
Microbiome of Unilateral Chronic Rhinosinusitis: A Controlled Paired Analysis

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Sample Collection
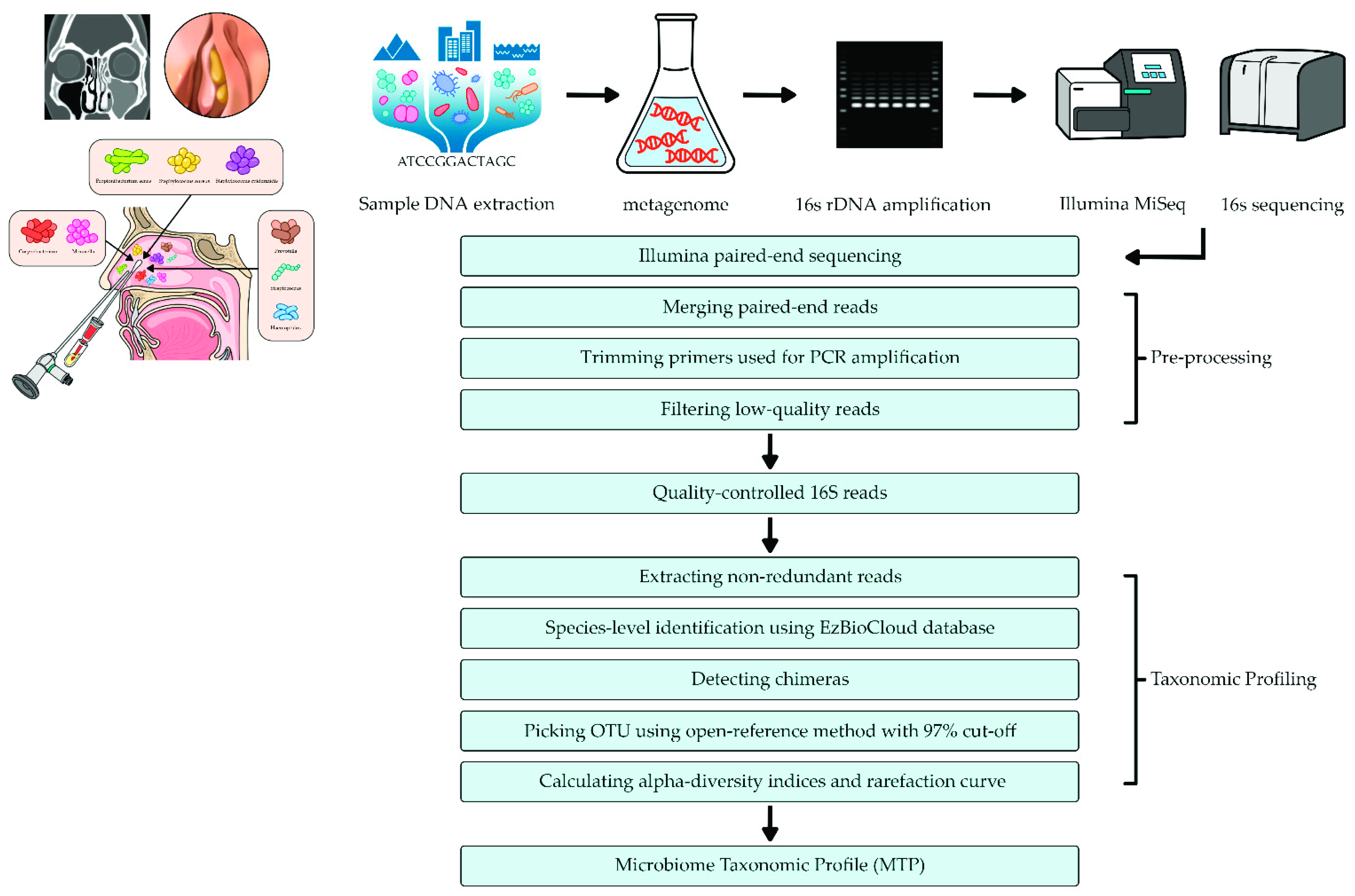
2.3. DNA Extraction and Metagenomic Analysis
2.4. Pyrosequencing Data Analysis
2.5. Bioinformatics Analysis
2.6. Statistical Analysis
3. Results
3.1. Sequence Read Counts and Taxonomic Assignments
3.2. Comparison of Richness and Alpha Diversity Indices
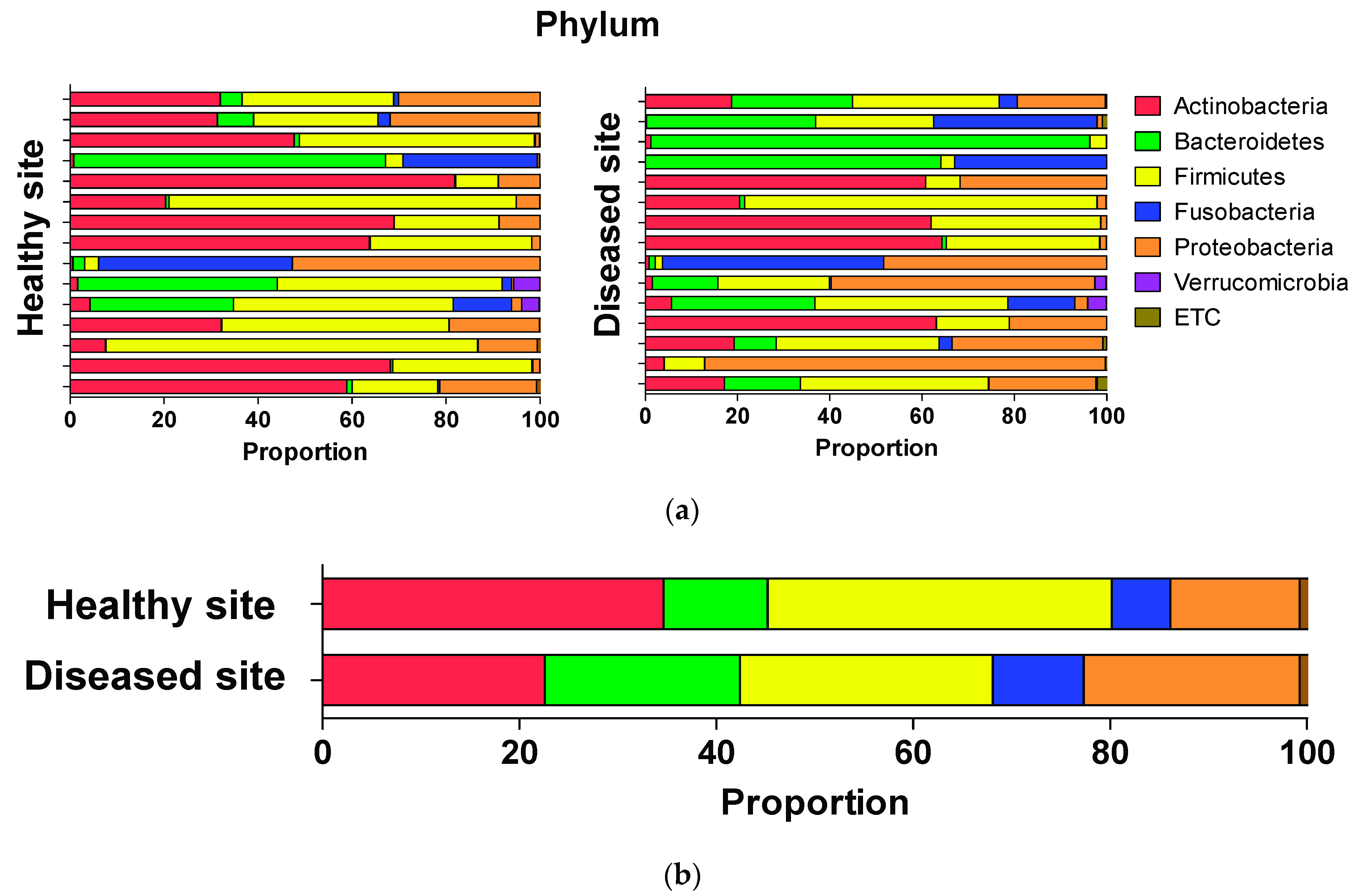
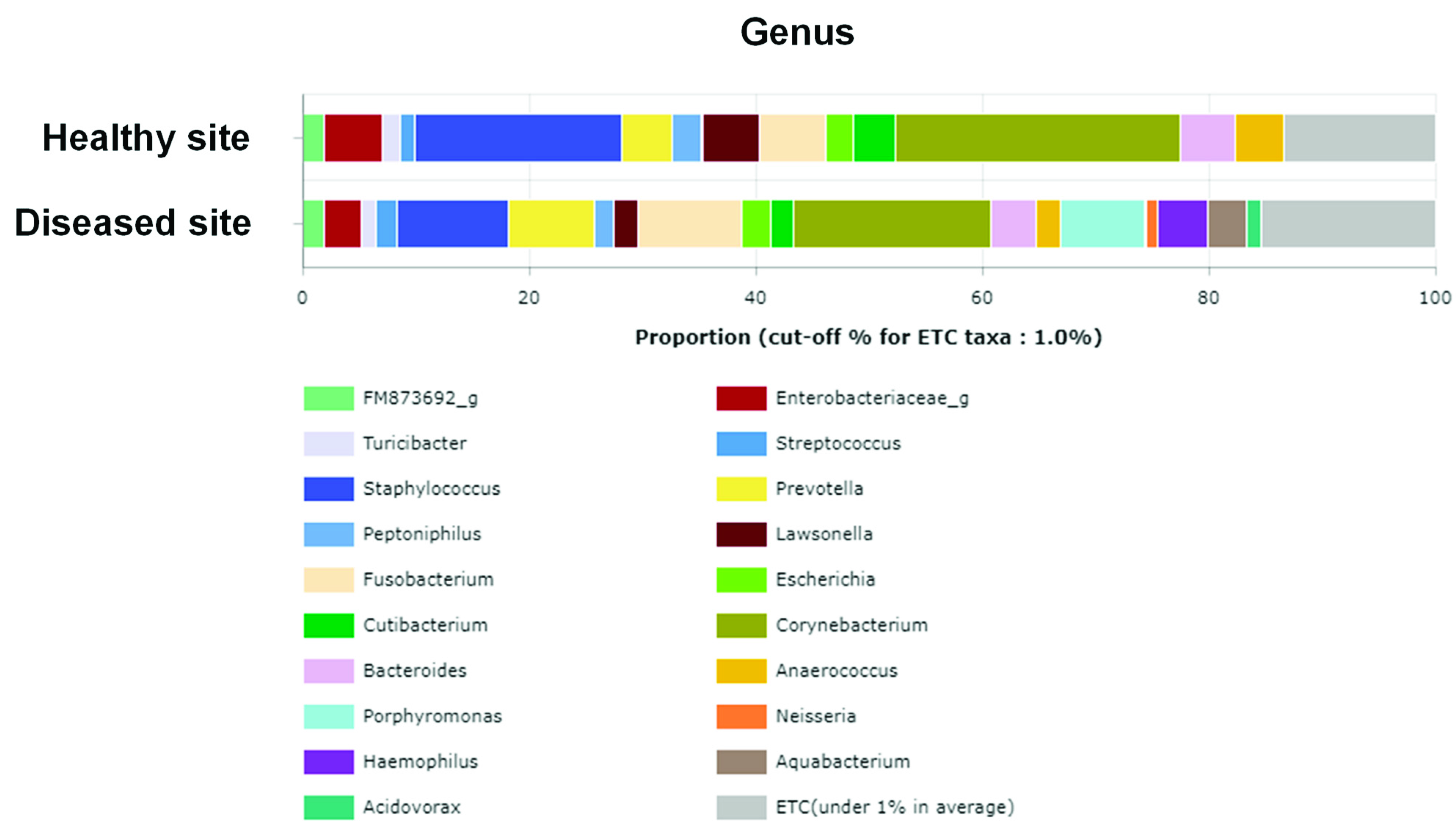
3.3. Average Composition of Microbiota at Phylum, Genus, and Species Levels
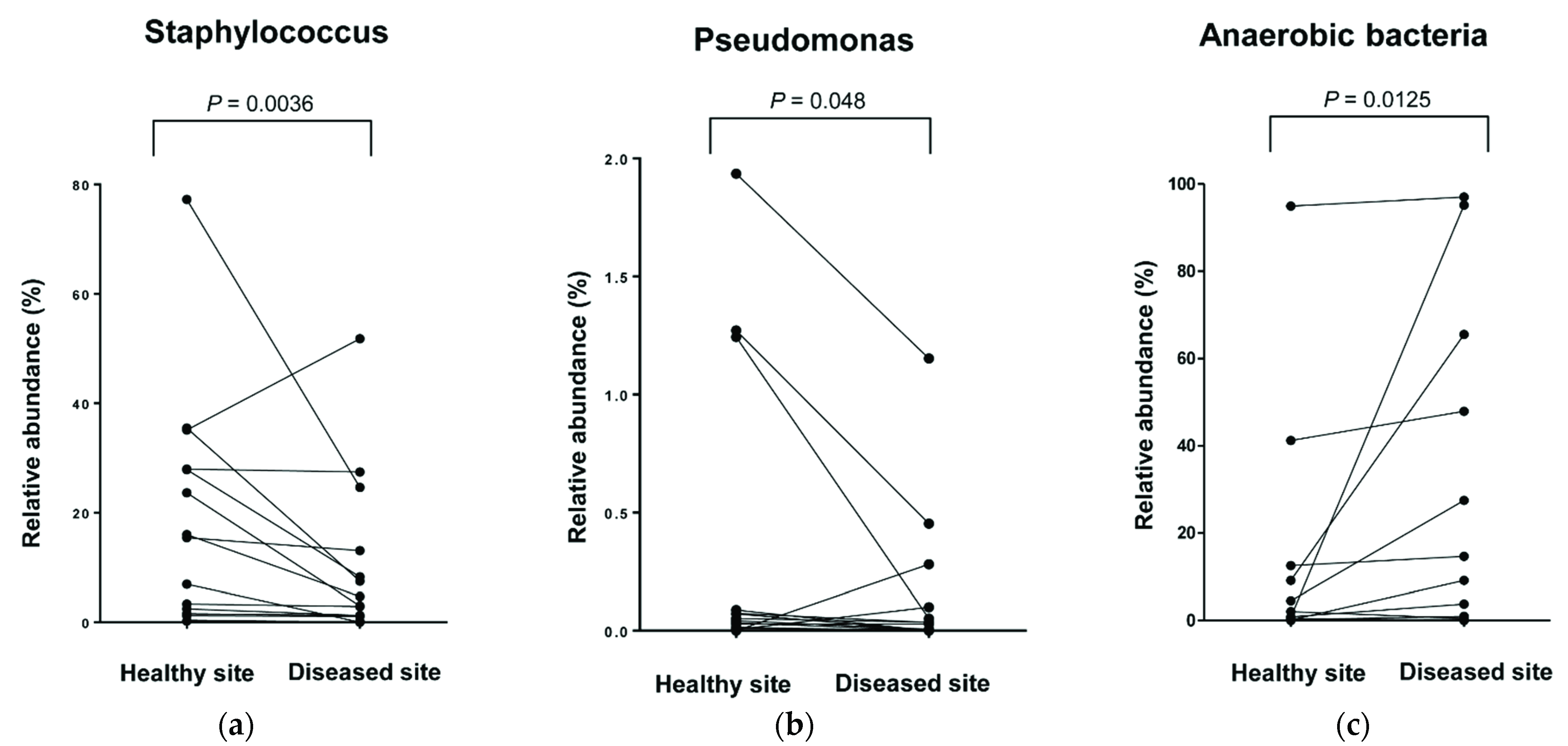
3.4. Comparison of Relative Abundance at Genus and Species Levels
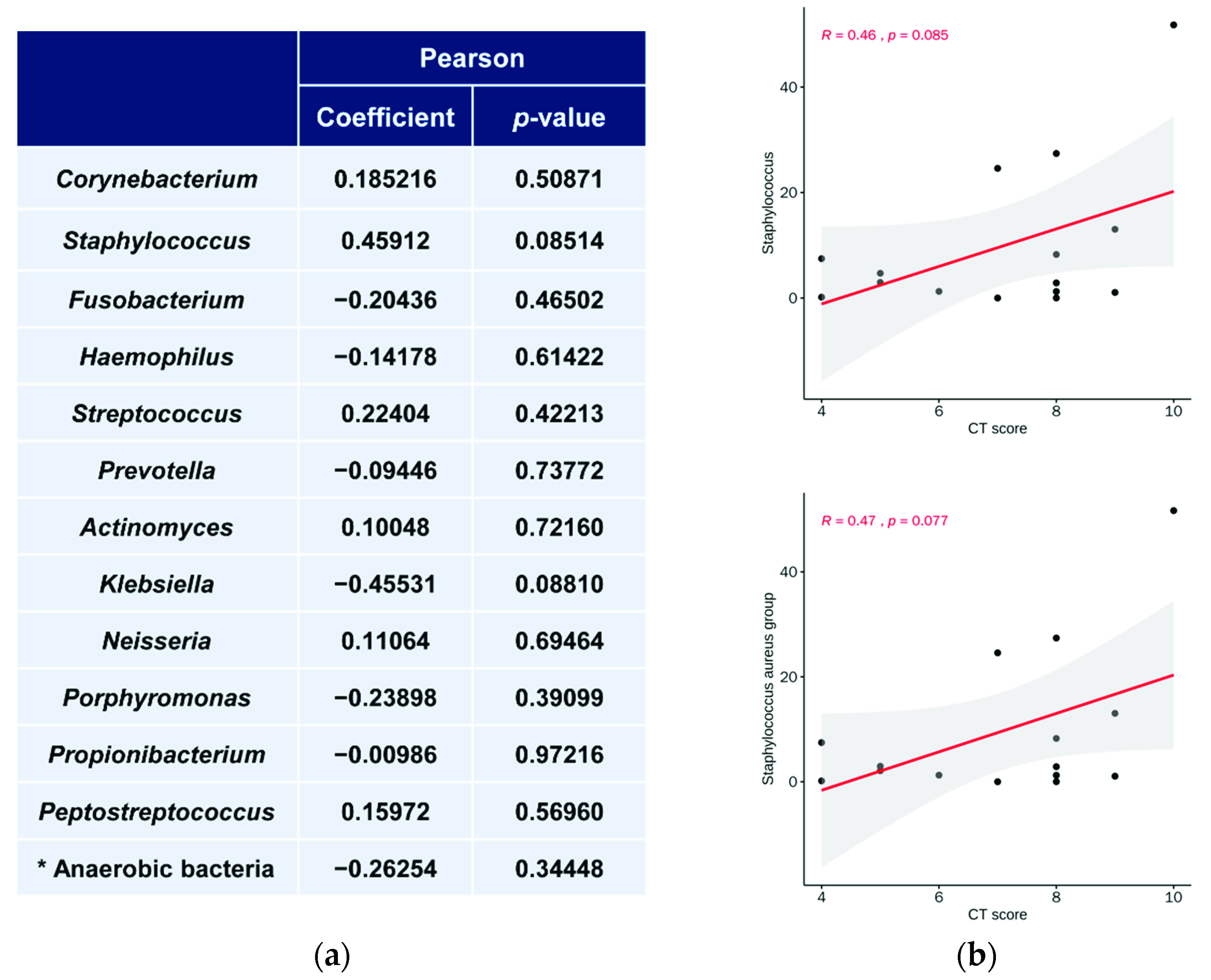
3.5. Relationship between Microbiota Abundance and LM CT Scores
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, S.; Karygianni, L.; Filippi, A.; Anderson, A.C.; Zürcher, A.; Hellwig, E.; Vach, K.; Macchiarelli, G.; Al-Ahmad, A. Combining culture and culture-independent methods reveals new microbial composition of halitosis patients’ tongue biofilm. MicrobiologyOpen 2020, 9, e958. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Sun, J.; Chang, E.B. Exploring gut microbes in human health and disease: Pushing the envelope. Genes Dis. 2014, 1, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.M.; Sadarangani, M.; Finlay, B.B. The role of the immune system in governing host-microbe interactions in the intestine. Nat. Immunol. 2013, 14, 660–667. [Google Scholar] [CrossRef]
- Negi, S.; Das, D.K.; Pahari, S.; Nadeem, S.; Agrewala, J.N. Potential Role of Gut Microbiota in Induction and Regulation of Innate Immune Memory. Front. Immunol. 2019, 10, 2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, K.F. Airway microbial dysbiosis in asthmatic patients: A target for prevention and treatment? J. Allergy Clin. Immunol. 2017, 139, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Hyun, D.W.; Min, H.J.; Kim, M.S.; Whon, T.W.; Shin, N.R.; Kim, P.S.; Kim, H.S.; Lee, J.Y.; Kang, W.; Choi, A.M.K.; et al. Dysbiosis of Inferior Turbinate Microbiota Is Associated with High Total IgE Levels in Patients with Allergic Rhinitis. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Glatz, M.; Horiuchi, K.; Kawasaki, H.; Akiyama, H.; Kaplan, D.H.; Kong, H.H.; Amagai, M.; Nagao, K. Dysbiosis and Staphylococcus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity 2015, 42, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Ren, H.; Lu, Y.; Fang, C.; Hou, G.; Yang, Z.; Chen, B.; Yang, F.; Zhao, Y.; Shi, Z.; et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine 2019, 47, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Roy Sarkar, S.; Banerjee, S. Gut microbiota in neurodegenerative disorders. J. Neuroimmunol. 2019, 328, 98–104. [Google Scholar] [CrossRef]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Fokkens, W.J.; Lund, V.J.; Hopkins, C.; Hellings, P.W.; Kern, R.; Reitsma, S.; Toppila-Salmi, S.; Bernal-Sprekelsen, M.; Mullol, J.; Alobid, I.; et al. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 2020, 58, 1–464. [Google Scholar] [CrossRef]
- Hopkins, C.; Surda, P.; Bast, F.; Hettige, R.; Walker, A.; Hellings, P.W. Prevention of chronic rhinosinusitis. Rhinology 2018, 56, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Mahdavinia, M.; Keshavarzian, A.; Tobin, M.C.; Landay, A.L.; Schleimer, R.P. A comprehensive review of the nasal microbiome in chronic rhinosinusitis (CRS). Clin. Exp. Allergy 2016, 46, 21–41. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Ponikau, J.U.; Sherris, D.A.; Congdon, D.; Frigas, E.; Homburger, H.A.; Swanson, M.C.; Gleich, G.J.; Kita, H. Chronic rhinosinusitis: An enhanced immune response to ubiquitous airborne fungi. J. Allergy Clin. Immunol. 2004, 114, 1369–1375. [Google Scholar] [CrossRef]
- Wagner Mackenzie, B.; Waite, D.W.; Hoggard, M.; Taylor, M.W.; Biswas, K.; Douglas, R.G. Moving beyond descriptions of diversity: Clinical and research implications of bacterial imbalance in chronic rhinosinusitis. Rhinology 2017, 55, 291–297. [Google Scholar] [CrossRef]
- Anderson, M.; Stokken, J.; Sanford, T.; Aurora, R.; Sindwani, R. A systematic review of the sinonasal microbiome in chronic rhinosinusitis. Am. J. Rhinol. Allergy 2016, 30, 161–166. [Google Scholar] [CrossRef]
- Kumpitsch, C.; Koskinen, K.; Schopf, V.; Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef] [Green Version]
- Park, I.H.; Lee, J.S.; Park, J.H.; Kang, S.H.; Hong, S.M.; Park, I.S.; Yoon, J.H.; Hong, S.J. Comparison of the human microbiome in adults and children with chronic rhinosinusitis. PLoS ONE 2020, 15, e0242770. [Google Scholar] [CrossRef] [PubMed]
- Hoggard, M.; Biswas, K.; Zoing, M.; Wagner Mackenzie, B.; Taylor, M.W.; Douglas, R.G. Evidence of microbiota dysbiosis in chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2017, 7, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.B.; Hong, S.W.; Kim, D.K.; Jeon, S.G.; Kim, K.R.; Cho, S.H.; Gho, Y.S.; Jee, Y.K.; Kim, Y.K. Decreased diversity of nasal microbiota and their secreted extracellular vesicles in patients with chronic rhinosinusitis based on a metagenomic analysis. Allergy 2014, 69, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Aurora, R.; Chatterjee, D.; Hentzleman, J.; Prasad, G.; Sindwani, R.; Sanford, T. Contrasting the microbiomes from healthy volunteers and patients with chronic rhinosinusitis. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 1328–1338. [Google Scholar] [CrossRef]
- Boase, S.; Foreman, A.; Cleland, E.; Tan, L.; Melton-Kreft, R.; Pant, H.; Hu, F.Z.; Ehrlich, G.D.; Wormald, P.J. The microbiome of chronic rhinosinusitis: Culture, molecular diagnostics and biofilm detection. BMC Infect. Dis. 2013, 13, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feazel, L.M.; Robertson, C.E.; Ramakrishnan, V.R.; Frank, D.N. Microbiome complexity and Staphylococcus aureus in chronic rhinosinusitis. Laryngoscope 2012, 122, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, V.R.; Hauser, L.J.; Feazel, L.M.; Ir, D.; Robertson, C.E.; Frank, D.N. Sinus microbiota varies among chronic rhinosinusitis phenotypes and predicts surgical outcome. J. Allergy Clin. Immunol. 2015, 136, 334–342. [Google Scholar] [CrossRef]
- Cope, E.K.; Goldberg, A.N.; Pletcher, S.D.; Lynch, S.V. Compositionally and functionally distinct sinus microbiota in chronic rhinosinusitis patients have immunological and clinically divergent consequences. Microbiome 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassiouni, A.; Paramasivan, S.; Shiffer, A.; Dillon, M.R.; Cope, E.K.; Cooksley, C.; Ramezanpour, M.; Moraitis, S.; Ali, M.J.; Bleier, B.S.; et al. Microbiotyping the Sinonasal Microbiome. Front. Cell Infect. Microbiol 2020, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Aversa, Z.; Atkinson, E.J.; Schafer, M.J.; Theiler, R.N.; Rocca, W.A.; Blaser, M.J.; LeBrasseur, N.K. Association of Infant Antibiotic Exposure With Childhood Health Outcomes. Mayo Clin. Proc. 2021, 96, 66–77. [Google Scholar] [CrossRef]
- Nimmagadda, S.N.; Midani, F.S.; Durand, H.; Reese, A.T.; Murdoch, C.C.; Nicholson, B.P.; Veldman, T.; Burke, T.W.; Zaas, A.K.; Woods, C.W.; et al. Nasal microbiota exhibit neither reproducible nor orderly dynamics following rhinoviral infection. medRxiv 2020. [Google Scholar] [CrossRef]
- Başal, Y.; Güleç, G.; Ertuğrul, B.; Eryilmaz, A.; Günel, C.; Başak, S. Changes in nasal flora one year after endoscopic dacryocystorhinostomy. B-ent 2015, 11, 129–134. [Google Scholar] [PubMed]
- Kim, A.S.; Willis, A.L.; Laubitz, D.; Sharma, S.; Song, B.H.; Chiu, A.G.; Le, C.H.; Chang, E.H. The effect of maxillary sinus antrostomy size on the sinus microbiome. Int. Forum Allergy Rhinol. 2019, 9, 30–38. [Google Scholar] [CrossRef]
- Cocuzza, S.; Maniaci, A.; Di Luca, M.; La Mantia, I.; Grillo, C.; Spinato, G.; Motta, G.; Testa, D.; Ferlito, S. Long-term results of nasal surgery: Comparison of mini-invasive turbinoplasty. J. Biol. Regul. Homeost. Agents 2020, 34, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Myers, E.W.; Miller, W. Optimal alignments in linear space. Comput. Appl. Biosci. 1988, 4, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [Green Version]
- Jervis Bardy, J.; Psaltis, A.J. Next Generation Sequencing and the Microbiome of Chronic Rhinosinusitis: A Primer for Clinicians and Review of Current Research, Its Limitations, and Future Directions. Ann. Otol. Rhinol. Laryngol. 2016, 125, 613–621. [Google Scholar] [CrossRef]
- Ramakrishnan, V.R.; Hauser, L.J.; Frank, D.N. The sinonasal bacterial microbiome in health and disease. Curr. Opin Otolaryngol. Head Neck Surg. 2016, 24, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Johansen, F.E.; Kaetzel, C.S. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Stevens, W.W.; Lee, R.J.; Schleimer, R.P.; Cohen, N.A. Chronic rhinosinusitis pathogenesis. J. Allergy Clin. Immunol. 2015, 136, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Bordin, A.; Sidjabat, H.E.; Cottrell, K.; Cervin, A. Chronic rhinosinusitis: A microbiome in dysbiosis and the search for alternative treatment options. Microbiol. Aust. 2016, 37, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Lee, K.; Yoo, J.B.; Song, J.W.; Yoon, J.H. Bacteriological findings and antimicrobial susceptibility in chronic sinusitis with nasal polyp. Acta Otolaryngol. 2006, 126, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Benninger, M.S.; Appelbaum, P.C.; Denneny, J.C.; Osguthorpe, D.J.; Stankiewicz, J.A. Maxillary sinus puncture and culture in the diagnosis of acute rhinosinusitis: The case for pursuing alternative culture methods. Otolaryngol. Head Neck Surg. 2002, 127, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Abreu, N.A.; Nagalingam, N.A.; Song, Y.; Roediger, F.C.; Pletcher, S.D.; Goldberg, A.N.; Lynch, S.V. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci. Transl. Med. 2012, 4, 151ra124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner Mackenzie, B.; Waite, D.W.; Hoggard, M.; Douglas, R.G.; Taylor, M.W.; Biswas, K. Bacterial community collapse: A meta-analysis of the sinonasal microbiota in chronic rhinosinusitis. Environ. Microbiol. 2017, 19, 381–392. [Google Scholar] [CrossRef]
- Hoggard, M.; Waldvogel-Thurlow, S.; Zoing, M.; Chang, K.; Radcliff, F.J.; Wagner Mackenzie, B.; Biswas, K.; Douglas, R.G.; Taylor, M.W. Inflammatory Endotypes and Microbial Associations in Chronic Rhinosinusitis. Front. Immunol. 2018, 9, 2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeller, K.; Herlemann, D.P.R.; Schuldt, T.; Ovari, A.; Guder, E.; Podbielski, A.; Kreikemeyer, B.; Olzowy, B. Microbiome and Culture Based Analysis of Chronic Rhinosinusitis Compared to Healthy Sinus Mucosa. Front. Microbiol. 2018, 9, 643. [Google Scholar] [CrossRef] [Green Version]
- Krismer, B.; Weidenmaier, C.; Zipperer, A.; Peschel, A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat. Rev. Microbiol. 2017, 15, 675–687. [Google Scholar] [CrossRef]
- Folino, F.; Fattizzo, M.; Ruggiero, L.; Oriano, M.; Aliberti, S.; Blasi, F.; Gaffuri, M.; Marchisio, P.; Torretta, S. Nasopharyngeal Microbiota Analysis in Healthy and Otitis-prone Children: Focus on History of Spontaneous Tympanic Membrane Perforation. Pediatr. Infect. Dis. J. 2021, 40, 16–21. [Google Scholar] [CrossRef]
- Saibene, A.M.; Vassena, C.; Pipolo, C.; Trimboli, M.; De Vecchi, E.; Felisati, G.; Drago, L. Odontogenic and rhinogenic chronic sinusitis: A modern microbiological comparison. Int. Forum Allergy Rhinol. 2016, 6, 41–45. [Google Scholar] [CrossRef]
- Taschieri, S.; Torretta, S.; Corbella, S.; Del Fabbro, M.; Francetti, L.; Lolato, A.; Capaccio, P. Pathophysiology of sinusitis of odontogenic origin. J. Investig. Clin. Dent. 2017, 8, e12202. [Google Scholar] [CrossRef]
- Brook, I. The role of anaerobic bacteria in sinusitis. Anaerobe 2006, 12, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Lal, D.; Keim, P.; Delisle, J.; Barker, B.; Rank, M.A.; Chia, N.; Schupp, J.M.; Gillece, J.D.; Cope, E.K. Mapping and comparing bacterial microbiota in the sinonasal cavity of healthy, allergic rhinitis, and chronic rhinosinusitis subjects. Int. Forum Allergy Rhinol. 2017, 7, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Flynn, M.J.; Rose, F.V.; Jousimies-Somer, H.; Jakielaszek, C.; McTeague, M.; Wexler, H.M.; Berkowitz, E.; Wynne, B. Bacteriologic findings associated with chronic bacterial maxillary sinusitis in adults. Clin. Infect. Dis. 2002, 35, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartog, B.; Degener, J.E.; Van Benthem, P.P.; Hordijk, G.J. Microbiology of chronic maxillary sinusitis in adults: Isolated aerobic and anaerobic bacteria and their susceptibility to twenty antibiotics. Acta Otolaryngol. 1995, 115, 672–677. [Google Scholar] [CrossRef]
- Erkan, M.; Aslan, T.; Ozcan, M.; Koc, N. Bacteriology of antrum in adults with chronic maxillary sinusitis. Laryngoscope 1994, 104, 321–324. [Google Scholar] [CrossRef]
- Brook, I. Microbiology of chronic rhinosinusitis. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1059–1068. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subjects | Sex | Age | Underlying Diseases | Diseased Site | LM CT Score at Diseased Site | Blood Eo (%) | Allergy | Bacterial Culture at Lesion Side | Bacterial Culture at Normal Side |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 35 | none | Rt | 5 | 1.9 | none | S. pneumoniae | no growth |
| 2 | F | 34 | DM, Pul.Tb | Rt | 8 | 0.9 | none | S. aureus, S. epidermidis | no growth |
| 3 | M | 54 | DM | Lt | 7 | 2.2 | D. farinae, house dust | P. aeruginosa, G (-) bacilli | no growth |
| 4 | M | 32 | none | Rt | 4 | 1.7 | none | E. aerogenes | no growth |
| 5 | F | 17 | none | Rt | 9 | 0.11 | none | H. parainfluenzae | E. aerogenes |
| 6 | M | 48 | none | Rt | 6 | 3.5 | none | S. aureus (MRSA) | S. aureus |
| 7 | M | 56 | none | Lt | 4 | 4.2 | none | S. epidermidis | S. epidermidis |
| 8 | M | 67 | HTN, Hepatitis B | Lt | 8 | 2.3 | Cladosporium | S. aureus, G (+) cocci | S. aureus, G (+) cocci, G (−) bacilli |
| 9 | M | 59 | none | Rt | 9 | 5.2 | none | S. epidermidis, G (+) bacilli, Corynebacterium Species | S. epidermidis |
| 10 | M | 34 | none | Rt | 10 | 2.3 | D. farinae, D. pteronyssinus | S. aureus | S. aureus |
| 11 | M | 54 | none | Lt | 8 | 1.8 | D. farinae, house dust, storage mite, Acarus siro, cockroach, Cladosporium, Aspergillus | E. aerogenes | S. epidermidis |
| 12 | M | 57 | none | Lt | 7 | 4.4 | D. farinae, D. pteronyssinus, house dust, storage mite, cockroach, multiple tree, grass, weed pollens | S. anginosus | S. aureus |
| 13 | M | 39 | none | Lt | 5 | 4.8 | none | S. epidermidis, S. aureus | no growth |
| 14 | M | 42 | hyperthyroidism | Rt | 8 | 0.12 | D. farinae, D. pteronyssinus, house dust, Acarus siro | E. aerogenes | no growth |
| 15 | M | 65 | DM, HTN | Rt | 8 | 0.5 | none | K. aerogenes | K. aerogenes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.C.; Park, I.-H.; Lee, J.S.; Park, S.M.; Kang, S.H.; Hong, S.-M.; Byun, S.-H.; Jung, Y.G.; Hong, S.J. Microbiome of Unilateral Chronic Rhinosinusitis: A Controlled Paired Analysis. Int. J. Environ. Res. Public Health 2021, 18, 9878. https://doi.org/10.3390/ijerph18189878
Park SC, Park I-H, Lee JS, Park SM, Kang SH, Hong S-M, Byun S-H, Jung YG, Hong SJ. Microbiome of Unilateral Chronic Rhinosinusitis: A Controlled Paired Analysis. International Journal of Environmental Research and Public Health. 2021; 18(18):9878. https://doi.org/10.3390/ijerph18189878
Chicago/Turabian StylePark, Sang Chul, Il-Ho Park, Joong Seob Lee, Sung Min Park, Sung Hun Kang, Seok-Min Hong, Soo-Hwan Byun, Yong Gi Jung, and Seok Jin Hong. 2021. "Microbiome of Unilateral Chronic Rhinosinusitis: A Controlled Paired Analysis" International Journal of Environmental Research and Public Health 18, no. 18: 9878. https://doi.org/10.3390/ijerph18189878