
Advancing Our Understanding of Corneal Herpes Simplex Virus-1 Immune Evasion Mechanisms and Future Therapeutics

 and
and 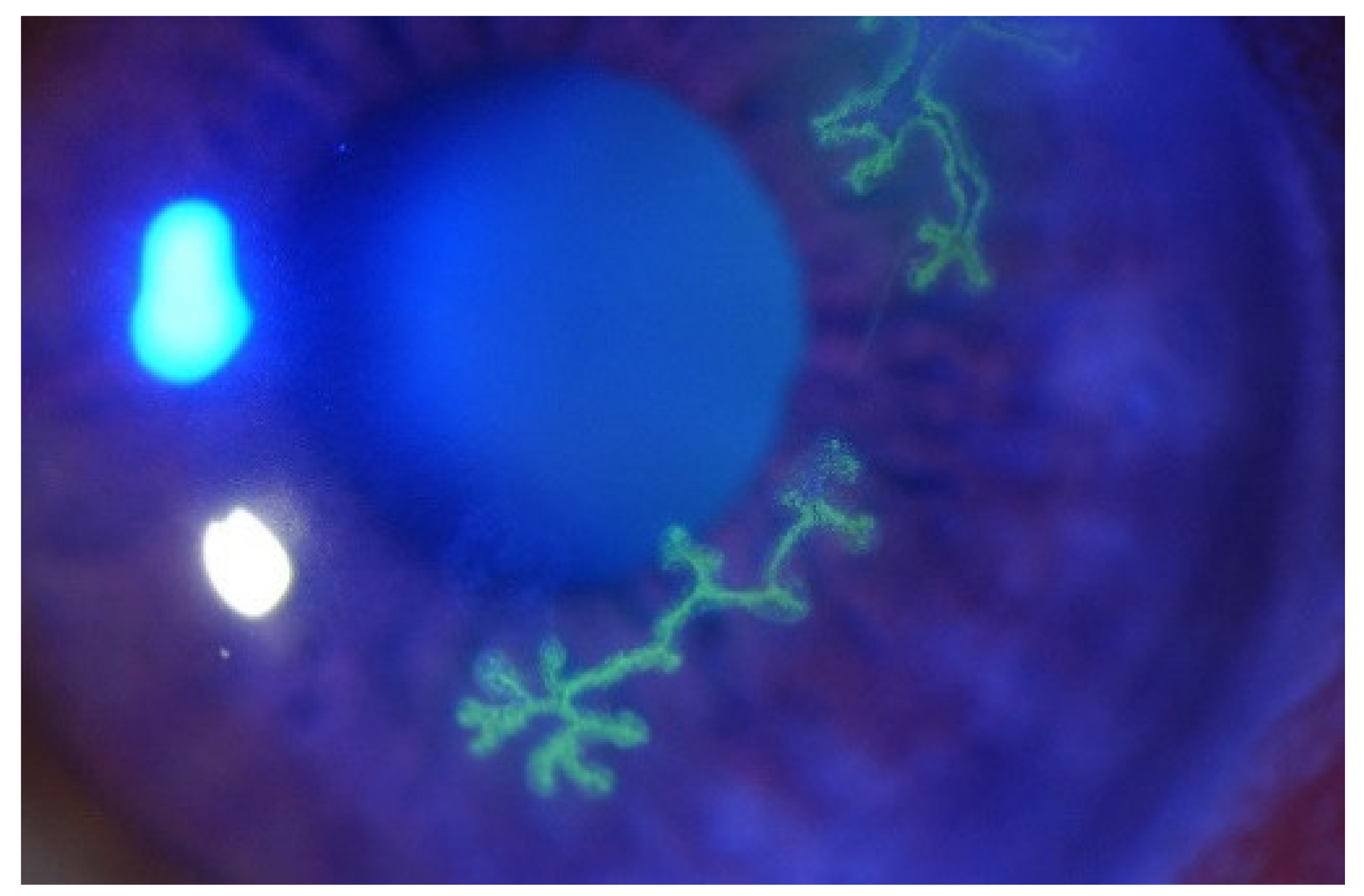{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background to the Disease
1.1. Epidemiology
1.2. Symptoms and Diagnosis
2. HSV-1 Replication Strategies and Mechanisms of Pathogenesis
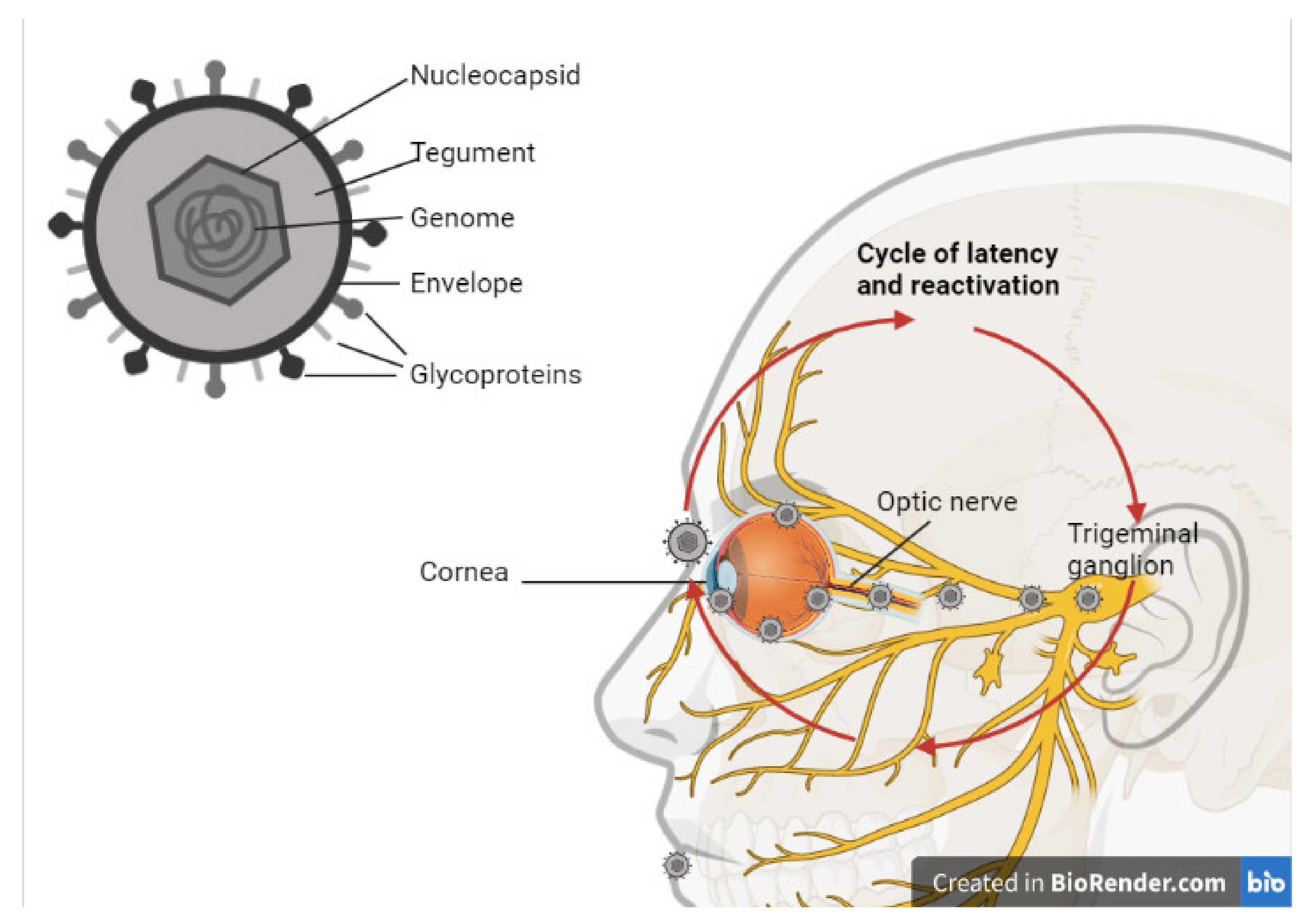
2.1. HSV-1 Host Cell Entry
2.2. HSV-1 Cycle of Latency and Infection
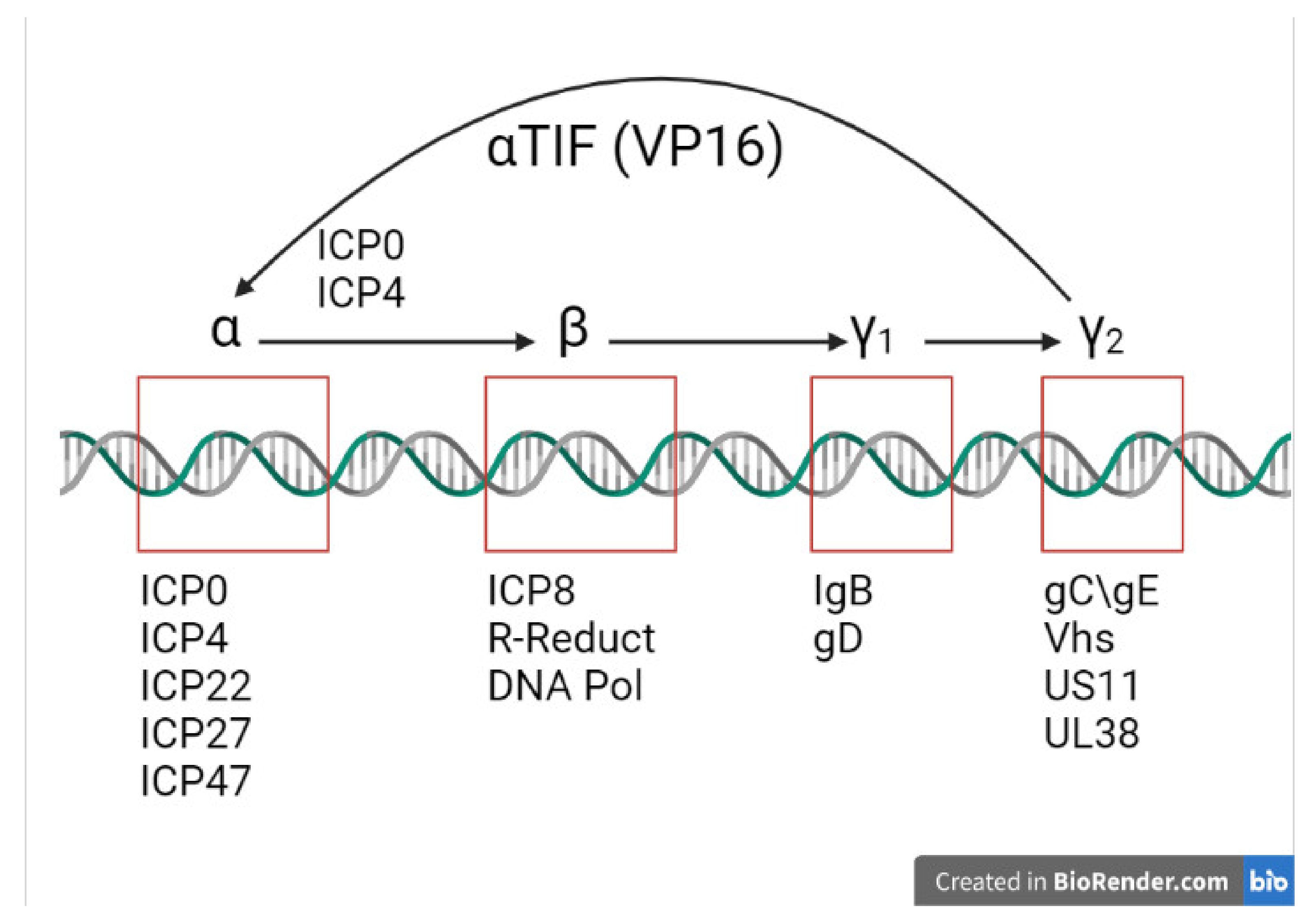
2.3. Immediate Early Proteins
2.4. Viral Early Phase
2.5. Late Phase
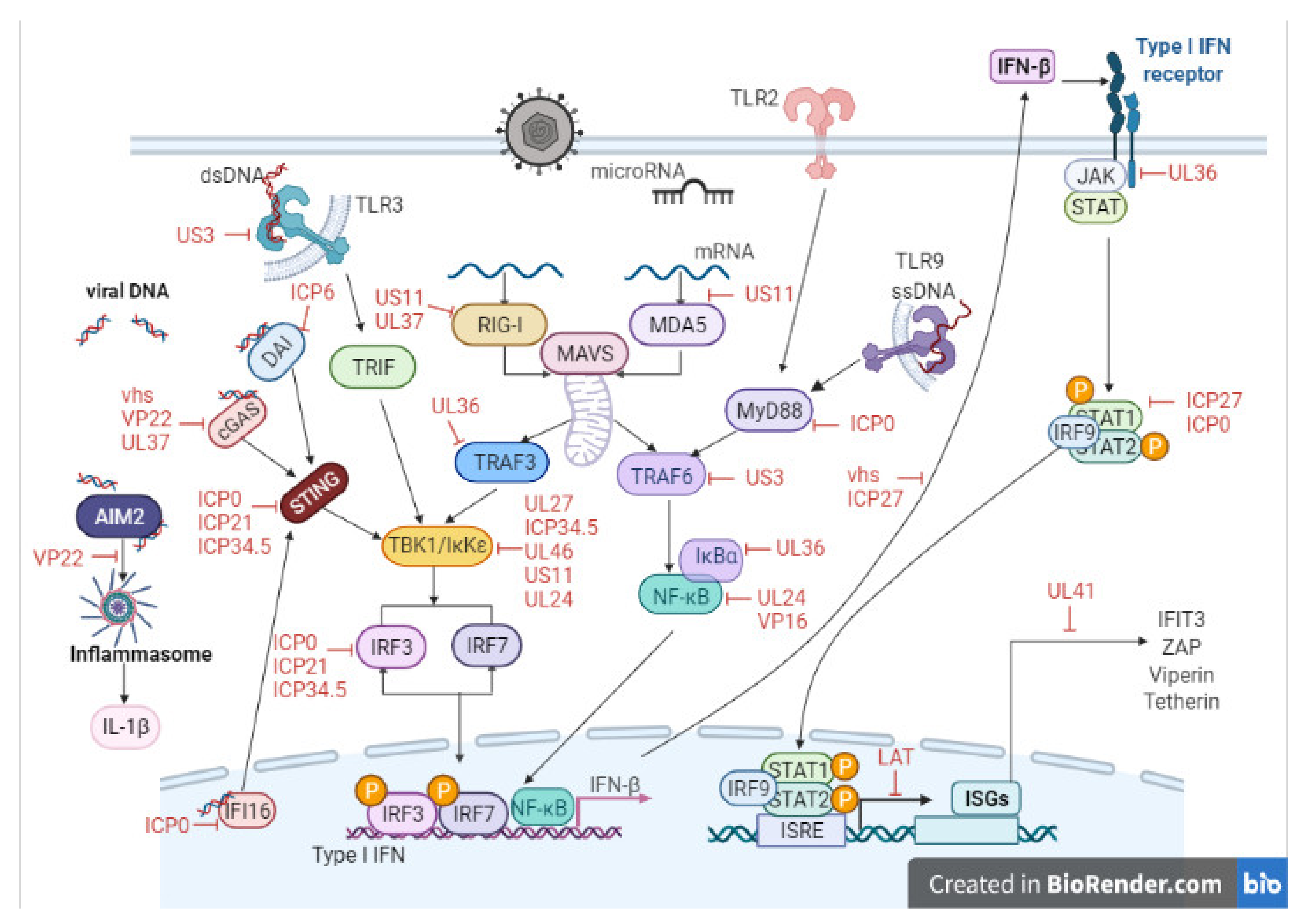
3. Host Cell–Virus Interaction
4. Key Viral Proteins
5. Current Treatments
6. Novel Treatments
6.1. Nucleic Acid Aptamers
6.2. Toll-like Receptors
6.3. Antibodies
6.4. MicroRNA
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| APC | Antigen-presenting cell |
| 3-OS-HS | 3-O-sulfated heparan sulfate proteoglycan |
| ATP | Adenosine triphosphate |
| ATRX | ATP-dependent helicase |
| CHO | Chinese hamster ovary |
| CD8 | Cluster of differentiation 8 |
| CoREST | Corepressor of RE1 silencing transcription factor |
| cGAS | cyclic GMP-AMP synthase |
| DDX41 | DEAD box helicase 41 |
| hDaxx | Death domain-associated protein |
| DC | Dendritic cells |
| DNA | Deoxyribonucleic acid |
| DAI | DNA-dependent activator of IFN regulatory factors |
| dsRNA | Double-stranded RNA |
| E | Early |
| ELISA | Enzyme-linked immunosorbent assay |
| EBV | Epstein–Barr virus |
| g | Glycoprotein |
| HSPGs | Heparan sulfate proteoglycans |
| HCV | Hepatitis C virus |
| HSK | Herpes stormal keratitis |
| HSV-1 | Herpes Simplex Virus type 1 |
| HSV-2 | Herpes simplex virus type-2 |
| HVEM | Herpesvirus Entry Mediator |
| pUL30 | HSV-1 polymerase |
| pUL29 | ICP8 |
| IE | Immediate Early |
| ICP | Infected cell proteins |
| IKK | Inhibitor of kB kinase |
| IFI16 | Interferon gamma inducible protein 16 |
| IRF | Interferon regulatory factor |
| IFN-α | Interferon-α |
| IFN-β | Interferon-β |
| IL | Interleukin |
| IRAK1 | Interleukin 1 receptor-associated kinase 1 |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| KO | Knockout |
| LGP2 | Laboratory of genetics and physiology 2 |
| LAT | Latency Associated Transcript |
| L | Latency Associated Transcript |
| MDA5 | Melanoma differentiation-associated gene-5 |
| MAVs | Mitochondrial antiviral signalling protein |
| mAB | Monoclonal antibody |
| Mal | MyD88 adaptor-like protein |
| MyD88 | Myeloid differentiation primary response protein 88 |
| NK | Natural killer |
| IKKε | NFκB kinase-epsilon |
| ND10 | Nuclear domain 10 |
| PAMPs | Pathogen associated molecular patterns |
| PRR | Pathogen recognition receptors |
| PCR | Polymerase chain reaction |
| PML | Promyelocytic leukemia protein |
| RING | Really Interesting New Gene |
| RIG-I | Retinoic acid inducible gene-I |
| RLRs | RIG-I-like receptors |
| Pol III | RNA polymerase III |
| SEMA6D | Semaphorin 6D |
| ssRNA | Single-stranded RNA |
| Sp100 | Speckled, 100 kDa |
| STING | Stimulator of IFN genes |
| TBK1 | TANK-binding kinase 1 |
| TAB3 | TGF-beta activated kinase 1 (MAP3K7) binding protein 3 |
| TRAF3 | TNF receptor-associated factor 3 |
| TLR | Toll-like receptors |
| TIRAP | Toll/interleukin 1 receptor domain-containing adaptor protein |
| TRIF | Toll/interleukin 1 receptor domain-containing adaptor-inducing IFN-β |
| TIR | Toll/interleukin-1 receptor |
| TRAM | TRIF-related adaptor molecule |
| IFN-I | Type 1 IFN |
| VZV | Varicella-zoster virus |
| UL41 | Virion-induced host shutoff protein, VHS |
| WT | Wild type |
| ZNF138 | Zinc finger protein 138 |
References
- Liesegang, T.J. Herpes Simplex Virus Epidemiology and Ocular Importance. Cornea 2001, 20, 1–13. [Google Scholar] [CrossRef]
- Cullen, B.R. Herpesvirus microRNAs: Phenotypes and functions. Curr. Opin. Virol. 2011, 1, 211–215. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.E.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Umene, K.; Sakaoka, H. Evolution of herpes simplex virus type 1 under herpesviral evolutionary processes. Arch. Virol. 1999, 144, 637–656. [Google Scholar] [CrossRef]
- Toma, H.S.; Murina, A.T.; Areaux, R.; Neumann, D.M.; Bhattacharjee, P.S.; Foster, T.P.; Kaufman, H.E.; Hill, J.M. Ocular HSV-1 Latency, Reactivation and Recurrent Disease. Semin. Ophthalmol. 2008, 23, 249–273. [Google Scholar] [CrossRef]
- Harris, K.D. Herpes Simplex Virus Keratitis. Home Healthc. Now 2019, 37, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Koelle, D.M.; Wald, A. Herpes simplex virus: The importance of asymptomatic shedding. J. Antimicrob. Chemother. 2000, 45, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.M.F.; Holland, E.J.; Huang, A.J. Bilateral herpetic keratoconjunctivitis. Ophthalmology 2003, 110, 493–496. [Google Scholar] [CrossRef]
- Rowe, M.A.; Leger, A.J.S.; Jeon, S.; Dhaliwal, D.K.; Knickelbein, J.E.; Hendricks, R.L. Herpes keratitis. Prog. Retin. Eye Res. 2013, 32, 88–101. [Google Scholar] [CrossRef]
- Farooq, A.V.; Shukla, D. Herpes Simplex Epithelial and Stromal Keratitis: An Epidemiologic Update. Surv. Ophthalmol. 2012, 57, 448–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabenau, H.F.; Buxbaum, S.; Preiser, W.; Weber, B.; Doerr, H.W. Seroprevalence of herpes simplex virus types 1 and type 2 in the Frankfurt am Main area, Germany. Med. Microbiol. Immunol. 2002, 190, 153–160. [Google Scholar] [CrossRef]
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in Herpes Simplex Virus Type 1 and Type 2 Seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Lobo, A.M.; Agelidis, A.M.; Shukla, D. Pathogenesis of herpes simplex keratitis: The host cell response and ocular sur-face sequelae to infection and inflammation. Ocul. Surf. 2019, 17, 40–49. [Google Scholar] [CrossRef]
- Reynaud, C.; Rousseau, A.; Kaswin, G.; M’Garrech, M.; Barreau, E.; Labetoulle, M. Persistent Impairment of Quality of Life in Patients with Herpes Simplex Keratitis. Ophthalmology 2017, 124, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Darougar, S.; Wishart, M.S.; Viswalingam, N.D. Epidemiological and clinical features of primary herpes simplex virus ocular infection. Br. J. Ophthalmol. 1985, 69, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.J.; Dreyer, E.B. Herpesvirus Infections of the Anterior Segment. Int. Ophthalmol. Clin. 1996, 36, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.; Choudhary, A. Herpes simplex keratitis. Prog. Retin. Eye Res. 2006, 25, 355–380. [Google Scholar] [CrossRef]
- Sharif, Z.; Sharif, W. Corneal neovascularization: Updates on pathophysiology, investigations & management. Rom. J. Ophthalmol. 2019, 63, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Baker, K. Herpes simplex keratitis. J. Med. Microbiol. 1996, 45, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, N.; Nishida, K.; Adachi, W.; Tei, M.; Honma, Y.; Dota, A.; Sotozono, C.; Yokoi, N.; Yamamoto, S.; Kinoshita, S. Detection of herpes simplex virus DNA in atypical epithelial keratitis using polymerase chain reaction. Br. J. Ophthalmol. 1999, 83, 957–960. [Google Scholar] [CrossRef] [PubMed]
- McBride, B.W.; Ward, K.A. Herpes simplex-specific IgG subclass response in herpetic keratitis. J. Med. Virol. 1987, 21, 179–189. [Google Scholar] [CrossRef]
- Wong, R.R.; Abd-Aziz, N.; Affendi, S.; Poh, C.L. Role of microRNAs in antiviral responses to dengue infection. J. Biomed. Sci. 2020, 27, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lian, Z.; Engström, M. Use of a flipped classroom in ophthalmology courses for nursing, dental and medical students: A quasi-experimental study using a mixed-methods approach. Nurse Educ. Today 2020, 85, 104262. [Google Scholar] [CrossRef]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Albecka, A.; Owen, D.J.; Ivanova, L.; Brun, J.; Liman, R.; Davies, L.; Ahmed, M.F.; Colaco, S.; Hollinshead, M.; Graham, S.C.; et al. Dual Function of the pUL7-pUL51 Tegument Protein Complex in Herpes Simplex Virus 1 Infection. J. Virol. 2017, 91, e02196-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettenleiter, T.C. Herpesvirus Assembly and Egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 1998, 72, 873–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campadelli-Fiume, G.; Cocchi, F.; Menotti, L.; Lopez, M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 2000, 10, 305–319. [Google Scholar] [CrossRef]
- Montgomery, R.; Warner, M.; Lum, B.J.; Spear, P.G. Herpes Simplex Virus-1 Entry into Cells Mediated by a Novel Member of the TNF/NGF Receptor Family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, R.J.; Krummenacher, G.H.; Cohen, R.; Eisenberg, J.; Spear, P.G.J.S. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A Novel Role for 3-O-Sulfated Heparan Sulfate in Herpes Simplex Virus 1 Entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.G.; Longnecker, R. Herpesvirus Entry Mediator and Ocular Herpesvirus Infection: More than Meets the Eye. J. Virol. 2017, 91, e00115-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummenacher, C.; Baribaud, F.; de Leon, M.P.; Baribaud, I.; Whitbeck, J.; Xu, R.; Cohen, G.H.; Eisenberg, R.J. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 2004, 322, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Manoj, S.; Jogger, C.R.; Myscofski, D.; Yoon, M.; Spear, P.G. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl. Acad. Sci. USA 2004, 101, 12414–12421. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ljubimov, A.V.; Jin, L.; Pfeffer, K.; Kronenberg, M.; Ghiasi, H. Herpes Simplex Virus 1 Latency and the Kinetics of Reactivation Are Regulated by a Complex Network of Interactions between the Herpesvirus Entry Mediator, Its Ligands (gD, BTLA, LIGHT, and CD160), and the Latency-Associated Transcript. J. Virol. 2018, 92, 01451-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, S.J.; Rhode-Kurnow, A.; Mott, K.R.; Jiang, X.; Carpenter, D.; Rodriguez-Barbosa, J.-I.; Jones, C.; Wechsler, S.L.; Ware, C.F.; Ghiasi, H. Interactions between Herpesvirus Entry Mediator (TNFRSF14) and Latency-Associated Transcript during Herpes Simplex Virus 1 Latency. J. Virol. 2013, 88, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.-J.; Akhtar, J.; Desai, P.; Shukla, D. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 2010, 391, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenheid, S.; Gatzke, L.; Meadows, H.; Tufaro, F. Herpes simplex virus infection and propagation in a mouse L cell mutant lacking heparan sulfate proteoglycans. J. Virol. 1993, 67, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; O’Donnell, C.; Copeland, R.J.; Scarlett, T.; Liu, J.; Shukla, D. Soluble 3-O-sulfated heparan sulfate can trigger herpes simplex virus type 1 entry into resistant Chinese hamster ovary (CHO-K1) cells. J. Gen. Virol. 2007, 88, 1075–1079. [Google Scholar] [CrossRef]
- O’Donnell, C.D.; Shukla, D. The importance of heparan sulfate in herpesvirus infection. Virol. Sin. 2008, 23, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Sharthiya, H.; Seng, C.; Van Kuppevelt, T.H.; Tiwari, V.; Fornaro, M. HSV-1 interaction to 3-O-sulfated heparan sulfate in mouse-derived DRG explant and profiles of inflammatory markers during virus infection. J. Neurovirol. 2017, 23, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Dollery, S.J.; Wright, C.C.; Johnson, D.C.; Nicola, A.V. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 2011, 85, 9964–9973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef]
- Nicola, A.V. Herpesvirus Entry into Host Cells Mediated by Endosomal Low pH. Traffic 2016, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 latent transcription and non-coding RNA: A critical retrospective. J. Neuroimmunol. 2017, 308, 65–101. [Google Scholar] [CrossRef]
- Kaufman, H.E.; Azcuy, A.M.; Varnell, E.D.; Sloop, G.D.; Thompson, H.W.; Hill, J.M. HSV-1 DNA in Tears and Saliva of Normal Adults. Investig. Opthalmol. Vis. Sci. 2005, 46, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Steiner, I. Human herpes viruses latent infection in the nervous system. Immunol. Rev. 1996, 152, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Hamrah, P.; Gate, D.; Mott, K.R.; Mantopoulos, D.; Zheng, L.; Town, T.; Jones, C.; Von Andrian, U.H.; Freeman, G.J.; et al. The Role of LAT in Increased CD8+ T Cell Exhaustion in Trigeminal Ganglia of Mice Latently Infected with Herpes Simplex Virus. J. Virol. 2011, 85, 4184–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.; Shukla, D. Recent advances in vaccine development for herpes simplex virus types I and II. Hum. Vaccines Immunother. 2013, 9, 729–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koganti, R.; Yadavalli, T.; Shukla, D. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms 2019, 7, 429. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Tscharke, D.C. Herpes Simplex Virus Latency Is Noisier the Closer We Look. J. Virol. 2020, 94, 01701-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoll, M.; Proença, J.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.R.; Connor, V.; Coleman, H.M.; Proença, J.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLOS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef] [PubMed]
- Azher, T.N.; Yin, X.-T.; Tajfirouz, D.; Huang, A.J.; Stuart, P.M. Herpes simplex keratitis: Challenges in diagnosis and clinical management. Clin. Ophthalmol. 2017, 11, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Poccardi, N.; Rousseau, A.; Haigh, O.; Takissian, J.; Naas, T.; Deback, C.; Trouillaud, L.; Issa, M.; Roubille, S.; Juillard, F.; et al. Herpes Simplex Virus 1 Replication, Ocular Disease, and Reactivations from Latency Are Restricted Unilaterally after Inoculation of Virus into the Lip. J. Virol. 2019, 93, e01586-19. [Google Scholar] [CrossRef]
- Fuller, A.O.; Lee, W.C. Herpes simplex virus type 1 entry through a cascade of virus-cell interactions requires different roles of gD and gH in penetration. J. Virol. 1992, 66, 5002–5012. [Google Scholar] [CrossRef] [Green Version]
- Campadelli-Fiume, G.; Menotti, L.; Avitabile, E.; Gianni, T. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr. Opin. Virol. 2012, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated Transport of Incoming Herpes Simplex Virus 1 Capsids to the Nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef]
- Roizman, B.; Gu, H.; Mandel, G. The First 30 Minutes in the Life of a Virus: unREST in the Nucleus. Cell Cycle 2005, 4, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of Herpesvirus Macromolecular Synthesis I. Cascade Regulation of the Synthesis of Three Groups of Viral Proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Batterson, W.; Roizman, B. Characterization of the herpes simplex virion-associated factor responsible for the induc-tion of alpha genes. J. Virol. 1983, 46, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.E.; Palfreyman, J.W.; Preston, C.M. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 1984, 180, 1–19. [Google Scholar] [CrossRef]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis: Sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA 1975, 72, 1276–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grondin, B.; DeLuca, N. Herpes Simplex Virus Type 1 ICP4 Promotes Transcription Preinitiation Complex Formation by Enhancing the Binding of TFIID to DNA. J. Virol. 2000, 74, 11504–11510. [Google Scholar] [CrossRef] [Green Version]
- Maruzuru, Y.; Shindo, K.; Liu, Z.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kato, A.; Kawaguchi, Y. Role of Herpes Simplex Virus 1 Immediate Early Protein ICP22 in Viral Nuclear Egress. J. Virol. 2014, 88, 7445–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri-Goldin, R.M.; E Mendoza, G. A herpesvirus regulatory protein appears to act post-transcriptionally by affecting mRNA processing. Genes Dev. 1992, 6, 848–863. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Patel, A.; Krause, P.R. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc. Natl. Acad. Sci. USA 2016, 113, 12256–12261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.-H.B.; Sciabica, K.S.; Sandri-Goldin, R.M. ICP27 Interacts with the RNA Export Factor Aly/REF To Direct Herpes Simplex Virus Type 1 Intronless mRNAs to the TAP Export Pathway. J. Virol. 2002, 76, 12877–12889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehmer, P.; Lehman, I.R. Herpes Simplex Virus DNA Replication. Annu. Rev. Biochem. 1997, 66, 347–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; A Schaffer, P. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 1992, 66, 2904–2915. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 1993, 67, 7501–7512. [Google Scholar] [CrossRef] [Green Version]
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069. [Google Scholar] [CrossRef]
- Maul, G.G.; Everett, R.D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP. J. Gen. Virol. 1994, 75, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING fin-ger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeoch, D.J.; Dalrymple, M.A.; Dolan, A.; McNab, D.; Perry, L.J.; Taylor, P.; Challberg, M.D. Structures of herpes sim-plex virus type 1 genes required for replication of virus DNA. J. Virol. 1988, 62, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.A.; Nelson, N.J.; McGeoch, D.J.; Challberg, M.D. Identification of herpes simplex virus type 1 genes required for origin-dependent DNA synthesis. J. Virol. 1988, 62, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.S.; Chiou, H.C.; Hall, J.D.; Mount, D.W.; Retondo, M.J.; Weller, S.; Coen, D.M. Sequence and mapping analyses of the herpes simplex virus DNA polymerase gene predict a C-terminal substrate binding domain. Proc. Natl. Acad. Sci. USA 1985, 82, 7969–7973. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, T.R.; Lehman, I.R. Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J. Biol. Chem. 1990, 265, 11227–11232. [Google Scholar] [CrossRef]
- Parris, D.S.; Cross, A.; Haarr, L.; Orr, A.; Frame, M.C.; Murphy, M.; McGeoch, D.J.; Marsden, H.S. Identification of the gene encoding the 65-kilodalton DNA-binding protein of herpes simplex virus type. J. Virol. 1988, 62, 818–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, J.B.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Amino Acids 143 to 150 of the Herpes Simplex Virus Type 1 Scaffold Protein Are Required for the Formation of Portal-Containing Capsids. J. Virol. 2008, 82, 6778–6781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.C.; Newcomb, W.W. Herpesvirus capsid assembly: Insights from structural analysis. Curr. Opin. Virol. 2011, 1, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whealy, M.E.; Card, J.P.; Meade, R.P.; Robbins, A.K.; Enquist, L.W. Effect of brefeldin A on alphaherpesvirus membrane protein glycosylation and virus egress. J. Virol. 1991, 65, 1066–1081. [Google Scholar] [CrossRef] [Green Version]
- Granzow, H.; Weiland, F.; Jons, A.; Klupp, B.G.; Karger, A.; Mettenleiter, T.C. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: A reassessment. J. Virol. 1997, 71, 2072–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crump, C. Virus Assembly and Egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Deatly, A.M.; Spivack, J.G.; Lavi, E.; Fraser, N.W. RNA from an immediate early region of the type 1 herpes simplex virus genome is present in the trigeminal ganglia of latently infected mice. Proc. Natl. Acad. Sci. USA 1987, 84, 3204–3208. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Sciences 1987, 235, 1056–1059. [Google Scholar] [CrossRef]
- Maillet, S.; Naas, T.; Crepin, S.; Roque-Afonso, A.-M.; Lafay, F.; Efstathiou, S.; Labetoulle, M. Herpes Simplex Virus Type 1 Latently Infected Neurons Differentially Express Latency-Associated and ICP0 Transcripts. J. Virol. 2006, 80, 9310–9321. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.A.; Preston, C.M. Establishment of latency in vitro by the herpes simplex virus type 1 mutant in. J. Gen. Virol. 1991, 72, 907–913. [Google Scholar] [CrossRef]
- Biswas, P.S.; Banerjee, K.; Kim, B.; Kinchington, P.R.; Rouse, B.T. Role of inflammatory cytokine-induced cyclooxygenase 2 in the ocular immunopathologic disease herpetic stromal keratitis. J. Virol. 2005, 79, 10589–10600. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, R.L.; Epstein, R.J.; Tumpey, T. The effect of cellular immune tolerance to HSV-1 antigens on the immuno-pathology of HSV-1 keratitis. Invest. Ophthalmol. Vis. Sci. 1989, 30, 105–115. [Google Scholar] [PubMed]
- Russell, R.G.; Nasisse, M.P.; Larsen, H.S.; Rouse, B.T. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Investig. Ophthalmol. Vis. Sci. 1984, 25, 938–944. [Google Scholar]
- Mosmann, T.R.; Coffman, R.L. Heterogeneity of Cytokine Secretion Patterns and Functions of Helper T Cells; Elsevier BV: Amsterdam, The Netherlands, 1989; Volume 46, pp. 111–147. [Google Scholar]
- Niemialtowski, M.G.; Rouse, B.T. Predominance of Th1 cells in ocular tissues during herpetic stromal keratitis. J. Immunol. 1992, 149, 3035–3039. [Google Scholar]
- Scott, P.; Kaufmann, S.H. The role of T-cell subsets and cytokines in the regulation of infection. Immunol. Today 1991, 12, 346–348. [Google Scholar] [CrossRef]
- Deshpande, S.; Zheng, M.; Lee, S.; Banerjee, K.; Gangappa, S.; Kumaraguru, U.; Rouse, B.T. Bystander Activation Involving T Lymphocytes in Herpetic Stromal Keratitis. J. Immunol. 2001, 167, 2902–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.S.; Granucci, F.; Yeh, L.; Schaffer, P.A.; Cantor, H. Molecular mimicry by herpes simplex virus-type 1: Autoim-mune disease after viral infection. Science 1998, 279, 1344–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verjans, G.M.; Remeijer, L.; Mooy, C.M.; Osterhaus, A.D. Herpes simplex virus-specific T cells infiltrate the cornea of patients with herpetic stromal keratitis: No evidence for autoreactive T cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2607–2612. [Google Scholar]
- Deshpande, S.P.; Lee, S.; Zheng, M.; Song, B.; Knipe, D.; Kapp, J.A.; Rouse, B.T. Herpes Simplex Virus-Induced Keratitis: Evaluation of the Role of Molecular Mimicry in Lesion Pathogenesis. J. Virol. 2001, 75, 3077–3088. [Google Scholar] [CrossRef] [Green Version]
- Verjans, G.M.G.M.; Remeijer, L.; Van Binnendijk, R.S.; Cornelissen, J.G.C.; Völker-Dieben, H.J.; Baarsma, S.G.; Osterhaus, A.D.M.E. Identification and Characterization of Herpes Simplex Virus-Specific CD4+T Cells in Corneas of Herpetic Stromal Keratitis Patients. J. Infect. Dis. 1998, 177, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.K.; Laycock, K.A.; Nash, M.M.; Pepose, J.S. Corneal Langerhans cell dynamics after herpes simplex virus reactivation. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2282–2290. [Google Scholar]
- Hendricks, R.L.; Janowicz, M.; Tumpey, T.M. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J. Immunol. 1992, 148, 2522–2529. [Google Scholar]
- Brikos, C.; O’Neill, L.A. Signalling of toll-like receptors. In Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2008; Volume 183, pp. 21–50. [Google Scholar]
- Philpott, D.J.; Girardin, S.E. The role of Toll-like receptors and Nod proteins in bacterial infection. Mol. Immunol. 2004, 41, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G. Translational Mini-Review Series on Toll-like Receptors: Recent advances in understanding the role of Toll-like receptors in anti-viral immunity. Clin. Exp. Immunol. 2007, 147, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef] [Green Version]
- Reuven, E.M.; Fink, A.; Shai, Y. Regulation of innate immune responses by transmembrane interactions: Lessons from the TLR family. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1586–1593. [Google Scholar] [CrossRef] [Green Version]
- Herbst-Kralovetz, M.; Pyles, R. Toll-like receptors, innate immunity and HSV pathogenesis. Herpes J. IHMF 2006, 13, 37–41. [Google Scholar]
- Hiscott, J. Triggering the Innate Antiviral Response through IRF-3 Activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Mansur, D.S.; Kroon, E.G.; Nogueira, M.L.; Arantes, R.M.; Rodrigues, S.C.; Akira, S.; Gazzinelli, R.T.; Campos, M.A. Lethal Encephalitis in Myeloid Differentiation Factor 88-Deficient Mice Infected with Herpes Simplex Virus. Am. J. Pathol. 2005, 166, 1419–1426. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Fujita, T. Function of RIG-I-like Receptors in Antiviral Innate Immunity. J. Biol. Chem. 2007, 282, 15315–15318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Eisenächer, K.; Krug, A. Regulation of RLR-mediated innate immune signaling—It is all about keeping the balance. Eur. J. Cell Biol. 2012, 91, 36–47. [Google Scholar] [CrossRef]
- Kumagai, Y.; Takeuchi, O.; Akira, S. Pathogen recognition by innate receptors. J. Infect. Chemother. 2008, 14, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.N.; Sen, G.C. Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol. Ther. 2004, 103, 245–259. [Google Scholar] [CrossRef]
- Cheng, G.; Zhong, J.; Chung, J.; Chisari, F.V. Double-stranded DNA and double-stranded RNA induce a common anti-viral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9035–9040. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein US11 Downmodulates the RLR Signaling Pathway via Direct Interaction with RIG-I and MDA. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- Szabo, A.; Rajnavolgyi, E. Collaboration of Toll-like and RIG-I-like receptors in human dendritic cells: tRIGgering an-tiviral innate immune responses. Am. J. Clin. Exp. Immunol. 2013, 2, 195–207. [Google Scholar]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Negishi, H.; Yanai, H.; Nakajima, A.; Koshiba, R.; Atarashi, K.; Matsuda, A.; Matsuki, K.; Miki, S.; Doi, T.; Aderem, A.; et al. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat. Immunol. 2012, 13, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Zhan, G.; Zheng, C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. 2016, 13, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Qin, Q.; Chen, W.; Qu, J. Expression of toll-like receptors in the healthy and herpes simplex virus-infected cornea. Cornea 2007, 26, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tokuyama, T.; Yamamoto, J.; Koide, M.; Yokota, N.; Namba, H. Potent Bystander Effect in Suicide Gene Therapy Using Neural Stem Cells Transduced with Herpes Simplex Virus Thymidine Kinase Gene. Oncology 2005, 69, 503–508. [Google Scholar] [CrossRef]
- Hayashi, K.; Lee, J.-B.; Maitani, Y.; Toyooka, N.; Nemoto, H.; Hayashi, T. The role of a HSV thymidine kinase stimulating substance, scopadulciol, in improving the efficacy of cancer gene therapy. J. Gene Med. 2006, 8, 1056–1067. [Google Scholar] [CrossRef]
- Unterholzner, L. The interferon response to intracellular DNA: Why so many receptors? Immunobiology 2013, 218, 1312–1321. [Google Scholar] [CrossRef]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- Conrady, C.D.; Jones, H.; Zheng, M.; Carr, D.J. A functional type I interferon pathway drives resistance to cornea herpes simplex virus type 1 infection by recruitment of leukocytes. J. Biomed. Res. 2011, 25, 111–119. [Google Scholar] [CrossRef]
- Zheng, C. Evasion of Cytosolic DNA-Stimulated Innate Immune Responses by Herpes Simplex Virus. J. Virol. 2018, 92, 00099–17. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; He, H.; Wang, H. Oncolytic herpes simplex virus and immunotherapy. BMC Immunol. 2018, 19, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes Simplex Virus 1 UL24 Abrogates the DNA Sensing Signal Pathway by Inhibiting NF-κB Activation. J. Virol. 2017, 91, e00025-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, S.; Wang, K.; Zheng, C. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-α-induced NF-κB activation by interacting with p65/RelA and p50/NF-κB1. Med. Microbiol. Immunol. 2013, 202, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-κB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes Simplex Virus 1 Serine/Threonine Kinase US3 Hyperphosphorylates IRF3 and Inhibits Beta Interferon Production. J. Virol. 2013, 87, 12814–12827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes Simplex Virus 1-Encoded Tegument Protein VP16 Abro-gates the Production of Beta Interferon (IFN) by Inhibiting NF-κB Activation and Blocking IFN Regulatory Factor 3 To Recruit Its Coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.D. A detailed mutational analysis of Vmw110, a trans-acting transcriptional activator encoded by herpes simplex virus type 1. EMBO J. 1987, 6, 2069–2076. [Google Scholar] [CrossRef]
- Everett, R.; O’Hare, P.; O’Rourke, D.; Barlow, P.; Orr, A. Point mutations in the herpes simplex virus type 1 Vmw110 RING finger helix affect activation of gene expression, viral growth, and interaction with PML-containing nuclear structures. J. Virol. 1995, 69, 7339–7344. [Google Scholar] [CrossRef] [Green Version]
- Lium, E.K.; Silverstein, S. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J. Virol. 1997, 71, 8602–8614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, R.D.; Rizzo, W.B.; Schulman, J.D.; Mukherjee, A.B. Construction and Characterization of Herpes Simplex Virus Type 1 Mutants with Defined Lesions in Immediate Early Gene. J. Gen. Virol. 1989, 70, 1185–1202. [Google Scholar] [CrossRef]
- Wilcox, C.L.; Smith, R.L.; Everett, R.D.; Mysofski, D. The herpes simplex virus type 1 immediate-early protein ICP0 is necessary for the efficient establishment of latent infection. J. Virol. 1997, 71, 6777–6785. [Google Scholar] [CrossRef] [Green Version]
- A Leib, D.; Coen, D.M.; Bogard, C.L.; A Hicks, K.; Yager, D.R.; Knipe, D.M.; Tyler, K.L.; A Schaffer, P. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 1989, 63, 759–768. [Google Scholar] [CrossRef] [Green Version]
- Halford, W.P.; Schaffer, P.A. ICP0 Is Required for Efficient Reactivation of Herpes Simplex Virus Type 1 from Neuronal Latency. J. Virol. 2001, 75, 3240–3249. [Google Scholar] [CrossRef] [Green Version]
- A Harris, R.; Everett, R.D.; Zhu, X.X.; Silverstein, S.; Preston, C.M. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 1989, 63, 3513–3515. [Google Scholar] [CrossRef] [Green Version]
- Preston, C.M.; Nicholl, M.J. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 1997, 71, 7807–7813. [Google Scholar] [CrossRef] [Green Version]
- Samaniego, L.A.; Neiderhiser, L.; DeLuca, N.A. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 1998, 72, 3307–3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Režuchová, I.; Kúdelová, M.; Ďurmanová, V.; Vojvodová, A.; Košovský, J.; Rajčáni, J. Transcription at Early Stages of Herpes Simplex Virus 1 Infection and during Reactivation. Intervirology 2003, 46, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during her-pes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Dejean, A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 pro-teins, correlating with nuclear body disruption. J. Virol. 1999, 73, 5137–5143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelbi-Alix, M.K.; de Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bod-ies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashchuk, V.; Everett, R.D. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 compo-nents ATRX and hDaxx. J. Virol. 2010, 84, 4026–4040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.; Zheng, Y.; Roizman, B. Interaction of Herpes Simplex Virus ICP0 with ND10 Bodies: A Sequential Process of Adhesion, Fusion, and Retention. J. Virol. 2013, 87, 10244–10254. [Google Scholar] [CrossRef] [Green Version]
- Andrés, M.E.; Burger, C.; Peral, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef] [Green Version]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and Its Corepressors Mediate Plasticity of Neuronal Gene Chromatin throughout Neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Lunyak, V.V.; Burgess, R.; Prefontaine, G.G.; Nelson, C.; Sze, S.H.; Chenoweth, J.; Schwartz, P.; Pevzner, P.A.; Glass, C.; Mandel, G.; et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science 2002, 298, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Paladino, P.; Collins, S.E.; Mossman, K.L. Cellular Localization of the Herpes Simplex Virus ICP0 Protein Dictates Its Ability to Block IRF3-Mediated Innate Immune Responses. PLoS ONE 2010, 5, e10428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes Simplex Virus 1 E3 Ubiquitin Ligase ICP0 Protein Inhibits Tumor Necrosis Factor Alpha-Induced NF-κB Activation by Interacting with p65/RelA and p50/NF-κB1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Keller, B.C.; Gale, M., Jr.; Diamond, M.S. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J. Virol. 2008, 82, 8465–8475. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nat. Cell Biol. 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain in-hibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.A.; Rosato, P.C.; Parker, Z.M.; Khalenkov, A.; Leib, D.A. Synergistic control of herpes simplex virus patho-genesis by IRF-3, and IRF-7 revealed through non-invasive bioluminescence imaging. Virology 2013, 444, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahnazaryan, D.; Khalil, R.; Wynne, C.; Jefferies, C.A.; Gabhann-Dromgoole, J.N.; Murphy, C.C. Herpes simplex virus 1 targets IRF7 via ICP0 to limit type I IFN induction. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Zhu, F.X.; King, S.M.; Smith, E.J.; Levy, D.E.; Yuan, Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits vi-rus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. 2002, 99, 5573–5578. [Google Scholar] [CrossRef] [Green Version]
- Hahn, A.M.; Huye, L.E.; Ning, S.; Webster-Cyriaque, J.; Pagano, J.S. Interferon Regulatory Factor 7 Is Negatively Regulated by the Epstein-Barr Virus Immediate-Early Gene, BZLF-1. J. Virol. 2005, 79, 10040–10052. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Kumar, A.; Ingle, H.; Kumar, H. The Interplay Between Viral-Derived miRNAs and Host Immunity During Infection. Front. Immunol. 2020, 10, 3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, U. Silencing Viral MicroRNA as a Novel Antiviral Therapy? J. Biomed. Biotechnol. 2009, 2009, 1–18. [Google Scholar] [CrossRef]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.J.J.; Whitley, R.J.; Gnann, J.W. Acyclovir: A Decade Later. N. Engl. J. Med. 1992, 327, 782–789. [Google Scholar] [CrossRef]
- Herpetic Eye Disease Study Group. Oral acyclovir for herpes simplex virus eye disease: Effect on prevention of epithelial keratitis and stromal keratitis. Arch. Ophthalmol. 2000, 118, 1030–1036. [Google Scholar] [CrossRef] [Green Version]
- Gnann, J.W.; Barton, N.H., Jr.; Whitley, R.J. Acyclovir: Mechanism of action, pharmacokinetics, safety and clinical applications. Pharmacotherapy 1983, 3, 275–283. [Google Scholar] [CrossRef]
- Lass, J.H.; Langston, R.H.; Foster, C.S.; Pavan-Langston, D. Antiviral medications and corneal wound healing. Antivir. Res. 1984, 4, 143–157. [Google Scholar] [CrossRef]
- Fleischer, R.; Johnson, M. Acyclovir Nephrotoxicity: A Case Report Highlighting the Importance of Prevention, Detection, and Treatment of Acyclovir-Induced Nephropathy. Case Rep. Med. 2010, 2010, 602783. [Google Scholar] [CrossRef] [Green Version]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes Simplex Virus Resistance to Acyclovir and Penciclovir after Two Decades of Antiviral Therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutner, K.R.; Friedman, D.J.; Forszpaniak, C.; Andersen, P.L.; Wood, M.J. Valaciclovir compared with acyclovir for im-proved therapy for herpes zoster in immunocompetent adults. Antimicrob. Agents Chemother. 1995, 39, 1546–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, J.; Hoh, H.B.; Easty, D.L.; Herbort, C.P.; Resnikoff, S.; Rigal, D.; Romdane, K. Ganciclovir Ophthalmic Gel (Virgan; 0.15%) in the Treatment of Herpes Simplex Keratitis. Cornea 1997, 16, 393–399. [Google Scholar] [CrossRef]
- Tyring, S.; Engst, R.; Corriveau, C.; Robillard, N.; Trottier, S.; Van Slycken, S.; A Crann, R.; A Locke, L.; Saltzman, R.; Palestine, A.G. Famciclovir for ophthalmic zoster: A randomised aciclovir controlled study. Br. J. Ophthalmol. 2001, 85, 576–581. [Google Scholar] [CrossRef] [Green Version]
- David, D.; Berkowitz, J. Ocular effects of topical and systemic corticosteroids. Lancet 1969, 294, 149–151. [Google Scholar] [CrossRef]
- Phulke, S.; Kaushik, S.; Kaur, S.; Pandav, S.S. Steroid-induced Glaucoma: An Avoidable Irreversible Blindness. J. Curr. Glaucoma Pract. 2017, 11, 67–72. [Google Scholar]
- Donshik, P.C.; Cavanaugh, H.D.; Boruchoff, S.A.; Dohlman, C.H. Posterior subcapsular cataracts induced by topical cor-ticosteroids following keratoplasty for keratoconus. Ann. Ophthalmol. 1981, 13, 29–32. [Google Scholar] [PubMed]
- Koujah, L.; Suryawanshi, R.K.; Shukla, D. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell. Mol. Life Sci. 2018, 76, 405–419. [Google Scholar] [CrossRef]
- Yadavalli, T.; Agelidis, A.; Jaishankar, D.; Mangano, K.; Thakkar, N.; Penmetcha, K.; Shukla, D. Targeting Herpes Simplex Virus-1 gD by a DNA Aptamer Can Be an Effective New Strategy to Curb Viral Infection. Mol. Ther. Nucleic Acids 2017, 9, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Gopinath, S.C.B.; Hayashi, K.; Kumar, P.K.R. Aptamer That Binds to the gD Protein of Herpes Simplex Virus 1 and Effi-ciently Inhibits Viral Entry. J. Virol. 2012, 86, 6732–6744. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.D.; Bunka, D.H.J.; Forzan, M.; Spear, P.G.; Stockley, P.; McGowan, I.; James, W. Generation of neutralizing aptamers against herpes simplex virus type 2: Potential components of multivalent microbicides. J. Gen. Virol. 2011, 92, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Li, M.; Wang, K.; Wang, S.; Lu, Q.; Yan, J.; Mossman, K.L.; Lin, R.; Zheng, C. The Herpes Simplex Virus 1-Encoded Envelope Glycoprotein B Activates NF-κB through the Toll-Like Receptor 2 and MyD88/TRAF6-Dependent Signaling Pathway. PLoS ONE 2013, 8, 54586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahanban-Esfahlan, R.; Seidi, K.; Majidinia, M.; Karimian, A.; Yousefi, B.; Nabavi, S.M.; Astani, A.; Berindan-Neagoe, I.; Gulei, D.; Fallarino, F.; et al. Toll-like receptors as novel therapeutic targets for herpes simplex virus infection. Rev. Med. Virol. 2019, 29, 2048. [Google Scholar] [CrossRef]
- Sarangi, P.P.; Kim, B.; Kurt-Jones, E.; Rouse, B.T. Innate recognition network driving herpes simplex virus-induced cor-neal immunopathology: Role of the toll pathway in early inflammatory events in stromal keratitis. J. Virol. 2007, 81, 11128–11138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabhann-Dromgoole, J.N.; De Chaumont, C.; Shahnazaryan, D.; Smith, S.; Malone, C.; Hassan, J.; De Gascun, C.F.; Jefferies, C.A.; Murphy, C.N.J. Systemic IL-1β production as a consequence of corneal HSV-1 infection-contribution to the development of herpes simplex keratitis. Int. J. Ophthalmol. 2019, 12, 1493–1497. [Google Scholar] [CrossRef]
- Staats, H.; Lausch, R.N. Cytokine expression in vivo during murine herpetic stromal keratitis. Effect of protective antibody therapy. J. Immunol. 1993, 151, 277–283. [Google Scholar]
- Lausch, R.N.; Chen, S.-H.; Tumpey, T.M.; Su, Y.-H.; Oakes, J.E. Early Cytokine Synthesis in the Excised Mouse Cornea. J. Interf. Cytokine Res. 1996, 16, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Dutt, J.; Arrunategui-Correa, V.; Baltatzis, S.; Foster, C.S. Cytokine mRNA in BALB/c mouse corneas infected with herpes simplex virus. Eye 1999, 13, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Lokensgard, S.H.J.R.; Hu, S.; Sheng, W.; Vanoijen, M.; Cox, D.; Cheeran, M.; Peterson, P.K. Robust expression of TNFa, IL-1ß, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 2001, 7, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, A.; Veiga-Parga, T.; Rajasagi, N.K.; Reddy, P.B.J.; Sehrawat, S.; Sharma, S.; Rouse, B.T. Role of IL-17 and Th17 Cells in Herpes Simplex Virus-Induced Corneal Immunopathology. J. Immunol. 2011, 187, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Miserocchi, E.; Modorati, G.; Galli, L.; Rama, P. Efficacy of Valacyclovir vs Acyclovir for the Prevention of Recurrent Herpes Simplex Virus Eye Disease: A Pilot Study. Am. J. Ophthalmol. 2007, 144, 547–551.e1. [Google Scholar] [CrossRef]
- Biswas, P.S.; Banerjee, K.; Kim, B.; Rouse, B.T. Mice Transgenic for IL-1 Receptor Antagonist Protein Are Resistant to Herpetic Stromal Keratitis: Possible Role for IL-1 in Herpetic Stromal Keratitis Pathogenesis. J. Immunol. 2004, 172, 3736–3744. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; Gitton, X.; Widmer, A.; Patel, N.; Hawkins, P.N. Use of Canakinumab in the Cryopyrin-Associated Periodic Syndrome. N. Engl. J. Med. 2009, 360, 2416–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, N.; De Meulemeester, M.; Pikhlak, A.; Yücel, A.E.; Richard, D.; Murphy, V.; Arulmani, U.; Sallstig, P.; So, A. Canakinumab relieves symptoms of acute flares and improves health-related quality of life in patients with difficult-to-treat Gouty Arthritis by suppressing inflammation: Results of a randomized, dose-ranging study. Arthritis Res. 2011, 13, R53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, A.; Arndt, M.A.E.; Grosse-Hovest, L.; Weichert, W.; Giebel, B.; Dittmer, U.; Hengel, H.; Jäger, D.; Schneweis, K.E.; Eis-Hübinger, A.M.; et al. Overcoming drug-resistant herpes simplex virus (HSV) infection by a humanized antibody. Proc. Natl. Acad. Sci. USA 2013, 110, 6760–6765. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Wang, L.; Xu, H.; Wang, Z.; Zhang, T.; Wang, M.; Ning, Y.; Deng, F.; Hu, Z.; Wang, H.; et al. A novel glycoprotein D-specific monoclonal antibody neutralizes herpes simplex virus. Antivir. Res. 2017, 147, 131–141. [Google Scholar] [CrossRef]
- Zhou, C.; Zhao, L.; Wang, K.; Qi, Q.; Wang, M.; Yang, L.; Sun, P.; Mu, H. MicroRNA-146a inhibits NF-κB activation and pro-inflammatory cytokine production by regulating IRAK1 expression in THP-1 cells. Exp. Ther. Med. 2019, 18, 3078. [Google Scholar] [CrossRef] [Green Version]
- Ike, A.C.; Onu, C.J.; Ononugbo, C.M.; Reward, E.E.; Muo, S.O. Immune Response to Herpes Simplex Virus Infection and Vaccine Development. Vaccines 2020, 8, 302. [Google Scholar] [CrossRef]
- Jaggi, U.; Wang, S.; Tormanen, K.; Matundan, H.; Ljubimov, A.V.; Ghiasi, H. Role of Herpes Simplex Virus Type 1 (HSV-1) Glycoprotein K (gK) Pathogenic CD8+ T Cells in Exacerbation of Eye Disease. Front. Immunol. 2018, 9, 2895. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.N.; Reynolds, G.M.; Waldron, E.E.; Ward, E.; Giannopoulos, K.; Murray, P.G. Monoclonal antibodies. Mol. Pathol. MP 2000, 53, 111–117. [Google Scholar] [CrossRef]
- Brdovčak, M.C.; Zubković, A.; Jurak, I. Herpes Simplex Virus 1 Deregulation of Host MicroRNAs. Non-Coding RNA 2018, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes sim-plex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Flores, O.; Umbach, J.L.; Pesola, J.M.; Bentley, P.; Rosato, P.C.; Leib, D.A.; Cullen, B.R.; Coen, D.M. A Neuron-Specific Host MicroRNA Targets Herpes Simplex Virus-1 ICP0 Expression and Promotes Latency. Cell Host Microbe 2014, 15, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Griffiths, A.; Li, G.; Silva, L.M.; Kramer, M.F.; Gaasterland, T.; Wang, X.-J.; Coen, D.M. Prediction and Identification of Herpes Simplex Virus 1-Encoded MicroRNAs. J. Virol. 2006, 80, 5499–5508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, J.; Sun, H.; Fan, H.; Wang, C.; Li, Y.; Liu, M.; Tang, H. MiR-23a facilitates the replication of HSV-1 through the suppression of interferon regulatory factor. PLoS ONE 2014, 9, e114021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar] [PubMed]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; Van Der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greenan, E.; Gallagher, S.; Khalil, R.; Murphy, C.C.; Ní Gabhann-Dromgoole, J. Advancing Our Understanding of Corneal Herpes Simplex Virus-1 Immune Evasion Mechanisms and Future Therapeutics. Viruses 2021, 13, 1856. https://doi.org/10.3390/v13091856
Greenan E, Gallagher S, Khalil R, Murphy CC, Ní Gabhann-Dromgoole J. Advancing Our Understanding of Corneal Herpes Simplex Virus-1 Immune Evasion Mechanisms and Future Therapeutics. Viruses. 2021; 13(9):1856. https://doi.org/10.3390/v13091856
Chicago/Turabian StyleGreenan, Emily, Sophie Gallagher, Rana Khalil, Conor C. Murphy, and Joan Ní Gabhann-Dromgoole. 2021. "Advancing Our Understanding of Corneal Herpes Simplex Virus-1 Immune Evasion Mechanisms and Future Therapeutics" Viruses 13, no. 9: 1856. https://doi.org/10.3390/v13091856