
CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients Cohort
2.2. Exome and Targeted-Exome Sequencing
2.3. Conventional CNV-Detection Methods—Microarray-Based Chromosome Analyses
2.4. Conventional Locus CNV-Detection Method—MLPA
2.5. NGS-Based CNV Calling, Annotation, and Filtering
2.6. Pathogenic CNVs
2.7. Samples Selection
3. Results
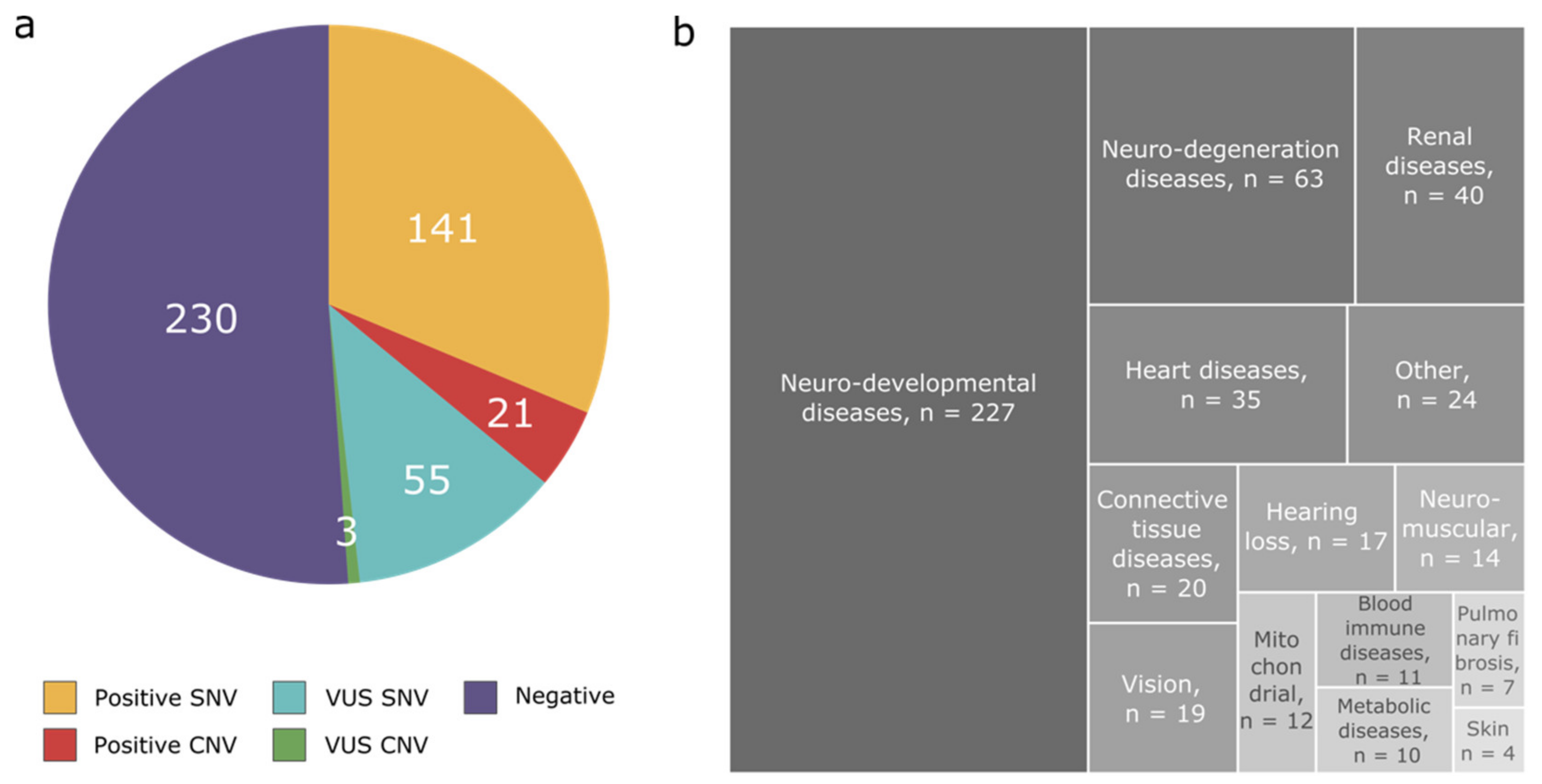
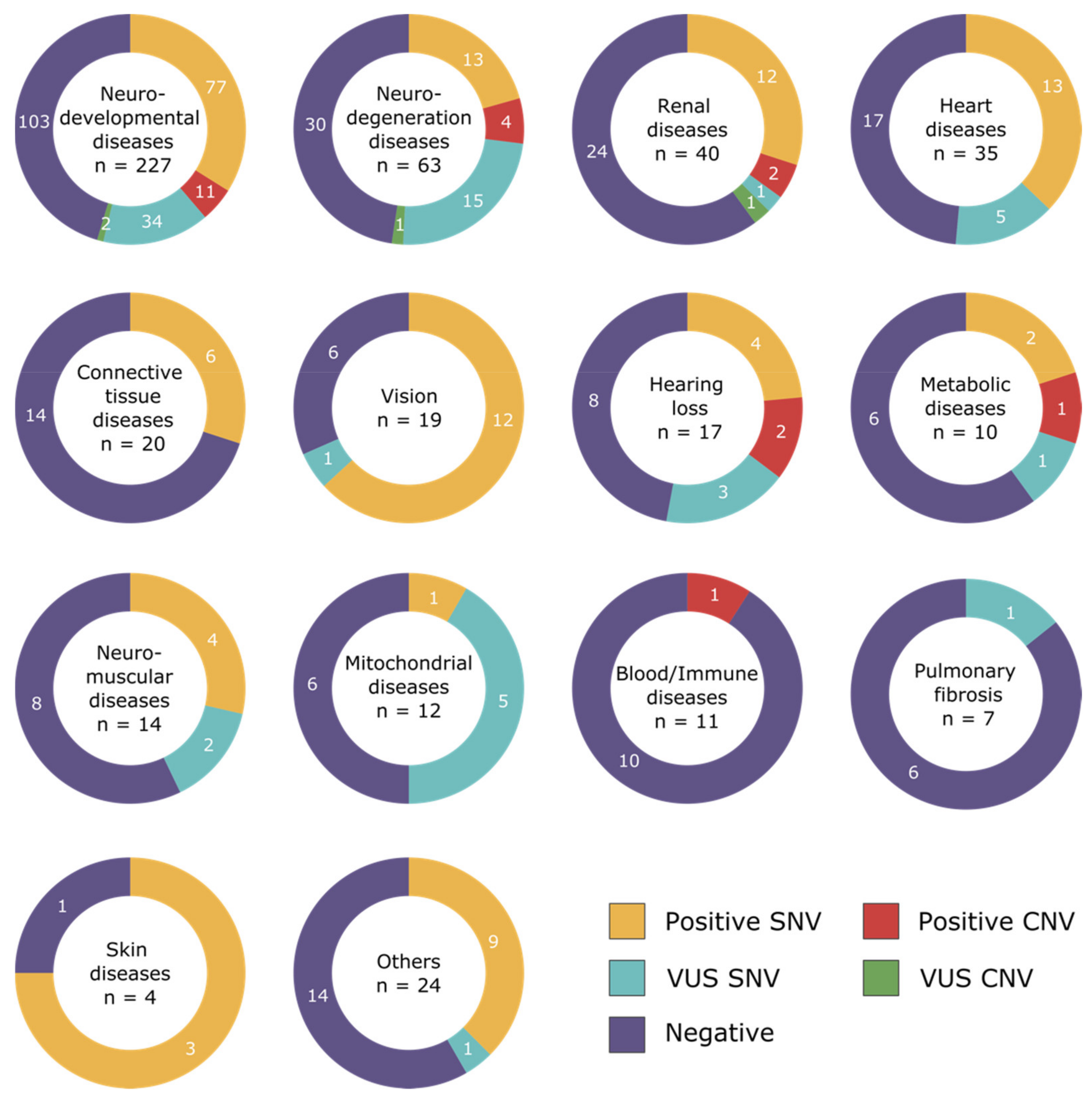
3.1. Disease Categories
3.2. Diagnostic Yield
3.3. Pathogenic CNVs per Disease Categories
3.4. Comparison of arrayCGH and NGS CNV Results
3.5. Pathogenic CNVs Undetected by the NGS Pipeline
3.6. Variants of Unknown Significance in arrayCGH Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boycott, K.M.; Rath, A.; Chong, J.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; Dunnen, J.T.D.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Boycott, K.M.; Hartley, T.; Biesecker, L.G.; Gibbs, R.A.; Innes, A.M.; Riess, O.; Belmont, J.; Dunwoodie, S.L.; Jojic, N.; Lassmann, T.; et al. A Diagnosis for All Rare Genetic Diseases: The Horizon and the Next Frontiers. Cell 2019, 177, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Hartley, T.; Lemire, G.; Kernohan, K.D.; Howley, H.E.; Adams, D.R.; Boycott, K.M. New Diagnostic Approaches for Undiagnosed Rare Genetic Diseases. Annu. Rev. Genom. Hum. Genet. 2020, 21, 351–372. [Google Scholar] [CrossRef] [Green Version]
- McAllister, M.; Davies, L.; Payne, K.; Nicholls, S.; Donnai, D.; MacLeod, R. The emotional effects of genetic diseases: Implications for clinical genetics. Am. J. Med Genet. Part A 2007, 143A, 2651–2661. [Google Scholar] [CrossRef]
- Boone, P.; Bacino, C.A.; Shaw, C.; Eng, P.A.; Hixson, P.M.; Pursley, A.N.; Kang, S.-H.L.; Yang, Y.; Wiszniewska, J.; Nowakowska, B.A.; et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010, 31, 1326–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahajpal, N.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Vinkšel, M.; Writzl, K.; Maver, A.; Peterlin, B. Improving diagnostics of rare genetic diseases with NGS approaches. J. Community Genet. 2021, 12, 247–256. [Google Scholar] [CrossRef]
- Monroe, G.R.; Frederix, G.W.; Savelberg, S.M.C.; De Vries, T.I.; Duran, K.J.; Van Der Smagt, J.J.; Terhal, P.A.; Van Hasselt, P.M.; Kroes, H.Y.; Verhoeven-Duif, N.M.; et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet. Med. 2016, 18, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstrand, A.; Eisfeldt, J.; Pettersson, M.; Carvalho, C.M.B.; Kvarnung, M.; Grigelioniene, G.; Anderlid, B.-M.; Bjerin, O.; Gustavsson, P.; Hammarsjö, A.; et al. From cytogenetics to cytogenomics: Whole-genome sequencing as a first-line test comprehensively captures the diverse spectrum of disease-causing genetic variation underlying intellectual disability. Genome Med. 2019, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: Features and perspectives. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef] [PubMed]
- Retterer, K.; Scuffins, J.; Schmidt, D.; Lewis, R.; Pineda-Alvarez, D.; Stafford, A.; Schmidt, L.; Warren, S.; Gibellini, F.; Kondakova, A.; et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet. Med. 2015, 17, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchuk, D.S.; Crooks, K.; Strande, N.; Kaiser-Rogers, K.; Milko, L.V.; Brandt, A.; Arreola, A.; Tilley, C.R.; Bizon, C.; Vora, N.L.; et al. Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS ONE 2018, 13, e0209185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadalayil, L.; Rafiq, S.; Rose-Zerilli, M.; Pengelly, R.; Parker, H.; Oscier, D.; Strefford, P.J.C.; Tapper, W.J.; Gibson, J.; Ennis, S.; et al. Exome sequence read depth methods for identifying copy number changes. Briefings Bioinform. 2015, 16, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.; Stebbings, E.; Grigoriadou, S.; Wood, N.; Hambleton, S.; Burns, S.; Thrasher, A.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformormatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Royer-Bertrand, B.; Tsouni, P.; Mullen, P.; Xavier, B.C.; Crettol, L.M.; Lobrinus, A.J.; Ghika, J.; Baumgartner, M.R.; Rivolta, C.; Superti-Furga, A.; et al. Peripheral neuropathy and cognitive impairment associated with a novel monoallelic HARS variant. Ann. Clin. Transl. Neurol. 2019, 6, 1072–1080. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Quinodoz, M.; Royer-Bertrand, B.; Cisarova, K.; Di Gioia, S.A.; Superti-Furga, A.; Rivolta, C. DOMINO: Using Machine Learning to Predict Genes Associated with Dominant Disorders. Am. J. Hum. Genet. 2017, 101, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Firth, H.V.; Richards, S.M.; Bevan, P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Nowakowska, B. Clinical interpretation of copy number variants in the human genome. J. Appl. Genet. 2017, 58, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Atallah, I.; Quinodoz, M.; Campos-Xavier, B.; Peter, V.G.; Fouriki, A.; Bonvin, C.; Bottani, A.; Kumps, C.; Angelini, F.; Enders, F.B.; et al. Immune deficiency, autoimmune disease and intellectual disability: A pleiotropic disorder caused by biallelic variants in the TPP2 gene. Clin. Genet. 2021, 99, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Amr, S.; Rehm, H.L.; Funke, B.H.; Pugh, T.; Gowrisankar, S.; Shakhbatyan, R.; Duffy, E.; Bowser, M.; Harrison, B.; et al. Comprehensive Diagnostic Testing for Stereocilin: An approach for analyzing medically important genes with high homology. J. Mol. Diagn. 2014, 16, 639–647. [Google Scholar] [CrossRef]
- Dolcetti, A.; Silversides, C.K.; Marshall, C.R.; Lionel, A.C.; Stavropoulos, D.J.; Scherer, S.W.; Bassett, A.S. 1q21.1 Microduplication expression in adults. Genet. Med. 2012, 15, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Xavier, J.; Zhou, B.; Bilan, F.; Zhang, X.; Gilbert-Dussardier, B.; Viaux-Savelon, S.; Pattni, R.; Ho, S.; Cohen, D.; Levinson, D.F.; et al. 1q21.1 microduplication: Large verbal–nonverbal performance discrepancy and ddPCR assays of HYDIN/HYDIN2 copy number. npj Genom. Med. 2018, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Benítez-Burraco, A.; Barcos-Martínez, M.; Espejo-Portero, I.; Fernández-Urquiza, M.; Torres-Ruiz, R.; Rodriguez-Perales, S.; Romero, M.S.J. Narrowing the Genetic Causes of Language Dysfunction in the 1q21.1 Microduplication Syndrome. Front. Pediatr. 2018, 6, 163. [Google Scholar] [CrossRef]
- Haverfield, E.V.; Whited, A.J.; Petras, K.S.; Dobyns, W.B.; Das, S. Intragenic deletions and duplications of the LIS1 and DCX genes: A major disease-causing mechanism in lissencephaly and subcortical band heterotopia. Eur. J. Hum. Genet. 2008, 17, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Mulhern, M.S.; Stumpel, C.; Stong, N.; Brunner, H.G.; Bier, L.; Lippa, N.; Riviello, J.; Rouhl, R.P.W.; Kempers, M.; Pfundt, R.; et al. NBEA: Developmental disease gene with early generalized epilepsy phenotypes. Ann. Neurol. 2018, 84, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Olson, H.E.; Jean-Marçais, N.; Yang, E.; Heron, D.; Tatton-Brown, K.; Van Der Zwaag, P.A.; Bijlsma, E.K.; Krock, B.L.; Backer, E.; Kamsteeg, E.-J.; et al. A Recurrent De Novo PACS2 Heterozygous Missense Variant Causes Neonatal-Onset Developmental Epileptic Encephalopathy, Facial Dysmorphism, and Cerebellar Dysgenesis. Am. J. Hum. Genet. 2018, 103, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Qian, X.; Wu, Y.; Li, X.; Chen, J.; Xu, J.; Qi, J. Analysis of gene mutations in PKD1/PKD2 by multiplex ligation-dependent probe amplification: Some new findings. Ren. Fail. 2015, 37, 366–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vona, B.; Hofrichter, M.; Neuner, C.; Schröder, J.; Gehrig, A.; Hennermann, J.B.; Kraus, F.B.; Shehata-Dieler, W.; Klopocki, E.; Nanda, I.; et al. DFNB16 is a frequent cause of congenital hearing impairment: Implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 2015, 87, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plevova, P.; Paprskarova, M.; Tvrda, P.; Turska, P.; Slavkovsky, R.; Mrazkova, E. STRC Deletion is a Frequent Cause of Slight to Moderate Congenital Hearing Impairment in the Czech Republic. Otol. Neurotol. 2017, 38, e393–e400. [Google Scholar] [CrossRef]
- Moteki, H.; Azaiez, H.; Sloan-Heggen, C.M.; Booth, K.T.; Nishio, S.-Y.; Wakui, K.; Yamaguchi, T.; Kolbe, D.L.; Iwasa, Y.-I.; Shearer, A.; et al. Detection and Confirmation of Deafness-Causing Copy Number Variations in the STRC Gene by Massively Parallel Sequencing and Comparative Genomic Hybridization. Ann. Otol. Rhinol. Laryngol. 2016, 125, 918–923. [Google Scholar] [CrossRef] [Green Version]
- Kucharík, M.; Budiš, J.; Hýblová, M.; Minárik, G.; Szemes, T. Copy Number Variant Detection with Low-Coverage Whole-Genome Sequencing Represents a Viable Alternative to the Conventional Array-CGH. Diagnostics 2021, 11, 708. [Google Scholar] [CrossRef]
- Mandelker, D.; Schmidt, R.J.; Ankala, A.; Gibson, K.M.; Bowser, M.; Sharma, H.; Duffy, E.; Hegde, M.; Santani, A.; Lebo, M.; et al. Navigating highly homologous genes in a molecular diagnostic setting: A resource for clinical next-generation sequencing. Genet. Med. 2016, 18, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Yao, R.; Zhang, C.; Yu, T.; Li, N.; Hu, X.; Wang, X.; Wang, J.; Shen, Y. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol. Cytogenet. 2017, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Choi, J.-W.; Lee, J.-Y.; Kong, G. Gene-based comparative analysis of tools for estimating copy number alterations using whole-exome sequencing data. Oncotarget 2017, 8, 27277–27285. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Pusey, B.N.; Adams, C.J.; Lau, C.C.; Bone, W.P.; Gahl, W.A.; Markello, T.C.; Adams, D.R. Explorations to improve the completeness of exome sequencing. BMC Med. Genom. 2016, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Zhang, M.; Li, X.; Wang, Z.; Pan, D.; Shi, Y. Performance comparison of four types of target enrichment baits for exome DNA sequencing. Hereditas 2021, 158, 10. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Nickerson, D.A.; Chong, J.X. Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am. J. Hum. Genet. 2019, 105, 448–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, J.E.; Genomics, C.F.M.; O’Donnell-Luria, A.H.; Chong, J.X.; Harel, T.; Jhangiani, S.N.; Akdemir, Z.H.C.; Buyske, S.; Pehlivan, D.; Carvalho, C.M.B.; et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet. Med. 2019, 21, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Guturu, H.; Bernstein, J.A.; Bejerano, G. Systematic reanalysis of clinical exome data yields additional diagnoses: Implications for providers. Genet. Med. 2016, 19, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Basel-Salmon, L.; Orenstein, N.; Markus-Bustani, K.; Ruhrman-Shahar, N.; Kilim, Y.; Magal, N.; Hubshman, M.W.; Bazak, L. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet. Med. 2018, 21, 1443–1451. [Google Scholar] [CrossRef]
- Fung, J.L.F.; Yu, M.H.C.; Huang, S.; Chung, C.C.Y.; Chan, M.C.Y.; Pajusalu, S.; Mak, C.C.Y.; Hui, V.C.C.; Tsang, M.H.Y.; Yeung, K.S.; et al. A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis. npj Genom. Med. 2020, 5, 37. [Google Scholar] [CrossRef]
- Tan, N.B.; Stapleton, R.; Stark, Z.; Delatycki, M.B.; Yeung, A.; Hunter, M.F.; Amor, D.J.; Brown, N.J.; Stutterd, C.A.; McGillivray, G.; et al. Evaluating systematic reanalysis of clinical genomic data in rare disease from single center experience and literature review. Mol. Genet. Genom. Med. 2020, 8, e1508. [Google Scholar] [CrossRef]
- Kalsner, L.; Chamberlain, S.J. Prader-Willi, Angelman, and 15q11-q13 Duplication Syndromes. Pediatr. Clin. N. Am. 2015, 62, 587–606. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Case (Phase) | Disease Category | Clinical Description | NGS Library | NGS CNV Coordinates (Grch37) + Size + Gene If Small CNV | CNV Validation | Acgh/MLPA CNV Results (Grch37) | Known Syndrome (+ Additional Comment) |
|---|---|---|---|---|---|---|---|
| Case 1 (2) | NDD | DD, mild ID | ES | chr22:44489809-51220722 x1 >6.7 Mb | aCGH 180k | 22q13.31q13.33(44481506_51186249)x1 | Phelan-McDermid syndrome (qTer) |
| Case 2 (2) | NDD | DD, language delay, hypotonia | ES | chr15:23609491-28632839 x3 >5 Mb | aCGH 180k | 15q11.2q13.1(22765628_28535051)x3 | 15q11.2-q13.2 microduplication syndrome |
| Case 3 (2) | BID | immunodeficiency and ID | ES | chr13:103298645-103301836 x0 >4.4 Kb–TPP2 (exons 20–22) | WGS | Not enough aCGH targets No MLPA available | (patient presented in [24]) |
| Case 4 (2) | NDD | DD, renale acidosis, SS, optic atrophy | ES | chr14:99640489-106236323 x3 >7 Mb | aCGH 180k | 14q32.2q32.33(99634561_107278770)x3 | 14q distal duplications (qTer) |
| Case 5 (2) | Renal | Renal kystes and DD | ES | chrX: 107224311-107979574 x0 >755 kb | aCGH 180k | Xq22.3(107182490_108105721)x0 | |
| Case 6 (2) | NDD | DD with pyramidal signs & facial dysmorphism | ES | chr2:237028837-242815426 x1 >5 Mb | aCGH 180k | 2q37.2q37.3(236980552_243041364)x1 | 2q37 deletion syndrome (qTer) |
| Case 7 (2) | NG | Ataxia | ES | chr10:125769666-128860040 x3 chr10:133747956-135379033 x1 >7 Mb | aCGH 180k | 10q26.13q26.2(125757754_128852954)x3 10q26.3(130764002_131513932)x3, 10q26.3(131528966_135434178)x1 | 10q26 deletion syndrome |
| Case 8 (2) | NDD | Progressive cognitive decline, epilepsy | ES | chr22:18834446-21414817 x1 >2.5 Mb | aCGH 180k | 22q11.21(18894835_21464119)x1 | 22q11.2 microdeletion syndrome |
| Case 9 (2) | NG | Myoclonic dystonia | ES | chr7:92818581-94540820 x1 >1.7 Mb–including SGCE | aCGH 180k | 7q21.2q21.3(92776146_94641008)x1 | |
| Case 10 (2) | NDD | Global DD, mild dysmorphic features | ES | chr17:1082962-1657828 x3 >574 Kb | aCGH 180k | 17p13.3(1071072_1658551)x3 | 17p13.3 microduplication syndrome |
| Case 11 (2) | NDD | DD, ASD | ES | chr1:146461120-147416212 x3 >955 Kb | aCGH 180k | 1q21.1q21.2(146542843-149243967)x3 | |
| Case 12 (3) | Hearing loss | Deafness | TsoE | chr15:43892159-43901532 x1 >9.4 Kb–STRC (exons 16–28) | MLPA | STRC (exon 19-3′UTR) Not enough aCGH targets | + STRC c.4552G>A (p.Gly1518Ser) |
| Case 13 (3) | NG | Neuroacanthocytosis | ES | chr9:79827886-79828230 x1 >344 bp–VPS13A (exons 8–9) chr9:80018153-80018237x1 >84 bp–VPS13A (exon 69) | Sanger | Not enough aCGH targets No MLPA available | |
| Case 14 (3) | Hearing loss | Deafness, neutropenia | TsoE | chr3:69928286-69988332 x1 >60 Kb–MITF (exons 1–3) | aCGH 1M | 3p13(69917276_69989173)x1 (MITF) | |
| Case 15 (3) | Metabolic disease | Hypophosphatemic rickets | TsoE | chrX:21958944-22151741 x1 >192 Kb–PHEX | aCGH 180k | Xp22.11(21950459_22180647)x1 (PHEX) | |
| Case 16 (3) | NDD | Early onset epilepsy | TsoE | chrX:99551276_99663595 x1 >112 Kb–PCDH19 | MLPA | PCDH19 fully deleted x1 | |
| Case 17 (3) | NG | Familial spastic paraparesia | TsoE | chr2:32352018_32353548 x1 >1.53 Kb–SPAST (exons 8–9) | MLPA | SPAST (exons 8–9) Not enough aCGH targets | |
| Case 18 (3) | NDD | DD, epilepsy, hypotonia, hair anomalies | ES | chr15:22742397-28772634 x4 >6 Mb | aCGH 180k | 15q11.1q13.1(20102541_28535051)x4 | maternal 15q duplication syndrome, (supernumerary chromosome) |
| Case (Phase) | Disease Category | Clinical Description | NGS Library | CNV Coordinates from NGS (Grch37) | CNV Validation | Acgh CNV Results (Grch37) Size + Gene of Interest | Reason CNV Missed by the CNV Pipeline |
|---|---|---|---|---|---|---|---|
| Case 19 (2) | NDD | Lissencephaly | ES | Not detected | aCGH 180k | 17p13.3(2433963_2548152)x1 >114 kb–PAFAH1B1 (exons 1–2) | PAFAH1B1: exon1 is not targeted by the ES library, exon 2 is targeted with low coverage (Figure S2). |
| Case 20 (2) | NDD | DD, Epilepsy | ES | chr13:35615071-35697711 x1 | aCGH 180k | 13q13.3(35522735_35696113)x1 >173 kb–NBEA (exons 2–17) | Gene not present in the gene panel analysed originally. |
| Case 21 (2) | NDD | DD, Epilepsy | ES | chr14:105814831-106370569 x1 | aCGH 180k | 14q32.33(105807673_107278770)x1 >1.4 Mb–including PACS2 | Gene not present in the gene panel analysed originally |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Royer-Bertrand, B.; Cisarova, K.; Niel-Butschi, F.; Mittaz-Crettol, L.; Fodstad, H.; Superti-Furga, A. CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations. Genes 2021, 12, 1427. https://doi.org/10.3390/genes12091427
Royer-Bertrand B, Cisarova K, Niel-Butschi F, Mittaz-Crettol L, Fodstad H, Superti-Furga A. CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations. Genes. 2021; 12(9):1427. https://doi.org/10.3390/genes12091427
Chicago/Turabian StyleRoyer-Bertrand, Beryl, Katarina Cisarova, Florence Niel-Butschi, Laureane Mittaz-Crettol, Heidi Fodstad, and Andrea Superti-Furga. 2021. "CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations" Genes 12, no. 9: 1427. https://doi.org/10.3390/genes12091427