with submicromolar IC50s. However, the derivatives 1a–c were tenfold less active compared to the reference compound JNJ-7706621. The electron-donating nature of the phenyl substituents does not seem to have an important effect on the inhibitory activity, since no significant difference was noted between non-substituted (1a), difluoro (1b), and methyl derivatives (1c). Nevertheless, the bulky trifluoromethyl group seems to cause unfavorable steric interactions.
2.3. Molecular Modeling
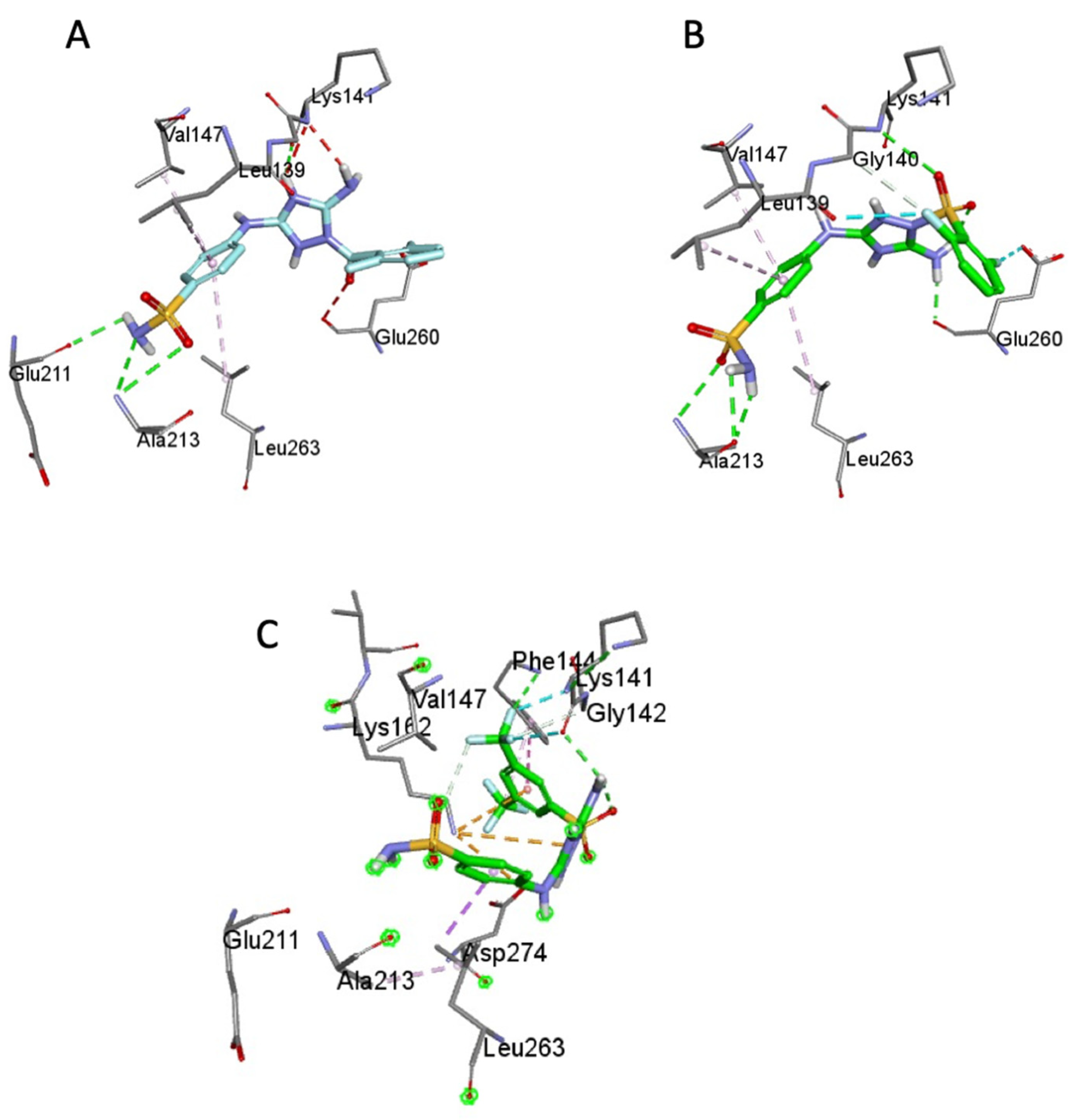
To obtain a better understanding of the differences in Aurora-A kinase inhibition by JNJ-7706621 versus its sulfonamide analogues, the compounds were docked in silico to the enzyme active site. All the compounds showed close affinity scores compared to JNJ-7706621, < −8.0 kcal/mol. Commensurate with JNJ-7706621, 1b interacted with key amino acids of ATP binding site, Val-147, Lys-141, Glu 260, Leu 263, and Leu-139). Nevertheless, 1b showed only one H-bond, with Ala-213, in contrast to JNJ-7706621 which displayed two H-bonds with Ala-213 and with Glu-211, both of which were reported as essential for optimal anti-Aurora-A kinase activity [12], Figure 3A,B. Moreover, the bulky trifluoromethyl groups of 1d changed the orientation of the external acidic sulfonamide away from the hinge region, affecting its interaction with Ala-213 and with Glu-211, Figure 3C.
3. Materials and Methods
General method for the synthesis of compounds (1a–d):
The corresponding benzenesulfonyl chloride (3.75 mmol) was added dropwise to a suspension of 4-((3-amino-1H-1,2,4-triazol-5-yl)amino)benzenesulfonamide (5) (3.75 mmol) in anhydrous pyridine (3 mL). The reaction was stirred at RT overnight. The compounds were then purified either by flash chromatography or by recrystallization from a suitable solvent system.
4-((5-amino-1-(phenylsulfonyl)-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (1a): column chromatography, Silica Gel, Hexane-EtOAc (30:70). Yield: 34%. white solid (MP: 229–231 °C). 1H NMR (300 MHz, DMSO-d6): 9.70 (s, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.78 (s, 1H), 7.69 (d, J = 8.6 Hz, 4H), 7.60–7.46 (m, 3H), 7.15 (s, 2H). 13C NMR (75 MHz, DMSO-d6): 159.77, 157.88, 143.97, 136.32, 135.68, 135.44, 130.19, 127.88, 127.24, 116.39. LC/MS (ESI+) m/z [M + 1] calculated: 395.06, found: 395.44. Elemental analysis, calculated (%): C, 42.63; H, 3.58; N, 21.31; S, 16.26, found (%): C, 43.01; H, 3.56; N, 21.12; S, 15.98.
4-((5-amino-1-((2,6-difluorophenyl)sulfonyl)-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (1b): column chromatography, Silica Gel, Hexane-EtOAc (30:70). Yield: 18.6%. White solid (MP: 224–226 °C). 1H NMR (300 MHz, DMSO-d6): 10.97 (s, 1H), 7.87 (d, J = 8.3 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H), 7.58 (d, J = 8.3 Hz, 2H), 7.52–7.39 (m, 1H), 7.08 (s, 2H), 5.63 (s, 2H). 13 C NMR (75 MHz, DMSO-d6): 159.87, 159.46 (d, J = 255.8 Hz), 157.75, 157.47, 143.92, 138.78, 135.81, 127.14, 116.36, 114.50, 114.20. LC/MS (ESI+) m/z [M + 1] calculated: 431.04, found: 431.46. Elemental analysis, calculated (%): C, 39.07; H, 2.81; F, 8.83; N, 19.53; S, 14.90, found (%): C,39.42; H, 2.76; N, 19.13; S, 14.51; F, 8.54.
4-((5-amino-1-tosyl-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (1c): column chromatography, Silica Gel, CH2Cl2 (100%). Yield: 3.4%. White solid 1H NMR (300 MHz, DMSO-d6): 9.68 (s, 1H), 7.86 (d, J = 9.0 Hz, 2H), 7.77 (d, J = 8.2 Hz, 2H), 7.56 (m, 6H), 7.13 (s, 2H), 6.10 (s, 2H), 2.36 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 159.70, 157.87, 146.35, 143.99, 135.63, 133.39, 130.60, 127.91, 127.23, 116.35, 21.58. LC/MS (ESI+) m/z [M + 1] calculated: 409.08, found: 409.47. Elemental analysis, calculated (%): C, 44.11; H, 3.95; N, 20.58; S, 15.70., found (%): C, 44.17; H, 3.89; N, 20.29; S, 15.55.
4-((5-amino-1-((3,5-bis(trifluoromethyl)phenyl)sulfonyl)-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (1d): recrystallized from acetone. Yield: 36.97%. White solid (MP: 241.6–243 °C). 1H NMR (300 MHz, DMSO-d6): 9.78 (s, 1H), 8.59 (d, J = 9.2 Hz, 2H), 7.67 (d, J = 8.9 Hz, 2H), 7.53 (d, J = 8.9 Hz, 1H), 7.16 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 160.41, 158.00, 143.67, 138.30, 136.10, 132.36, 131.91, 127.19, 124.55 (q, J = 255.8 HZ), 116.5. LC/MS (ESI+) m/z [M + 1] calculated: 530.04, found: 531.51 elemental analysis, calculated (%): C, 36.23; H, 2.28; F, 21.49; N, 15.84; S, 12.09, found (%): C, 36.29; H, 2.14; F, 21.40; N, 15.97; S, 11.97.
Biochemical assay:
JNJ-7706621and compounds 1a–c were prepared as a 5× stock solution in DMSO. The compounds were then tested in a 10-dose IC50 mode, with 3-fold serial dilutions at a starting concentration of 100 μM. The assay kinase buffer was used to dilute the Aurora-A kinase to a final concentration of 1.25 nM. The assay reaction was initiated by the addition of freshly prepared master mix to all wells (reaction, control, and blank) to achieve the following final concentrations: 1.25 nM Aurora-A kinase, 10 µM CSox-peptide substrate, 15 µM ATP, and 1 mM DTT in a final reaction volume of 50 µL per well. The reaction wells were mixed, and the relative fluorescence unit (RFU) data were collected every 3 min for 30 min at 30 °C. The used λEx/Em of the chelated Mg2+ with the Sox was 360/485 nm.
Molecular docking:
In this study, Aurora-A kinase co-crystallized with 4-fluoro-N-(3-(5-(morpholinomethyl)-1H-benzo[d]imidazol-2-yl)-1I-pyrazole-4-yl) benzamide (PDB ID 2W1C) was used. This pyrazole-benzimidazole derivative displayed the main binding interactions with the ATP-pocket required to design a classical Aurora-A kinase inhibitor. The re-docking studies were performed for the crystal structure 2W1C as a validation method. AutoDock tools [13] were used to process the enzyme. PyRx [14] was used to perform the docking. Discovery Studio Visualizer [15] was used to visualize and access the docking results.
4. Conclusions
This work reported the development of new triazole derivatives as Aurora-A kinase inhibitors. The new compounds displayed important inhibitory activity, as attested by their IC50s in the low to submicromolar range. These new derivatives represent promising inhibitors that can be considered for further investigations.
Funding
This research was funded by Deanship of Scientific Research at Umm Al-Qura University, Grant Number 19-MED-1-01-0041.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within this article.
Acknowledgments
The author would like to thank the deanship of Scientific Research at Umm Al-Qura University for supporting this work (Grant Code: 19-MED-1-01-0041).
Conflicts of Interest
The author declare that they have no competing interests.
Sample Availability
Not available.
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Neuse, C.J.; Lomas, O.C.; Schliemann, C.; Shen, Y.J.; Manier, S.; Bustoros, M.; Ghobrial, I.M. Genome instability in multiple myeloma. Leukemia 2020, 34, 2887–2897. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Assoro, A.B.; Haddad, T.; Galanis, E. Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front. Oncol. 2015, 5, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitzen, J.J.; de Jonge, M.J.; Verweij, J. Aurora kinase inhibitors. Crit. Rev. Oncol. Hematol. 2010, 73, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, S.; Rugg, C.A.; Gruninger, R.H.; Lin, R.; Fuentes-Pesquera, A.; Connolly, P.J.; Wetter, S.K.; Hollister, B.; Kruger, W.W.; Napier, C.; et al. The in vitro and in vivo effects of JNJ-7706621: A dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res. 2005, 65, 9038–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodland, G.E.; Melhus, K.; Generalov, R.; Gilani, S.; Bertoni, F.; Dahle, J.; Syljuasen, R.G.; Patzke, S. The Dual Cell Cycle Kinase Inhibitor JNJ-7706621 Reverses Resistance to CD37-Targeted Radioimmunotherapy in Activated B Cell Like Diffuse Large B Cell Lymphoma Cell Lines. Front. Oncol. 2019, 9, 1301. [Google Scholar] [CrossRef] [PubMed]
- Vijayadas, K.N.; Davis, H.C.; Kotmale, A.S.; Gawade, R.L.; Puranik, V.G.; Rajamohanan, P.R.; Sanjayan, G.J. An unusual conformational similarity of two peptide folds featuring sulfonamide and carboxamide on the backbone. Chem. Commun. 2012, 48, 9747–9749. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Connolly, P.J.; Huang, S.; Wetter, S.K.; Lu, Y.; Murray, W.V.; Emanuel, S.L.; Gruninger, R.H.; Fuentes-Pesquera, A.R.; Rugg, C.A.; et al. 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: Synthesis and evaluation of biological activities. J. Med. Chem. 2005, 48, 4208–4211. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.R.; Peterson, L.B.; Imperiali, B.; Stains, C.I. Quantification of protein kinase enzymatic activity in unfractionated cell lysates using CSox-based sensors. Curr. Protoc. Chem. Biol. 2014, 6, 135–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, A.; Wang, L.; Xu, S.; Xu, J. Aurora-A kinase inhibitor scaffolds and binding modes. Drug Discov. Today 2011, 16, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
Figure 1.
Chemical structure of JNJ-7706621 and its designed bioisosteric analogues (1).

Figure 2.
Chemical synthesis of compounds 1a–d. Reagents and conditions: (a) iPrOH, heated in a sealed tube at 90 °C, 3 h. (b) NH2NH2, THF, RT, 12 h. (c) Pyridine, substituted sulfonyl chlorides, RT, overnight.
Figure 2.
Chemical synthesis of compounds 1a–d. Reagents and conditions: (a) iPrOH, heated in a sealed tube at 90 °C, 3 h. (b) NH2NH2, THF, RT, 12 h. (c) Pyridine, substituted sulfonyl chlorides, RT, overnight.

Figure 3.
The molecular interactions of JNJ-7706621 (A), 1b (B) and 1d (C) with the ATP-binding site of Aurora-A kinase (PDB ID: 2W1C).
Figure 3.
The molecular interactions of JNJ-7706621 (A), 1b (B) and 1d (C) with the ATP-binding site of Aurora-A kinase (PDB ID: 2W1C).


{kind=link}
{kind=link}
{kind=link}
Table 1.
Inhibitory activity of the developed compounds against Aurora-A kinase.
 | |||
| Compound | X | R | IC50 (µM) 1 |
| JNJ-7706621 | CO | 2,6-difluoro | 0.016 ± 0.00 |
| 1a | SO2 | H | 0.13 ± 0.06 |
| 1b | SO2 | 2,6-difluoro | 0.21 ± 0.02 |
| 1c | SO2 | 4-Me | 0.23 ± 0.02 |
| 1d | SO2 | 2,6-ditrifluoromethyl | 1.78 ± 0.34 |
1 Data are presented as Mean ± SEM of 3 independent experiments.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Abdullah, O. Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors. Molecules 2021, 26, 5678. https://doi.org/10.3390/molecules26185678
AMA Style
Abdullah O. Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors. Molecules. 2021; 26(18):5678. https://doi.org/10.3390/molecules26185678
Chicago/Turabian StyleAbdullah, Omeima. 2021. "Design, Synthesis, and Biochemical Evaluation of New Triazole Derivatives as Aurora-A Kinase Inhibitors" Molecules 26, no. 18: 5678. https://doi.org/10.3390/molecules26185678