
De novo Assembly, Annotation, and Analysis of Transcriptome Data of the Ladakh Ground Skink Provide Genetic Information on High-Altitude Adaptation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Ethics Statement
2.2. RNA Isolation, Library Preparation, and Sequencing
2.3. Assembly and Assessment of Transcriptome Quality and Completeness
2.4. Functional Annotation
2.5. Positively Selected Genes Related to Mechanisms of High-Altitude Adaptation
2.6. Data Availability
3. Results
3.1. Sequencing and Transcriptome Assembly
3.2. Assembly Completeness
3.3. Functional Annotation
3.4. Positive Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mittermeier, R.A.; Robles-Gil, P.; Homan, M.; Pilgrim, J.; Brooks, T.; Mittermeier, C.G.; Lamoreux, J.; da Fonseca, G.A.B. Hotspots Revisited; CEMEX: Mexico City, Mexico, 2004; p. 392. [Google Scholar]
- Hofmann, S.; Stoeck, M.; Zheng, Y.; Ficetola, F.G.; Li, J.T.; Scheidt, U.; Schmidt, J. Molecular Phylogenies Indicate a Paleo-Tibetan Origin of Himalayan Lazy Toads (Scutiger). Sci. Rep. 2017, 7, 3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Qi, Y.; Fu, J. Genetic signals of high-altitude adaptation in amphibians: A comparative transcriptome analysis. BMC Genet. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-D.; Zhang, B.-L.; Zhou, W.-W.; Li, Y.-X.; Jin, J.-Q.; Shao, Y.; Yang, H.-C.; Liu, Y.-H.; Yan, F.; Chen, H.-M.; et al. Selection and Environmental Adaptation Along a Path to Speciation in the Tibetan Frog Nanorana parkeri. Proc. Natl. Acad. Sci. USA 2018, 115, E5056–E5065. [Google Scholar] [CrossRef] [Green Version]
- Che, J.; Zhou, W.-W.; Hu, J.-S.; Yan, F.; Papenfuss, T.J.; Wake, D.B.; Zhang, Y.-P. Spiny frogs (Paini) illuminate the history of the Himalayan region and Southeast Asia. Proc. Natl. Acad. Sci. USA 2010, 107, 13765–13770. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Jin, H.; Fu, J. Molecular convergent and parallel evolution among four high-elevation anuran species from the Tibetan region. BMC Genom. 2020, 21, 839. [Google Scholar] [CrossRef]
- Sun, Y.B.; Fu, T.T.; Jin, J.Q.; Murphy, R.W.; Hillis, D.M.; Zhang, Y.P.; Che, J. Species Groups Distributed across Elevational Gradients Reveal Convergent and Continuous Genetic Adaptation to High Elevations. Proc. Natl. Acad. Sci. USA 2018, 115, E10634–E10641. [Google Scholar] [CrossRef] [Green Version]
- Dorge, T.; Hofmann, S.; Wangdwei, M.; Duoje, L.; Solhøy, T.; Miehe, G. The ecological specialist, Thermophis baileyi (Wall, 1907)—New records, distribution, and biogeographic conclusions. Herpetol. Bull. 2007, 101, 8–12. [Google Scholar]
- Jin, Y.T.; Brandt, D.Y.C.; Li, J.; Wo, Y.; Tong, H.; Shchur, V. Elevation as a selective force on mitochondrial respiratory chain complexes of the Phrynocephalus lizards in the Tibetan plateau. Curr. Zool. 2020, 67, 191–199. [Google Scholar] [CrossRef]
- Baig, K.J.; Wagner, P.; Ananjeva, N.B.; Böhme, W. A morphology-based taxonomic revision of Laudakia Gray, 1845 (Squamata: Agamidae). Vertebr. Zool. 2012, 62, 213–260. [Google Scholar]
- Gangloff, E.J.; Telemeco, R.S. High Temperature, Oxygen, and Performance: Insights from Reptiles and Amphibians. Integr. Comp. Biol. 2018, 58, 9–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, D.C. Temperature and hypoxia in ectothermic tetrapods. J. Thermal. Biol. 2007, 32, 125–133. [Google Scholar] [CrossRef]
- Yang, W.J.; Qi, Y.; Bi, K.; Fu, J. Toward Understanding the Genetic Basis of Adaption to High-Elevation Life in Poikilothermic Species: A Comparative Transcriptomic Analysis of Two Ranid Frogs, Rana chensinensis and R. kukunoris. BMC Genom. 2012, 13, 588. [Google Scholar] [CrossRef] [Green Version]
- Li, J.T.; Gao, Y.-D.; Xie, L.; Deng, C.; Shi, P.; Guan, M.-L.; Huang, S.; Ren, J.-L.; Wu, D.-D.; Ding, L.; et al. Comparative genomic investigation of high-elevation adaptation in ectothermic snakes. Proc. Natl. Acad. Sci. USA 2018, 115, 8406–8411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Qi, Y.; Fu, J. Exploring the genetic basis of adaptation to high elevations in reptiles: A comparative transcriptome analysis of two toad-headed agamas (genus Phrynocephalus). PLoS ONE 2014, 9, e112218. [Google Scholar] [CrossRef]
- Hofmann, S.; Kuhl, H.; Baniya, C.B.; Stock, M. Multi-Tissue Transcriptomes Yield Information on High-Altitude Adaptation and Sex-Determination in Scutiger cf. sikimmensis. Genes 2019, 10, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosbrugger, V.; Favre, L.; Müllner-Riehl, A.; Päckert, M.; Mulch, A. Cenozoic Evolution of Geo-Biodiversity in the Tibeto-Himalayan Region. In Mountains, Climate and Biodiversity; Hoorn, C., Perrigo, A., Antonelli, A., Eds.; Wiley-Blackwell: Oxford, UK, 2018; pp. 429–449. [Google Scholar]
- Pacifici, M.; Foden, W.B.; Visconti, P.; Watson, J.E.M.; Butchart, S.H.M.; Kovacs, K.M.; Scheffers, B.R.; Hole, D.G.; Martin, T.G.; Akçakaya, H.R.; et al. Assessing species vulnerability to climate change. Nat. Clim. Chang. 2015, 5, 215–225. [Google Scholar] [CrossRef]
- Xu, J.; Badola, R.; Chettri, N.; Chaudhary, R.P.; Zomer, R.; Pokhrel, B.; Hussain, S.A.; Pradhan, S.; Pradhan, R. Sustaining Biodiversity and Ecosystem Services in the Hindu Kush Himalaya. In The Hindu Kush Himalaya Assessment; Wester, P., Mishra, A., Mukherji, A., Shrestha, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 127–165. [Google Scholar]
- Kouyoumdjian, L.; Gangloff, E.; Souchet, J.; Cordero, G.A.; Dupoué, A.; Aubret, F. Transplanting gravid lizards to high elevation alters maternal and embryonic oxygen physiology, but not reproductive success or hatchling phenotype. J. Exp. Biol. 2019, 222, jeb206839. [Google Scholar] [CrossRef] [Green Version]
- Banasiak, K.J.; Xia, Y.; Haddad, G.G. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol. 2000, 62, 215–249. [Google Scholar] [CrossRef]
- Bickler, P.E.; Donohoe, P.H. Adaptive responses of vertebrate neurons to hypoxia. J. Exp. Biol. 2002, 205, 3579–3586. [Google Scholar] [CrossRef] [PubMed]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef] [Green Version]
- Linkem, C.W.; Diesmos, A.C.; Brown, R.M. Molecular systematics of the Philippine forest skinks (Squamata: Scincidae: Sphenomorphus): Testing morphological hypotheses of interspecific relationships. Zool. J. Linn. Soc. Lond. 2011, 163, 1217–1243. [Google Scholar] [CrossRef] [Green Version]
- Shea, G.M.; Greer, A.E. From Sphenomorphus to Lipinia: Generic reassignment of two poorly known New Guinea skinks. J. Herpetol. 2002, 36, 148–156. [Google Scholar] [CrossRef]
- Borkin, L.J.; Litvinchuk, S.N.; Melnikov, D.A.; Skorinov, D.V. Altitudinal distribution of skinks of the genus Asymblepharus in the Western Himalaya, India (Reptilia: Sauria: Scincidae). In Biodiversität und Naturausstattung im Himalaya VI; Hartmann, R., Weipert, J., Barclay, M., Eds.; Verein der Freunde & Förderer des Naturkundemuseums Erfurt e.V.: Erfurt, Germany, 2018; pp. 163–167. [Google Scholar]
- Hartmann, M.; Weipert, J.; Weigel, A. Die zoologischen Nepal-Expeditionen des Naturkundemuseums Erfurt. Veröffentlichungen des Nat. Erf. 1998, 17, 15–30. [Google Scholar]
- Hartmann, M.; Weipert, J. Biodiversität und Naturausstattung im Himalaya V; Verein der Freunde und Förderer des Naturkundemuseums Erfurt e.V.: Erfurt, Germany, 2015; p. 580. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 July 2021).
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Mac Manes, M.D. The Oyster River Protocol: A multi assembler and Kmer approach for de novo transcriptome assembly. PEERJ 2018, 6, e5428. [Google Scholar] [CrossRef]
- Hoelzer, M.; Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. Gigascience 2019, 8, 1–16. [Google Scholar]
- Lu, B.; Zeng, Z.; Shi, T. Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq. Sci. China Life Sci. 2013, 56, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 2015, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.; Bowden, J.; Couger, M.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef]
- Kannan, S.; Hui, J.; Mazooji, K.; Pachter, L.; Tse, D. Shannon: An Information-Optimal de novo RNA-Seq Assembler. BioRxiv 2016. [Google Scholar] [CrossRef]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction. Methods in Molecular Biology; Kollmar, M., Ed.; Humana: Louisville, KY, USA, 2019; Volume 1962. [Google Scholar]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Suvorov, V.; Prjibelski, A.D. rnaQUAST: A quality assessment tool for de novo transcriptome assemblies. Bioinformatics 2016, 32, 2210–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Aligment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Litvinchuk, S.N.; Borkin, L.J.; Hofmann, S. Rediscovery of the high-altitude Lazy Toad, Scutiger occidentalis Dubois, 1978, in India. Russ. J. Herpetol. 2019, 26, 17–22. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5—A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielman, S.J.; Weaver, S.; Shank, S.D.; Magalis, B.R.; Li, M.; Kosakovsky Pond, S.L. Evolution of Viral Genomes: Interplay Between Selection, Recombination, and Other Forces. In Evolutionary Genomics. Methods in Molecular Biology; Anisimova, M., Ed.; Humana: Louisville, KY, USA, 2019; Volume 1910. [Google Scholar]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained Bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Pond, S.L.K. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky, P.S.L.; Murrell, B.; Fourment, M.; Frost, S.D.; Delport, W.; Scheffler, K. 2011 A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 2011, 28, 3033–3043. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Santillan, D.D.; Machain-Williams, C.; Hernandez-Montes, G.; Ortega, J. De Novo Transcriptome Assembly and Functional Annotation in Five Species of Bats. Sci. Rep. 2019, 9, 6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theissinger, K.; Falckenhayn, C.; Blande, D.; Toljamoc, A.; Gutekunstb, J.; Makkonenc, J.; Jussilac, J.; Lykob, F.; Schrimpfa, A.; Schulza, R.; et al. De novo assembly and annotation of the freshwater crayfish Astacus astacus transcriptome. Mar. Genom. 2016, 28, 7–10. [Google Scholar] [CrossRef]
- Waits, D.S.; Simpson, D.Y.; Sparkman, A.M.; Bronikowski, A.M.; Schwartz, T.S. The utility of reptile blood transcriptomes in molecular ecology. Mol. Ecol. Resour. 2020, 20, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, M.; Yurchenko, A.A.; Augley, J.J.; Adams, C.E.; Herzyk, P.; Elmer, K.R. De novo transcriptome assembly, annotation and comparison of four ecological and evolutionary model salmonid fish species. BMC Genom. 2018, 19, 32. [Google Scholar]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gates, K.; Sandoval-Castillo, J.; Bernatchez, L.; Beheregaray, L.B. De novo transcriptome assembly and annotation for the desert rainbowfish (Melanotaenia splendida tatei) with comparison with candidate genes for future climates. Mar. Genom. 2017, 35, 63–68. [Google Scholar] [CrossRef]
- Murphy, W.J.; Pringle, T.H.; Crider, T.A.; Springer, M.S.; Miller, W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007, 17, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquesi, G.I.M.; Adams, R.H.; Card, D.C.; Schield, D.R.; Corbin, A.B.; Perry, B.W.; Reyes-Velasco, J.; Ruggiero, R.P.; Vandewege, M.W.; Shortt, J.A.; et al. Squamate reptiles challenge paradigms of genomic repeat element evolution set by birds and mammals. Nat. Commun. 2018, 9, 2774. [Google Scholar] [CrossRef] [Green Version]
- Childebayeva, A.; Goodrich, J.M.; Léon-Velarde, F.; Rivera-Chira, M.; Kiyamu, M.; Brutsaert, T.; Dolinoy, D.; Bigham, A. Genome-wide DNA methylation changes associated with high-altitude acclimatization during an Everest base camp trek. Front. Physiol. 2021, 12, 660906. [Google Scholar] [CrossRef]
- Solari, K.A.; Ramakrishnan, U.; Hadly, E.A. Gene expression is implicated in the ability of pikas to occupy Himalayan elevational gradient. PLoS ONE 2018, 13, e0207936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, P.; Sharma, A.; Sodhi, M.; Thakur, K.; Kataria, R.; Niranjan, S.K.; Bharti, V.; Kumar, P.; Giri, A.; Kalia, S.; et al. Transcriptome analysis of circulating PBMCs to understand mechanism of high-altitude adaptation in native cattle of Ladakh region. Sci. Rep. 2018, 8, 7681. [Google Scholar] [CrossRef] [PubMed] [Green Version]



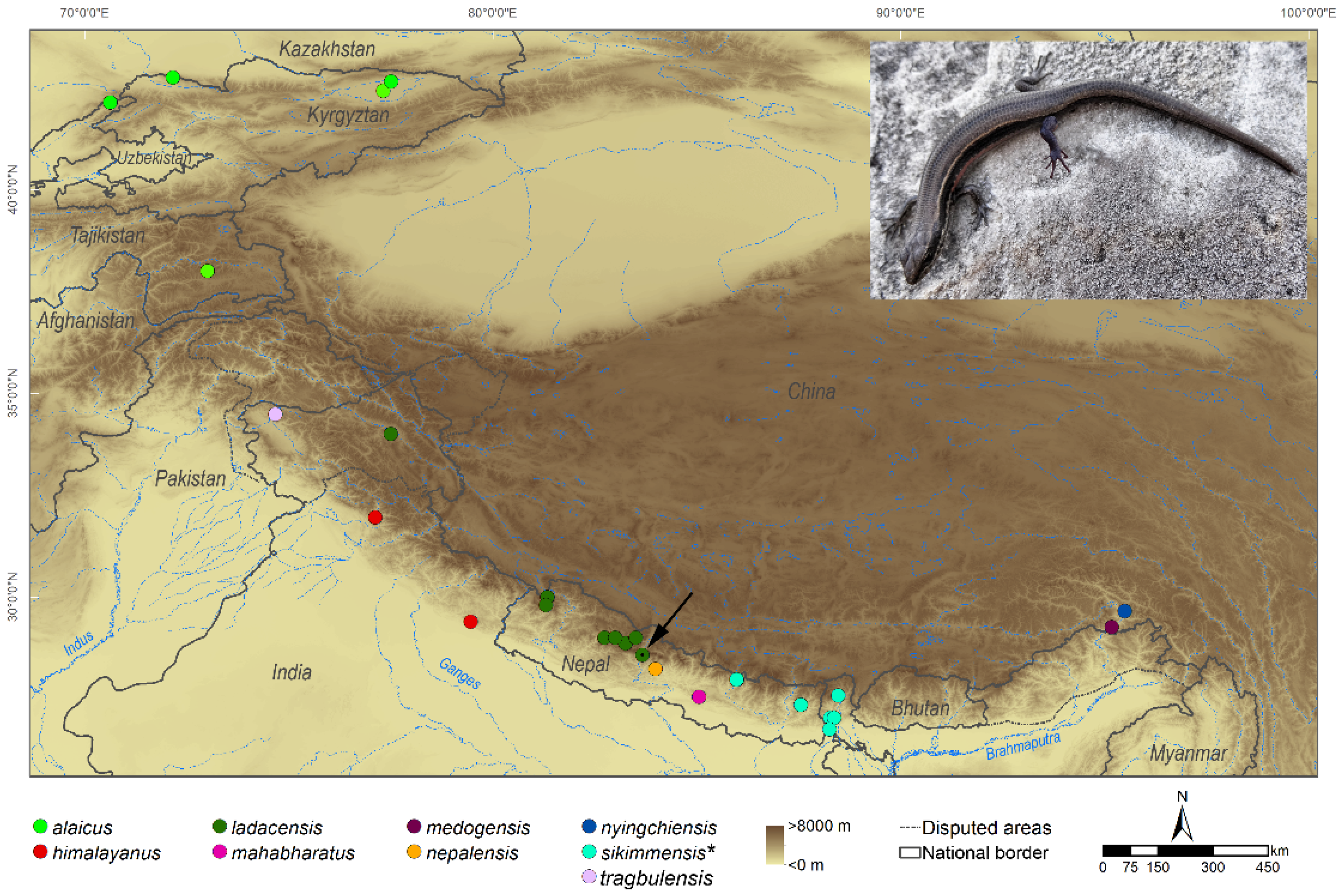{kind=link}
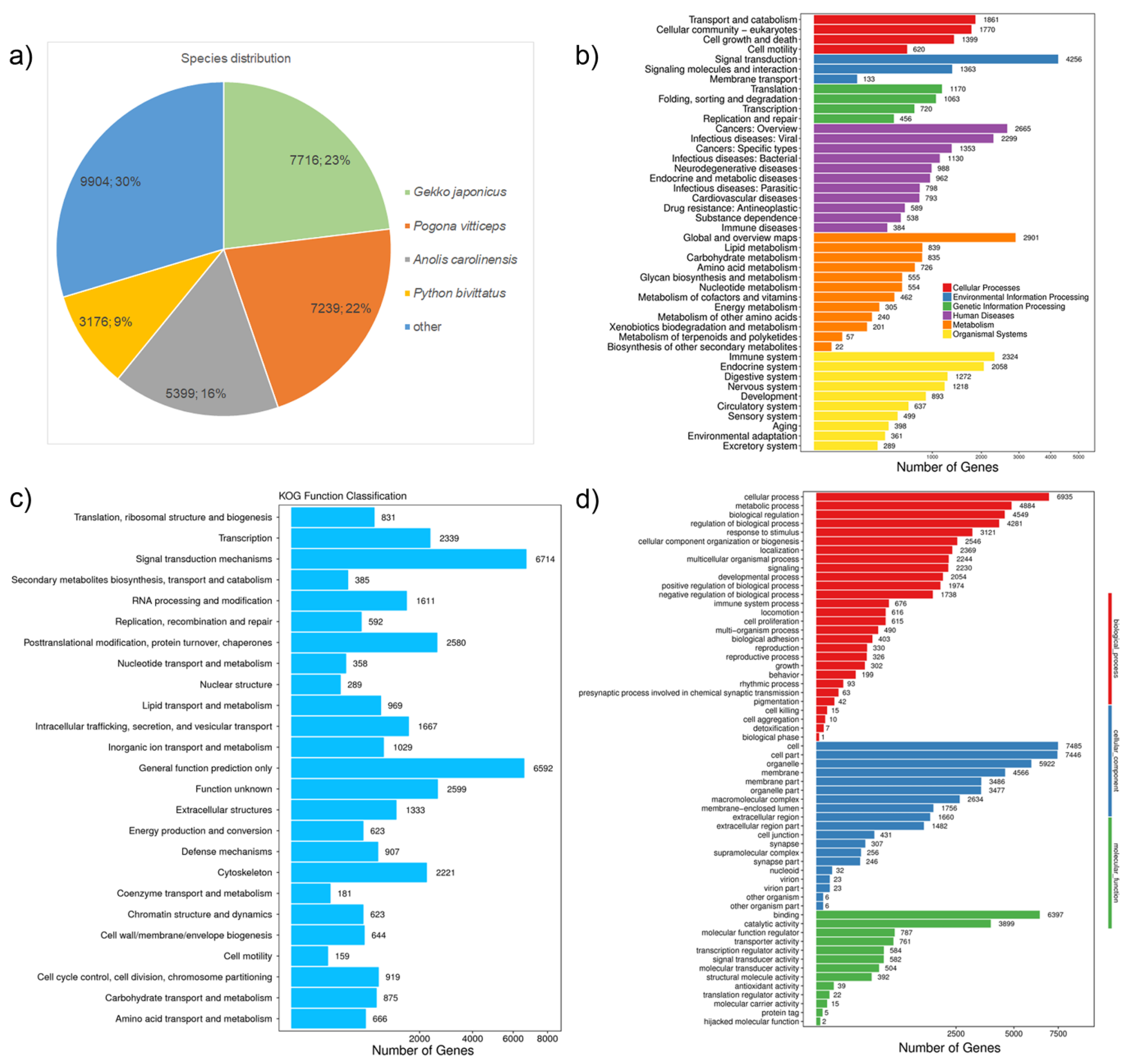{kind=link}
| Brain Tissue | Embryonic Disc Tissue | Pooled Tissue | |
|---|---|---|---|
| Number of paired-end raw reads | 74,780,628 | 73,557,120 | 64,195,486 |
| Number of cleaned reads | 73,139,294 | 71,887,892 | 61,397,610 |
| Number of base pairs in final assembly | 102,605,079 | 98,917,807 | 47,613,446 |
| Number of transcripts in final assembly | 151,718 | 105,133 | 66,696 |
| Average transcript length (bp) | 676 | 940 | 712 |
| Minimum transcript length (bp) | 131 | 131 | 131 |
| Maximum transcript length (bp) | 17,543 | 18,168 | 15,866 |
| N50 | 1215 | 2052 | 1194 |
| N90 | 257 | 311 | 278 |
| GC% content of the final ORP assembly | 0.48 | 0.48 | 0.48 |
| BUSCO Statistics | Brain | Embryonic Disc | Pooled Tissues | ||||||
|---|---|---|---|---|---|---|---|---|---|
| EU | VB | TP | EU | VB | TP | EU | VB | TP | |
| Complete | 220/255 (86.3%) | 2052/3354 (61.1%) | 2696/5310 (50.8%) | 250/255 (98.0%) | 2743/3354 (81.8%) | 3778/5310 (71.1%) | 178/255 (69.9%) | 1405/3354 (41.9%) | 1750/5310 (33.0%) |
| Single-copy | 189/255 (74.1%) | 1759/3354 (52.4%) | 2305/5310 (43.4%) | 185/255 (72.5%) | 1871/3354 (55.8%) | 2555/5310 (48.1%) | 148/255 (58.0%) | 1150/3354 (34.3%) | 1421/5310 (26.8%) |
| Duplicated | 31/255 (12.2%) | 293/3354 (8.7%) | 391/5310 (7.4%) | 65/255 (25.5%) | 872/3354 (26.0%) | 1223/5310 (23.0%) | 30/255 (11.8%) | 255/3354 (7.6%) | 329/5310 (6.2%) |
| Fragmented | 23/255 (9.0%) | 589/3354 (17.6%) | 709/5310 (13.4%) | 3/255 (1.2%) | 221/3354 (6.6%) | 323/5310 (6.1%) | 47/255 (18.4%) | 654/3354 (19.5%) | 645/5310 (12.1%) |
| Missing | 12/255 (4.7%) | 713/3354 (21.3%) | 1905/5310 (35.8%) | 2/255 (0.8%) | 390/3354 (11.6%) | 1209/5310 (22.8%) | 30/255 (11.8%) | 1295/3354 (38.6%) | 2915/5310 (54.9%) |
| Total | NR | NT | SwissProt | KEGG | KOG | InterPro | GO | Intersection | Overall | |
|---|---|---|---|---|---|---|---|---|---|---|
| N | 76,968 | 33,444 | 34,114 | 30,994 | 28,961 | 26,608 | 27,013 | 11,010 | 7292 | 39,975 |
| % | 100 | 43.45 | 44.32 | 40.27 | 37.63 | 34.57 | 35.10 | 14.30 | 9.47 | 51.94 |
| GeneID | Gene | p-Value [15] | Gene Description | Transcript | B | FB | aB |
|---|---|---|---|---|---|---|---|
| 000773 | IL1RAP | 3.58 × 10−2 | Interleukin 1 receptor accessory protein | 000813 | <0.00 × 10−5 | 2 | A. |
| 000907 | MICU1 | 1.79 × 10−2 | Mitochondrial calcium uptake 1 | 000909 | 1.30 × 10−3 | 1 | yes |
| 001142 | TARBP1 | 1.39 × 10−6 | TAR RNA binding protein 1 | 001104 | <0.00 × 10−5 | 1 | yes |
| 002254 | MIA3 | 1.83 × 10−2 | Melanoma inhibitory activity family member 3 | 002276 | 1.93 × 10−2 | 1 | yes |
| 002549 | RPS2 | 1.99 × 10−2 | Ribosomal protein S2 | 002541 | 5.00 × 10−4 | 2 | yes |
| 002995 | RNF10 | 4.38 × 10−2 | Ring finger protein 10 | 003046 | 2.63 × 10−2 | 5 | yes |
| 003987 | NUP107 | 1.54 × 10−4 | Nucleoporin 107kDa | 004158 | <0.00 × 10−5 | 1 | yes |
| 006133 | GRK6 | 1.07 × 10−2 | G protein-coupled receptor kinase 6 | 006252 | 1.16 × 10−2 | 2 | A. |
| 007074 | SMC4 | 1.56 × 10−3 | Structural maintenance of chromosomes 4 | 007191 | 1.00 × 10−4 | 1 | yes |
| 015860 | SH3RF1 | 4.55 × 10−3 | SH3 domain containing ring finger 1 | 015968 | 8.90 × 10−3 | 1 | yes |
| Lowland | Gene Description | Transcript | B | FB | aB | |
|---|---|---|---|---|---|---|
| GeneID | Gene | |||||
| 000146 | PCSK9 | Proprotein convertase subtilisin/kexin type 9 | 000163 | 4.40 × 10−3 | 1 | 2 |
| 000201 | NPC1 | NPC intracellular cholesterol transporter 1 | 000261 | 5.10 × 10−3 | 2 | 1 |
| 000264 | LAMP1 | Lysosomal associated membrane protein 1 | 000247 | 2.90 × 10−3 | 8 | 2 |
| 000531 | SEPT12 | Septin 12 | 000602 | 1.29 × 10−2 | 1 | 1 |
| 000768 | SYK | Spleen associated tyrosine kinase | 000802 | <0.00 × 10−5 | 1 | 3 |
| 000798 | WBP4 | WW domain binding protein 4 | 000804 | 2.00 × 10−2 | 3 | 1 |
| 000955 | QSOX1 | Quiescin sulfhydryl oxidase 1 | 000962 | 9.20 × 10−3 | 3 | 1 |
| 001070 | CADM1 | Cell adhesion molecule 1 | 001188 | 1.00 × 10−4 | 1 | 1 |
| 002270 | KANK1 | KN motif and ankyrin repeat domains 1 | 002294 | 8.00 × 10−4 | 2 | 2 |
| 003015 | VLDLR | Very low-density lipoprotein receptor | 003096 | 1.66 × 10−2 | 3 | 1 |
| 003460 | ANKRD12 | Ankyrin repeat domain 12 | 003484 | 1.80 × 10−3 | 1 | 1 |
| 003908 | MDM1 | Mdm1 nuclear protein | 003923 | 2.30 × 10−3 | 2 | 2 |
| 005884 | CCDC66 | Coiled-coil domain containing 66 | 025744 | <0.00 × 10−5 | 1 | 1 |
| 006279 | GLYR1 | Glyoxylate reductase 1 homolog | 006325 | <0.00 × 10−5 | 1 | 3 |
| 007694 | ACOX1 | Acyl-CoA oxidase 1 | 007823 | <0.00 × 10−5 | 8 | 1 |
| 008005 | PHACTR2 | Phosphatase and actin regulator 2 | 008047 | 4.30 × 10−3 | 5 | 1 |
| 008420 | VTA1 | Vesicle trafficking 1 | 008463 | 3.36 × 10−2 | 1 | 1 |
| 009938 | 010074 | <0.00 × 10−5 | 6 | 3 | ||
| 013301 | COL1A2 | Collagen type I α 2 chain | 013614 | 1.10 × 10−3 | 17 | 3 |
| 013917 | MSH2 | MutS homolog 2 | 014076 | 1.11 × 10−2 | 1 | 1 |
| 013984 | LRRCC1 | Leucine-rich repeat and coiled-coil centrosomal protein 1 | 014100 | 2.50 × 10−2 | 1 | 2 |
| 016683 | TENT2 | Terminal nucleotidyltransferase 2 | 016777 | <0.00 × 10−5 | 2 | 2 |
| 017936 | ATP6V0A1 | ATPase H+ transporting V0 subunit a1 | 018008 | 3.50 × 10−3 | 1 | 1 |
| Up to 2000 m | ||||||
| Gene ID | Gene | Gene description | Transcript | B | FB | aB |
| 000608 | FBXL3 | F-box and leucine-rich repeat protein 3 | 000548 | 3.47 × 10−2 | 2 | 1 |
| 000768 | SYK | Spleen associated tyrosine kinas | 000802 | <0.00 × 100 | 1 | 3 |
| 000837 | KATNB1 | Katanin p80 (WD repeat containing) subunit B 1 | 000896 | 4.96 × 10−2 | 1 | 1 |
| 000955 | QSOX1 | Quiescin sulfhydryl oxidase 1 | 000962 | 8.90 × 10−3 | 3 | 1 |
| 002090 | KIAA0232 | KIAA0232 | 002069 | <0.00 × 100 | 5 | 2 |
| 002091 | RANBP2 | RAN binding protein 2 | 002100 | 7.00 × 10−3 | 2 | 1 |
| 002556 | SLC25A1 | Solute carrier family 25 member 1 | 002543 | 5.00 × 10−3 | 2 | 1 |
| 002948 | RSPH1 | Radial spoke head component 1 | 002963 | 1.59 × 10−2 | 2 | 1 |
| 003975 | TOGARAM1 | TOG array regulator of axonemal microtubules 1 | 004004 | <0.00 × 10−5 | 1 | 3 |
| 003987 | NUP107 | Nucleoporin 107 | 004158 | <0.00 × 10−5 | 1 | 2 |
| 004938 | RARS1 | Arginyl-tRNA synthetase 1 | 005003 | <0.00 × 10−5 | 3 | 2 |
| 005569 | CCT4 | Chaperonin containing TCP1 subunit 4 | 005683 | 2.00 × 10−4 | 2 | 1 |
| 006189 | PARP1 | Poly(ADP-ribose) polymerase 1 | 006356 | 4.11 × 10−2 | 4 | 1 |
| 006926 | CTNND1 | Catenin delta 1 | 007006 | 3.00 × 10−4 | 1 | 1 |
| 007100 | ZNF277 | Zinc finger protein 277 | 007173 | 1.17 × 10−2 | 3 | 3 |
| 007489 | PRDX4 | Peroxiredoxin 4 | 007498 | 2.20 × 10−3 | 1 | 1 |
| 007985 | POLR3A | RNA polymerase III subunit A | 008077 | 1.37 × 10−2 | 1 | 1 |
| 008005 | PHACTR2 | Phosphatase and actin regulator 2 | 008047 | 3.90 × 10−3 | 5 | 1 |
| 008475 | SNTA1 | Syntrophin α 1 | 008544 | 5.20 × 10−3 | 5 | 1 |
| 008991 | KEAP1 | Kelch-like ECH-associated protein 1 | 009010 | 2.00 × 10−4 | 3 | 1 |
| 009213 | SWAP70 | Switching B cell complex subunit SWAP70 | 009242 | 8.70 × 10−3 | 1 | 1 |
| 013059 | JPH1 | Junctophilin 1 | 013093 | 1.09 × 10−2 | 4 | 1 |
| 013301 | COL1A2 | Collagen type I α 2 chain | 013614 | 1.10 × 10−3 | 17 | 3 |
| 013326 | ADGRF5 | Adhesion G protein-coupled receptor F5 | 013405 | 4.00 × 10−4 | 1 | 1 |
| 015062 | COL3A1 | Collagen type III α 1 chain | 015539 | 3.00 × 10−2 | 11 | 4 |
| 015374 | RANBP17 | RAN binding protein 17 | 015630 | 7.90 × 10−3 | 3 | 2 |
| 015422 | BAIAP2 | BAR/IMD-domain-containing adaptor protein 2 | 015540 | 4.96 × 10−2 | 2 | 2 |
| 016662 | PGS1 | Phosphatidylglycerophosphate synthase 1 | 016740 | <0.00 × 10−5 | 1 | 1 |
| 017208 | FLOT1 | Flotillin 1 | 017291 | <0.00 × 10−5 | 4 | 2 |
| 017228 | SLC4A1 | Solute carrier family 4 member 1 (Diego blood group) | 017345 | 4.36 × 10−2 | 2 | 1 |
| 017316 | PTBP3 | Polypyrimidine tract binding protein 3 | 017407 | 6.00 × 10−4 | 2 | 3 |
| 018003 | FGFR1 | Fibroblast growth factor receptor 1 | 018080 | 6.00 × 10−4 | 2 | 1 |
| 2000–3500 m | ||||||
| Gene ID | Gene | Gene description | Transcript | B | FB | aB |
| 000306 | ZNF622 | Zinc finger protein 622 | 000291 | 8.50 × 10−3 | 5 | 1 |
| 000907 | MICU1 | Mitochondrial calcium uptake 1 | 000909 | 1.30 × 10−3 | 1 | 1 |
| 001396 | ABCC3 | ATP-binding cassette subfamily C member 3 | 001480 | <0.00 × 10−5 | 1 | 1 |
| 002254 | MIA3 | Melanoma inhibitory activity family member 3 | 002276 | 2.22 × 10−2 | 1 | 2 |
| 002779 | CDH1 | Cadherin 1 | 003031 | 1.50 × 10−3 | 1 | 1 |
| 004137 | STARD13 | StAR-related lipid transfer domain containing 13 | 004235 | 2.00 × 10−4 | 3 | 1 |
| 005084 | COL1A1 | Collagen type I α 1 chain | 005298 | 5.00 × 10−4 | 18 | 2 |
| 006739 | RALGAPB | Ral GTPase-activating protein, β subunit | 006803 | 9.40 × 10−3 | 1 | 1 |
| 006920 | NEO1 | Neogenin 1 | 007074 | <0.00 × 10−5 | 1 | 1 |
| 006926 | CTNND1 | Catenin delta 1 | 007006 | 3.00 × 10−4 | 1 | 1 |
| 007694 | ACOX1 | Acyl-CoA oxidase 1 | 007823 | 0.00 × 100 | 8 | 1 |
| 007907 | FLOT2 | Flotillin 2 | 008015 | 0.00 × 100 | 1 | 1 |
| 008206 | ADGRG6 | Adhesion G protein-coupled receptor G6 | 008405 | 1.21 × 10−2 | 4 | 1 |
| 009366 | PLEKHG3 | Pleckstrin homology and RhoGEF domain containing G3 | 026649 | 1.43 × 10−2 | 1 | 2 |
| 010640 | MYLK | Myosin light chain kinase | 010735 | 3.00 × 10−4 | 2 | 1 |
| 011707 | FYN | FYN proto-oncogene, Src family tyrosine kinase | 011760 | 5.00 × 10−3 | 1 | 1 |
| 013938 | SPEG | SPEG complex locus | 023342 | 3.99 × 10−2 | 2 | 2 |
| 014232 | RBBP5 | RB binding protein 5 | 014319 | <0.00 × 10−5 | 5 | 2 |
| 014373 | CD82 | Tetraspanin | 014458 | 2.92 × 10−2 | 5 | 1 |
| 015062 | COL3A1 | Collagen type III α 1 chain | 015539 | 2.92 × 10−2 | 11 | 4 |
| 015121 | SLC26A4 | Solute carrier family 26 member 4 | 015204 | 3.02 × 10−2 | 2 | 1 |
| 015894 | PNN | Pinin, desmosome associated protein | 015973 | 1.56 × 10−2 | 1 | 1 |
| 016077 | 016133 | 1.00 × 10−3 | 4 | 3 | ||
| 017036 | NIF3L1 | NGG1 interacting factor 3 like 1 | 017110 | 8.50 × 10−3 | 4 | 1 |
| 3500–4500 m | ||||||
| GeneID | Gene | Gene description | Transcript | B | FB | aB |
| 000773 | IL1RAP | Interleukin 1 receptor accessory protein | 000813 | <0.00 × 10−5 | 2 | 5 |
| 001545 | ZCCHC8 | Zinc finger CCHC-type containing 8 | 001573 | 2.78 × 10−2 | 1 | 2 |
| 002034 | CCDC138 | Coiled-coil domain containing 138 | 002022 | 4.00 × 10−4 | 1 | 1 |
| 003612 | NR3C2 | Nuclear receptor subfamily 3 group C member 2 | 003696 | 4.97 × 10−2 | 7 | 1 |
| 004231 | COL6A3 | Collagen type VI α 3 chain | 004512 | <0.00 × 10−5 | 6 | 2 |
| 005058 | RPL11 | Ribosomal protein L11 | 005076 | 1.67 × 10−2 | 1 | 1 |
| 005562 | PFKM | Phosphofructokinase, muscle | 005957 | 4.83 × 10−2 | 3 | 2 |
| 006776 | FUS | FUS RNA-binding protein | 006895 | 6.70 × 10−3 | 1 | 1 |
| 006926 | CTNND1 | Catenin delta 1 | 007006 | 3.00 × 10−4 | 1 | 1 |
| 007250 | ATP11B | ATPase phospholipid transporting 11B (putative) | 007392 | 1.40 × 10−3 | 12 | 1 |
| 007887 | ABHD3 | Abhydrolase domain containing 3, phospholipase | 007896 | 4.00 × 10−3 | 2 | 2 |
| 009164 | NADK2 | NAD kinase 2, mitochondrial | 009211 | 1.18 × 10−2 | 1 | 1 |
| 009700 | 009686 | 5.50 × 10−3 | 1 | 1 | ||
| 009800 | FLNA | Filamin A | 010200 | 0.00 × 100 | 2 | 2 |
| 011843 | SENP7 | SUMO specific peptidase 7 | 011850 | 1.43 × 10−2 | 1 | 1 |
| 013301 | COL1A2 | Collagen type I α 2 chain | 013614 | 1.20 × 10−3 | 17 | 3 |
| 013313 | AGAP2 | ArfGAP with GTPase domain, ankyrin repeat, PH domain 2 | 013455 | 3.40 × 10−3 | 1 | 1 |
| 014695 | RFWD2 | COP1 E3 ubiquitin ligase | 014789 | 1.00 × 10−3 | 2 | 2 |
| 014919 | ALDOA | Aldolase, fructose-bisphosphate A | 014984 | 2.99 × 10−2 | 2 | 1 |
| 015785 | CDK14 | Cyclin-dependent kinase 14 | 015872 | 6.10 × 10−3 | 1 | 1 |
| 016662 | PGS1 | Phosphatidylglycerophosphate synthase 1 | 016740 | <0.00 × 10−5 | 1 | 1 |
| 017054 | NFIX | Nuclear factor I X | 017136 | 9.50 × 10−3 | 1 | 1 |
| 017166 | FARSA | Phenylalanyl-tRNA synthetase subunit α | 017239 | 2.58 × 10−2 | 1 | 2 |
| 017936 | ATP6V0A1 | ATPase H + transporting V0 subunit a1 | 018008 | 3.50 × 10−3 | 1 | 1 |
| Frogs and lizards, common genes at similar elevation | ||||||
| Gene ID | Gene | Gene description | Transcript | B | FB | aB |
| 016662 | PGS1 | Phosphatidylglycerophosphate synthase 1 | 016740 | <0.00 × 10−5 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofmann, S.; Baniya, C.B.; Stöck, M.; Podsiadlowski, L. De novo Assembly, Annotation, and Analysis of Transcriptome Data of the Ladakh Ground Skink Provide Genetic Information on High-Altitude Adaptation. Genes 2021, 12, 1423. https://doi.org/10.3390/genes12091423
Hofmann S, Baniya CB, Stöck M, Podsiadlowski L. De novo Assembly, Annotation, and Analysis of Transcriptome Data of the Ladakh Ground Skink Provide Genetic Information on High-Altitude Adaptation. Genes. 2021; 12(9):1423. https://doi.org/10.3390/genes12091423
Chicago/Turabian StyleHofmann, Sylvia, Chitra Bahadur Baniya, Matthias Stöck, and Lars Podsiadlowski. 2021. "De novo Assembly, Annotation, and Analysis of Transcriptome Data of the Ladakh Ground Skink Provide Genetic Information on High-Altitude Adaptation" Genes 12, no. 9: 1423. https://doi.org/10.3390/genes12091423