
Abstract
At the end of December 2019, the COVID-19 pandemic began in Wuhan of China. COVID-19 affects different people with a wide spectrum of clinical manifestations, ranging from asymptomatic with recovery without hospitalization up to a severe acute respiratory syndrome (SARS). The innate and adaptive immunity appears responsible for the defense against the virus and recovery from the disease. The innate immune system, as the first line of defense, is essential for the detection of virus and subsequent activation of acquired immunity. The innate immune response is carried out by sentinel cells such as monocytes/macrophages and dendritic cells and by receptors known as pattern recognition receptors (PRR). These receptors can recognize various components of the virus, which lead to intracellular signaling and subsequently the synthesis of various cytokines. These cytokines then recruit other immune cells, activate adaptive immune responses, and inhibit viral spreading. The most common receptors include Toll-like receptors, C-type lectin receptors, and RIG-I like receptors. This review describes the current knowledge about the interplay between innate immune responses and SARS-CoV-2 with a focus on the innate immune cells and the role of their receptors in viral RNA recognition, as well as their mechanisms for recognizing SARS-CoV-2.
Similar content being viewed by others

INTRODUCTION
Coronaviruses are RNA viruses, which can infect different species of animals such as humans, pigs, cats, rats, mice, cows, camels, and bats. The name of these viruses was adapted from their crown-like shape in the electron microscope pictures. Coronaviruses have been introduced as viruses with a high proliferation rate [1]. This characteristic was attributed to the development of transcription errors and RNA related to RNA polymerase jumps of coronaviruses [2].
Human coronaviruses have been reported to involve respiratory, cardiovascular, gastrointestinal, renal, and nervous systems [3]. The people of the world confronted outbreaks of two types of these viruses in 2002–2003 and 2011. The first was named severe acute respiratory syndrome (SARS) [4], and the second type was introduced as Middle East respiratory syndrome (MERS) [5]. The SARS was transmitted from bat origin to humans through an intermediate host, palm civet cats, in 2002. The first case involved with SARS belonged to Guangdong State of China [6]. MERS was observed in Saudi Arabia for the first time. This virus with bat origin was transmitted to humans via camel. MERS sickened 2494 people and killed 858 of them [3].
At the end of December 2019, a case with acute respiratory syndrome in Wuhan of China was reported [7]. After an investigation of respiratory samples, the infection of a new type of coronavirus was indicated. The World Health Organization named it Coronavirus disease 2019 (COVID-19). This virus was named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses [8]. Epidemiologic reports reveal that the human-to-human transmission rate of this virus is higher than SARS and MERS, whereas its mortality rate is lower [9]. It has been indicated that COVID-19 mostly infects adult people (30–65 years old), whereas the infection percentage of individuals under 20 years of age is low.
Several research studies have tried to investigate the interaction between the immune system and the virus. The innate immunity and adaptive immunity are two arms of the immune system against the virus; the innate immune system, as the first line of defense, initiates the immune responses and triggers the adaptive immunity.
The innate immune response is carried out by sentinel cells such as monocytes/macrophages and dendritic cells and by receptors known as pattern recognition receptors.
These components can recognize various components of the virus, which lead to intracellular signaling and subsequently the synthesis of various cytokines. These cytokines subsequently recruit other immune cells, activate adaptive immune responses, and inhibit viral spreading.
In this review, the recent advances about the innate immune responses to SARS-CoV-2 virus will be discussed with a focus on the innate immune cells and the role of their receptors in the recognition of coronaviruses, as well as their mechanisms for recognizing SARS-CoV-2.
THE SEVERE ACUTE RESPIRATORY SYNDROME CORONAVIRUS 2 (SARS-CoV-2)
The SARS-CoV-2 is a virus that belongs to the Nidovirales order. The Nidovirales order consists of enveloped viruses that have a large single-strand positive RNA genome. The Nidovirales enter their host cell by binding to the surface receptors. After uncoating from the nucleocapsid, the RNA genome uses various intracellular organs of the host cell to produce nonstructural and structural proteins such as the nucleocapsid (N) protein, the membrane (M) protein, the envelope (E) protein, and the spike (S) protein (Fig. 1a). Based on different genomic and immunologic properties, the Nidovirales is classified into four virus families including Roniviridae, Arterividae, Mesoniviridae, and Coronaviridae. Torovirinae and Coronavirinae are two sub-families of Coronaviridae. Four genera of Alpha, Beta, Gamma, and Delta coronavirus are categorized in the Coronaviridae sub-family [10, 11]. The hosts of Alpha and Beta coronaviruses are mammals like bats, pigs, cats, mice, and humans, and this is while Gamma and Delta coronaviruses mainly infect birds and even mammals in rare cases [12]. Seven found human coronaviruses cause aggressive or mild diseases. HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1 create self-limiting respiratory diseases while SARS-CoV and MERS-CoV have a mortality rate of almost 10% and 40%, respectively [13, 14]. SARS-CoV-2 is the latest species of coronaviruses causing another severe acute respiratory syndrome and makes a clade in the subgenus of sarbecovirus [15]. The result of the whole-genome phylogenetic shows that SARS-CoV-2 has much more genetic similarity to SARS-CoV than MERS-CoV. These similarities are mainly at M, N, and E proteins than S protein [16]. Despite many similarities between SARS-CoV-2 and SARS-CoV, they have sufficient differences to consider SARS-CoV-2 as a new human-infecting coronavirus [17].

Typical SARS-CoV-2 structure and its genome. a SARS-CoV-2 is an RNA virus; its genome encodes structural proteins including spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins. b Schematic diagram of the SARS-CoV-2 genome. (i) The viral genome contains nonstructural genes (ORF1a and ORF1b) which constitute two-thirds of the genome and structural genes; spike (S), envelope (E), membrane (M), and nucleocapsid (N) as well as accessory genes (in yellow) interspersed within the structural genes. (ii) Schematic representation of SARS-CoV-2 gene encoding spike protein. The spike protein is composed of two subunits (S1 and S2) which are responsible for host cell fusion and virus entry. SP: signal peptide; NTD: N-terminal domain; RBM: receptor-binding motif; RBD: receptor-binding domain; FP: fusion peptide; HR1 and HR2: heptad repeat regions 1 and 2; TM: transmembrane; CP: cytoplasmic tail region.
The genome structure of coronaviruses has a similar organization. Besides 5′-cap and 3′-polyA at both sides, their genome started with the 5′ untranslated region (5′UTR) and ends at 3′UTR. Coronaviruses’ genome has at least ten open reading frames (ORFs) (Fig. 1b). The first two ones (ORF1a/b) occupy two-thirds of the genome and encode two polyproteins pp1a and pp1ab. By cleaving these two polyproteins, 16 non-structural proteins (nsp) are created. These proteins build the replication‐transcription complex (RTC) in double‐membrane vesicles (DMVs). Other ORFs are needed for producing structural proteins. Besides all the above, the genes of some accessory proteins like the hemagglutinin esterase, HE protein, 3a/b protein, and 4a/b protein exist in just some coronavirus genera [18]. Among structural proteins, Spike protein is a crucial surface glycoprotein of COVs for binding, fusion, and entering the host cell. The virus binds to the host cell receptor through its receptor-binding domain (RBD) in the S1 subunit and then fuses its genome and membrane by the S2 subunit (Fig. 1b). The attachment of S1 protein to its receptor causes the S2 conformation changes, reveal, and interaction of the heptad repeat 1 (HR1) and heptad repeat 2 (HR2) domains with each other and forms a six-helical bundle (6-HB) that results in membrane fusion between virus and target cell. The RBDs of SARS-CoV and SARS-CoV-2 identify angiotensin-converting enzyme 2 (ACE2) as their target whereas the specific receptor for the RBD of MERS-COV is dipeptidyl peptidase 4 (DPP4). Studies show that the affinity of RBD to ACE2 is ten times higher in SARS-CoV-2 compared with SARS-CoV [19]. Due to the vital role of virus attachment in pathogenesis, many researchers are trying to find treatment by blocking the different subunits of RBD and ACE2 [20]. Another proven target for drug discovery is viral 3-chymotrypsin-like cysteine protease 4 (3CLpro) enzyme that has an essential role in virus replication. The 3CLpro sequence of SARS-CoV-2 is highly conserved and has a 96.08% and 87% equality sequence with SARS-CoV and MERS-CoV, respectively. Despite the high similarity sequence of SARS-CoV-2 and SARS-CoV, twelve-point mutations have been reported between SARS-CoV and SARS-CoV-2 3CLpro enzymes. These mutations mostly affect the 3CLpro structure [21].
Altogether, despite the conserved sequences of the SARS-CoV-2, the recombination rates of this virus are so high that special consideration is needed for finding treatments and care should be taken regarding the future of this virus family [6].
INNATE IMMUNE COMPONENTS AND COVID-19
The innate immune system is the first line of defense against pathogens, and it participates in antigen detection and restriction of pathogens as well as activation of effector adaptive immune responses, significantly [22].
The main components of innate immunity are physical barriers such as skin and mucosal surfaces; innate immune cells including monocytes, macrophages, dendritic cells, natural killer cells, and neutrophils; and several molecules in the plasma and cells to combat against invading pathogens. In this system, innate responses to pathogens are non-specific with no memory, but these responses are immediate and occur within hours after exposure to the microbe.
This system recognizes conserved molecular substances that are produced by microbial pathogens [23]. These microbial structures that are often essential for microbial survival are called pathogen-associated molecular patterns (PAMPs) [23].
The most important PAMPs are viral PAMPs, and infections derived from these microbes and resulting symptoms are the major challenges of the World Health Organization (WHO) nowadays.
In viral infections, the innate immune system uses several types of receptors such as pattern recognition receptors (PRRs) that present in different locations in the cells, and cellular components in the blood and tissues, to recognize viral PAMPs [24].
Based on the recent investigations, some PRRs play an essential role in SARS-CoV-2 detection and restriction. The roles played by PRRs and mechanisms used by the virus to overcome them are presented in detail as follows.
Pattern Recognition Receptors (PRRs)
PRRs are the cellular receptors for PAMPs that are expressed on the surface, in phagocytic vesicles, and in the cytosol of various innate immune cells, all of which are locations where microbes may be present [25].
The main PRRs are toll-like receptors (TLRs), nucleotide oligomerization domain (NOD)–like receptors (NLRs), retinoic acid–inducible gene 1 (RIG-I)–like receptors (RLRs), C-type lectin receptors (CLRs), scavenger receptors, N-formylmethionyl-leucyl-phenylalanine (FMLP), and DNA-dependent activator of IFN regulatory factors (DAI) [26].
Of which, the most well-characterized are TLRs, RLR, and NLRs, which are involved in viral product detection.
Toll-Like Receptors
The most efficient PRRs are TLRs, which are divided into 10 members in humans, named TLR1 through TLR10 [27]. Among them, TLR1, 2, 4, 5, and TLR6 are cell surface receptors, while TLR3, 7, 8, and 9 are expressed on the surface of the endosomal compartment and found to be involved in viral nucleic acid detection [27, 28]. TLR3 recognizes dsRNA, which is produced during viral RNA replication, while TLR7 and TLR8 detect ssRNA that presents in SARS-CoV and TLR9 activated by unmethylated CpG DNA present in DNA viruses [29, 30].
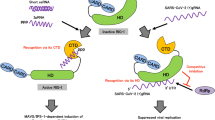
It is well known that SARS-CoV-2 is an RNA virus; hence, it can be recognized by either endosomal RNA receptors, i.e., TLR3 and TLR7/8, or by cytosolic RNA sensors, RLRs, such as RIG-I and MDA-5 (Fig. 2) [31].

Role of pattern recognition receptors in response to SARS-CoV2 infection. Internalization of virus through membrane fusion or binding to a surface receptor (ACE-2) leads to the release of genomic RNA into the cytoplasm. TLR7 and TLR8 in the endosome can recognize ssRNA from the virus, while dsRNA sensors (TLR3, RIG-I, and MDA-5) can detect the virus during SARS-CoV replication. These receptors can induce the activation of IRF3/7 and NF-kB signaling pathways. Activated IRFs and NF-kB translocate to the nucleus and, together with other transcription factors, induce the expression of IFN-I, ISGs, and pro-inflammatory cytokines/chemokines (IL-1, TNF-α, CCL-2, and CXCL-8).
Recognition of viral ligands by TLRs results in the activation of several downstream signaling pathways and ultimately their nuclear transcription factors. The main transcription factors that are activated by TLRs are nuclear factor κB (NF-κB), interferon response factor 3 (IRF3), and IRF7 [32, 33]. NF-κB stimulates the expression of genes encoding inflammatory cytokines (tumor necrosis factor [TNF] and IL-1, IL-6), chemokines (e.g., CCL2 and CXCL8), and IRF3/IRF7 which induces the production of type I interferons (IFN-α and IFN-β), which initiate innate immune responses against viral infection at the entry sites [34].
It is well established that TLRs can recognize SARS-CoV and MERS-CoV, which are antigenically similar to SARS-CoV-2 [18]. One report delineates that human coronaviruses such as SARS-CoV-2 are recognized by RNA sensors including TLR3 and TLR7 [35].
In experimental models of coronavirus infection, the studies have reported the increased expression of TLR3 and downstream signaling molecules and subsequently the production of type I IFN and pro-inflammatory cytokines [36]. The research also revealed that inhibition of adaptor proteins downstream of TLRs (MYD88 and TRIF) had different results. The study found out MYD88 −/− mice had reduced production of inflammatory cytokines, while TRIF−/− mice showed a reduction in the level of inflammatory cytokines in the 2nd day post-infection but an increase in the same cytokines in 4 days post-infection [36]. Since TRIF is associated with the expression of type-I interferons, and the cytokine can modulate immune responses, and based on the fact that production of several pro-inflammatory cytokines is induced by the MYD88 pathway, it appears that MYD88 can be considered as the main response to the SARS-CoV-2. Moreover, due to the fact that cytokine storm by the pro-inflammatory cytokines is the main complication of SARS-CoV-2 infected patients, MYD88 can be considered as the main pathway that participates in the SARS-CoV-2 complications.
Due to the fact that TLR4, which uses both MYD88 and TRIF pathways, recognizes several ligands, it has been hypothesized that TLR4 may participate in the recognition of SARS-CoV-2. Shon and colleagues reported that the expression of TLR4 was increased in COVID-19 patients [37]. Based on the potential roles played by MYD88 in response to SARS-CoV-2, it may be concluded that TLR4 fights against COVID-19 in a MYD88-dependent manner.
The research also demonstrated that the activation of TLR4 is due to the high expression of oxidized phospholipids in lung tissue samples from mice infected with respiratory viruses such as SARS and H1N1. This phospholipid, similar to LPS, activates TLR4 and subsequently downstream signaling molecules MyD88, which in turn leads to the overproduction of inflammatory cytokines [38].
An in vitro study also showed the expression of TLR4, and its downstream proteins were increased in PBMC of patients infected with SARS-CoV-2 [37].
Interestingly, the results of this study also showed that there was no significant difference in the expression of TLR3/7/8 and IRF3 between SARS-CoV-2-infected patients and healthy controls [37]. In parallel with this result, Imai et al. showed that a mutation in TLR4 in an animal model of ARDS (acute respiratory distress syndrome) induced by SARS-CoV was associated with decreased lung injury, while deletion of TLR3 did not show a significant change in lung injury [39].
Taken together, these results demonstrate that the TLR4 signaling via MYD88 pathway in COVID-19 patients leads to pro-inflammatory responses; thus, it seems that although TLR4 plays an essential role in the pathogenesis of COVID 19, its role in the SARS-CoV-2 infection is indirect.
Data from bioinformatics analysis also demonstrated that TLR7 and TLR8 can detect ssRNA fragments from the SARS-CoV-2 genome [40]. TLR7 and TLR8 upon stimulation by ssRNA from virus induce pDC to produce type I IFN, which in turn exerts a protective antiviral response [40].
According to the findings of a study by Van Der Made et al. on four patients with severe COVID-19, there was a loss-of-function mutation in the TLR7 gene, which in turn led to decreased IFN production [41]. Angelopoulou et al. also revealed that imiquimod, another TLR7 agonist, can stimulate innate immunity against pulmonary infections and is able to inhibit SARS-CoV-2 infection in the early stage of the disease [42].
Some studies also revealed that the membrane proteins of SARS such as M protein can induce IFN-β production through TLR signaling [43].
Collectively, it seems that the TLRs using MYD88 as the adaptor recognize SARS-CoV-2, and its related molecules can be considered as the immune responses to the virus to defend against human cells and also to induce pro-inflammatory complications. The main mechanisms that participate in the outcome of the TLR responses to the SARS-CoV-2 are yet to be clarified. The important roles played by TLRs were also documented by the investigation, since there was no alteration in the expression of RLRs such as RIG-I (retinoic acid–inducible gene-I), MDA-5 (melanoma differentiation–associated gene-5), and MAVS (mitochondrial antiviral signaling molecule), while the expression of TLR adaptor proteins (MyD88, TRAM/TICAM2, and TIRAP) was increased [43].
In general, this evidence indicates that the MYD88-dependent pathway may be the main factor involved in the induction of pro-inflammatory responses and pathogenesis of SARS-CoV-2 via NF-kB activation, while TLR7 and TLR8 induce IRF3/7 and antiviral responses, as the studies on TLR5, TLR7, and TLR8 agonists showed a protective immunity in COVID-19 patients. However, it needs to be clarified by further in vivo investigations.
RLRs (Retinoic Acid–Inducible Gene-I–Like Receptors)
RLRs are a family of cytosolic sensors that respond against viruses by inducing the production of antiviral type I interferons [44]. This family comprises three members: retinoic acid–inducible gene 1 (RIG-I), melanoma differentiation–associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) [45].
Unlike TLRs, RLRs are located throughout the cytosol; hence, the viruses that enter the cytosol of infected cells by direct membrane fusion and without entering the endosome can still be detected by RLRs [46, 47].
During SARS-CoV infection, the virus fuses its membrane with the host cell membrane through an interaction between the S protein of coronavirus with ACE2 receptor of host cells [48]. Then, the virus releases its RNA into the cytosol. Therefore, the viral RNA can be recognized by cytosolic sensors of host cells such as RLRs (Fig. 2) [48].
RLRs recognize different viral PAMPs, as RIG-I detects RNA with a 5′-triphosphate residue [49], while MDA5 can interact with long dsRNA, which is crucial for innate immune recognition of RNA viruses [50].
Activation of RIG-I or MDA-5 leads to signaling through an adaptor protein, mitochondrial antiviral signaling molecule (MAVS), which subsequently stimulates the activation of TBK1, IKKi, IKKα/β kinases and ultimately activates the transcription factor IRF3/7 and NF-κB for the expression of IFNs and interferon-stimulated genes (ISGs) as well as inflammatory cytokines (Fig. 2) [51].
Several studies have reported that RIG-I and MDA-5 are essential cellular sensors to recognize RNA viruses [52]. An in vivo study by Zalinger et al. indicated that MDA5 is able to recognize the virus and promote the host defense during murine coronavirus infection [53].
The findings of an in vitro research have also reported that RLRs have potential in MERS-CoV detection, as the results of this study demonstrated that RIG-I and MDA5 were increased in macrophages infected with MERS-CoV. This research also revealed that depletion of RLR downstream molecule, MAVS, decreased the production of inflammatory cytokines such as IL-6, TNF-α, IFN-γ, MIP-1α, RANTES, and IP-10 [54].
The vital role of RLRs in viral infection has also been shown in MDA5-deficient mice. These studies indicated that the mentioned animals are highly susceptible to infection with ssRNA viruses such as SARS coronavirus [55], Rhinovirus [56], and Coxsackie B virus [57].
Regarding the role of RLRs, some in vitro studies have also indicated the upregulation of MDA-5 and RIG-I during SARS-CoV and MERS-CoV infection [58, 59].
Recently, a study by Felsenstein et al. showed that MDA-5 and RIG-I as the cytosolic sensors are able to detect novel coronavirus, SARS-CoV-2, in the cytoplasm of infected cells [35].
However, there are some studies which reported that MERS-CoV inhibits RLRs to escape from immune responses. This research showed that MERS-CoV is able to suppress MDA-5 signaling through inhibition of the NF-kB pathway, while it had no effect on RIG-I-mediated signaling [60].
It is noteworthy that activation of MDA-5 is also associated with the upregulation of interferon-stimulated genes (ISGs), especially ISG15 that is essential for MDA5-to inhibit viral replication. Recent evidence reported that SARS-CoV-2 has a novel mechanism to overcome ISGylation of MDA-5. The virus produces a papain-like protease (PLpro), which has been demonstrated to have a de-ISGylation effect. PLpro of SARS-CoV-2 abolishes MDA-5 ISGylation to escape from innate immune responses [61].
It is well known that NSP8, a nonstructural protein of SARS-CoV-2, plays a role in viral evasion from antiviral immune responses. NSP8 interacts with MDA-5 and subsequently inhibits the recruitment of MAVS, an adaptor protein upstream of MDA-5, to form a signalosome with MDA-5. Indeed, NSP8 of SARS-CoV-2 diminishes the expression of IFN-I, ISGs, and proinflammatory cytokines [62].
Furthermore, M protein of SARS-CoV-2 may play a role in viral evasion. This protein can interact with MAVS and suppresses signaling through RIG-I and MDA5. Importantly, M protein does not affect the downstream molecules of MAVS such as TBK1, IRF3, and TRAF3 [63].
In general, research indicates that the innate immune system can recognize SARS-CoV-2 by a membrane or cytosolic receptor. When the coronavirus escapes from membrane receptors, it can be trapped by MDA-5 or RIG-I in the cytoplasm of infected cells; however, SARS-CoV-2 uses diverse mechanisms to escape from innate immune responses.
DC-SIGN
DC-SIGN (dendritic cell–specific intercellular adhesion molecule-3 grabbing non-integrin) is an innate immune receptor that belongs to C-type lectin receptors, which are highly expressed on DCs [64]. In addition to DC-SIGN, there is a DC-SIGN homolog named L-SIGN (liver/lymph node–specific SIGN) [65]. This protein is expressed in the liver, lymph node, and placenta and leads to SARS-CoV infection of these tissues [66]. Although these markers are specific for DCs, their expression has also been reported on monocytes, alveolar macrophages, and endothelial cells [67].
Carbohydrate residues are DC-SIGN ligands, since a wide range of pathogens express this ligand; hence, DC-SIGN can recognize a variety of pathogens.
DC-SIGN not only plays a role in antigen recognition but also mediates intercellular adhesion of DCs with T cells, as well as signaling to facilitate cell activation and migration to the other tissues [68]. In addition, these molecules can also deliver their cargo to other cells through a cross-presentation route [68].
DC-SIGN recognizes several PAMPs from pathogens; therefore, binding PAMPs to DC-SIGN leads to activation of several downstream molecules. The most important molecule is rapidly accelerated fibrosarcoma (Raf-1). However, studies have shown that the activation of Raf-1 still depends on the upstream molecules such as Ras. Activated Ras binds to Raf-1, and this binding induces the conformational changes in Raf-1. However, full activation of Raf-1 requires the phosphorylation of specific serine and tyrosine residues. Gringhuis et al. demonstrated the phosphorylation of serine 338 and tyrosine 340/341 carried out by p21-activated kinase (Pak) and an Src family tyrosine kinase, respectively. Subsequently, activated Raf-1 phosphorylated the p65 subunit of NFkB and ultimately activates the p50/p65 NFkB dimer. This modification activates NF-kB and then stimulates the expression of cytokines which in turn initiate an immune response against invading pathogen (Fig. 2) [27].
Zhang et al. in a study on avian coronavirus concluded that DC-SIGN may be a marker in the expansion of the virus from one organ to another [69].
It is now well established that coronaviruses, including SARS-CoV and MERS-CoV, use multiple receptors to infect the host cells. The most important receptors are ACE-2, DC-SIGN, and L-SIGN [70].
Recently, a research by Amraei revealed that L-SIGN and DC-SIGN are able to act as an alternative receptor for SARS-CoV-2 entry and infection. These receptors can bind to the RBD domain of spike protein; however, the affinity of these receptors for SARS-CoV-2 is lower than for ACE-2 [71]. Nevertheless, the neck domain of DC-SIGN leads to an oligomerization process, which in turn results in tetramerization of DC-SIGN that recognizes ligands with high affinity [44].
A previous study reported that ACE-2 is the main receptor for SARS infection and DC-SIGN acts as a cofactor for ACE-2 receptor. They showed that DC-SIGN exerts its role through uptake and transfer of viruses to target cells expressing ACE-2 [72].
Findings of an in vitro study on mouse 3T3 cell lines expressing hDC-SIGN established that DC-SIGN may play a role as an alternative receptor for feline coronavirus infection [73].
In contrast, a number of studies have indicated that not only does DC-SIGN serve as a receptor for SARS-CoV infection but also its function is independent of ACE-2 [74].
Further studies have also indicated the entry of human coronaviruses, HCoV-229E and HCoV-NL63, is mediated via ACE2-independent pathways using L-SIGN and DC-SIGN, respectively [75, 76].
Similarly, Han et al. found out that glycans on S protein of SARS-CoV determine the type of receptor-mediated infection. The findings of this research indicated that different glycosylation sites on S protein are important for virus entry through DC-SIGN or ACE-2 receptors [77].
For instance, glycosylation sites such as N109, N118, N119, N158, N227, N589, and N699 in S protein of SARS-COV were also demonstrated to be essential for virus entry through DC-SIGN or L-SIGN [55].
Because of different S glycoproteins on coronaviruses, it can be concluded that DC-SIGN, L-SIGN, and ACE-2 are the main receptors for SARS-CoV-2 infection, but their role is synergistic and depends on glycosylation sites on viral protein S.
Although DC-SIGN is considered as a receptor for expansion of SARS-CoV-2 infection, it may participate in the phagocytosis of the virus to be presented by MHC-II, and because the pathway presentation is the main inducer of adaptive immunity against SARS-CoV-2, it appears that the roles played by DC-SIGN need to be explored in the SARS-CoV-2-infected human cautiously.
INTERFERON RESPONSES
Interferons are well described as the first line of innate immune defense, especially in response to viral infections [78]. These molecules are secreted by infected cells, but affect neighboring cells and inhibit the spread of virus. As mentioned in the previous sections, upon viral infection, PRRs, including TLRs and RLRs, detect nucleic acids of the virus, then trigger downstream signaling molecules and ultimately lead to activation of type I interferons (IFN-I), such as IFN-α and IFN-β [79]. The antiviral activity of IFNs is induced through binding of the IFN-α/β to IFN-α/β receptor (IFNAR) on other cells [79].
Interaction of IFN-I with IFNAR induces several interferon-stimulated genes (ISGs) including double-stranded RNA-dependent protein kinase (PKR) that inhibits viral protein synthesis, and also RNase L, which promotes degradation of viral RNA [80].
Analysis of bronchoalveolar lavage fluid (BALF) from COVID-19 patients showed that the expression of ISGs was increased [81]. In addition, the high expression of ISGs was detected in the PBMCs from severe COVID-19 patients [82]. These results demonstrated that interferon has an essential role in the disease.
Previously, the role of IFN-I in SARS-CoV suppression was demonstrated during the 2003 SARS outbreak. The study on Rhesus monkey revealed that recombinant IFN-α2b can suppress SARS-CoV infection in infected animals [83]. In vitro and in vivo studies also showed that treatment of cell lines or animals with IFN inhibits the pathogenesis and spread of virus [84, 85]. A cohort study on patients with MERS-CoV infection reported that treatment with IFN-α improves the survival rate of patients [86]. It has been reported that in addition to anti-viral activities of IFN-I, it also acts as immunomodulatory molecule; hence, it is used for down-regulation of pro-inflammatory responses in some disorders, such as multiple sclerosis [87].
Therefore, it seems that IFN could possibly be a new choice for 2019-nCov treatment, not only for its anti-viral functions, but also for its immunomodulatory roles during cytokine storm of the COVID-19 infected patients.
In contrast, some in vitro studies have revealed SARS-CoV infection does not induce IFN production in cell cultures [88, 89]. These researches suggest that coronavirus is able to block the IFN induction by immune cells. The studies on tissue and blood samples also revealed there was no increase in the level of IFN-I in SARS-CoV-infected patients [83, 90].
Moreover, other researches indicated that SARS-CoV does not affect IFN and related signaling molecules (IRF-3 and NF-κB) in infected cells [90, 91], but in vivo studies on SARS-CoV-infected macaques have shown that the expression of IFN-I was increased during the early phase of the infection [92].
In Vero cells infected with SARS-CoV, treatment with IFN-I leads to control of virus growth. The data also showed that IFN-β is more effective than IFN-α in the reduction of virus titers [93]. Therefore, it appears that SARS-CoV avoids appropriate immune responses via suppression of IFN-I production, and then leads to activation of severe pro-inflammatory responses.
Furthermore, a retrospective cohort study in China showed that IFN-α2b treatment in the early phase of SARS-CoV-2 infection was associated with reduced mortality in hospitalized patients [94].
In vitro studies have also reported that SARS-CoV-2 is sensitive to IFN-I responses, as treatment of SARS-CoV-2 infected cells with IFN-I led to a decrease in viral load. Interestingly, this reduction was more significant in SARS-CoV-2 than SARS-CoV-infected cells [95]. Furthermore, it has been documented that IFN response at the early phase of COVID-19 elicits an antiviral state and restricts the virus growth, while delayed IFN-I responses were associated with over-activation of pro-inflammatory responses [96].
On the other hand, dysregulation of IFN-I responses may affect on the outcome of COVID-19.
Taken together, the data on the role of IFN in SARS-CoV infection is controversial, but this discrepancy can be explained by the fact that the virus can inhibit the IFN response in vitro, whereas in vivo researches showed that the immune system produces IFN-I during SARS-CoV infection. The other reason for this challenge may be due to the sampling which was done late during the infection, while IFN-I acts on primary immune responses.
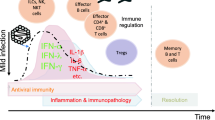
Even though antiviral effects of IFN-I are well demonstrated in several viral infections, including SARS-CoV and MERS-CoV, it seems that the role of IFN-I in COVID-19 patients is time-dependent (Fig. 3), as early IFN-I administration is associated with reduced viral load and disease severity, but delayed IFN-I response promoted viral load and disease severity in SARS-CoV-infected patients [97]. Moreover, in animal models of MERS-CoV and SARS-CoV, delayed IFN-I administration was associated with a high level of the pro-inflammatory cytokine, destruction of epithelial and endothelial cells, and a defect in control of viral infection [98].

Interplay between SARS-CoV-2 and the type I interferon response. The effect of IFN-I in COVID-19 outcome is time dependent. Early IFN-I response can control viral growth yields an antiviral state, so leading to a mild form of disease, while delayed IFN-I expression is associated with rapid SARS-CoV-2 replication, so the patients represent more extensive disease and poorer clinical outcomes.
In general, these data demonstrate that early IFN-I response orchestrates the protective effects against COVID-19, whereas delayed expression of IFN-I leads to the induction of an inflammatory response and ultimately pulmonary pathology during SARS-CoV-2 infection (Fig. 3).
CELLULAR COMPONENTS OF INNATE IMMUNITY AND SARS-CoV-2
SARS-CoV-2 is an obligate intracellular particle that needs a cell to initiate the infection cycle in the human body.
The magnitude of the immune response is related to how the virus invades the body and how immune cells interact with the virus. The immune system usually responds to the virus by activating innate (non-specific) and adaptive (specific) immune responses.
During the infection with SARS-CoV-2, the innate immunity initiates a non-specific immunity against the virus which is followed by specific immune responses through T and B cells [99].
The innate immune cells recognize a wide range of PAMPs that are commonly found in bacteria and viruses [100]. The innate immune cells also play an important role in shaping the downstream adaptive immune response; however, an overactive innate immune response can also result in immune pathology and subsequent tissue destruction [101, 102].
The studies have established that innate immune cells such as dendritic cells (DCs) and macrophages play an essential role in the inhibition of viral infections [103].
Dendritic Cell
DCs are the body sentinels, able to link innate and adaptive immunity. These cells can serve as professional antigen-presenting cells (APC) that trigger T cell stimulation and proliferation [104]. DCs are divided into two major subpopulations, namely, “conventional” (cDCs) and plasmacytoid DCs (pDCs) [105]. cDCs are subdivided into cDC1 and cDC2 based on their ontogeny and functions. These cells are the professional APCs that search our body and sample antigens and then migrate to the T cell area of lymph nodes (LNs).
Upon viral infection, DCs respond to the virus through production of large amounts of IFN-I [106]. IFN-I induces an antiviral state in infected and neighboring cells and stimulates NKC and CD8 + T cells to kill the virus.
It is well known that DCs as professional antigen-presenting cells could recognize SARS-CoV antigens in the periphery by receptors including TLRs. Upon recognition, these cells migrate from peripheral tissue to local lymph nodes to stimulate T cells in a chemokine-dependent manner [107]. DCs express a wide range of PRRs such as TLR2 and TLR4 to recognize SARS/MERS-CoV proteins and TLR3, 7 and TLR8 to detect viral RNA [108].
Researches have revealed that the role of DCs in SARS/MERS-CoV infection is controversial.
Some studies have shown that macrophages and DCs are the foci for MERS/SARS-CoV replication [109, 110], while some groups did not see SARS-CoV replication in macrophages or DCs [111, 112].
The research has reported that DCs expressing ACE2 especially lung DCs, as well as SARS-CoV-2-infected DCs, present a high level of DC-SIGN, suggesting that SARS-CoV-2 can infect DCs to facilitate infection [113].
In addition, during the SAR-CoV outbreak in 2003, the researchers showed that DCs are unable to eradicate SARS-CoV infection [111, 114]. In vitro studies also showed that infection of DCs by SARS-CoV produced low levels of IFN, while inflammatory cytokines and chemokines production were enhanced [112].
Evidence from in vitro studies also revealed that SARS-CoV could infect DCs or macrophages and replicate its genome, but there was no increase in viral load and viral protein synthesis [115].
The findings from SARS-CoV-infected DCs revealed that there was no relationship between the expression of TLRs and SARS-CoV infection, while an increase in chemokine expression was observed (i.e., CCR1, CCR3, CCR5, and CCR7) [116].
A recent in vitro study on SARS-CoV-2-infected DCs reported that, although DCs have been infected with SARS-CoV-2 and some viral proteins were expressed, there was no viral replication in infected DCs [117]. This study also showed a low level of IFN production, while the levels of some pro and inflammatory cytokines were increased [117].
Together, these data suggest that the SARS-CoV-2-infected dendritic cells have a role in the pathogenesis of COVID-19 through pro-inflammatory cytokine production. However, activation of adaptive immunity is a main immune response against COVID-19, which is controlled by dendritic cells.
Additionally, as in previous sections, DC-SIGN, which is expressed on the dendritic cells, plays important roles in the distribution of SARS-CoV-2 in the infected patients, the main mechanisms used by HIV to distribute in the human body. Taken together, dendritic cell function during COVID-19 infection is complicated and needs additional in vitro and in vivo experiments.
Monocyte/Macrophage
Monocytes and macrophages are defined as common phagocytic cells and the first line of defense against invading pathogens. These cells are present in blood and all tissues and play an essential role in the control of infections [118].
Monocytes were initially divided into two subsets based on CD14 and CD16 expression. The most important subsets are classical monocytes, which express CD14 high and CD16 low, and non-classical monocytes with low CD14 and high CD16 expression [119].
Recently, studies have reported the third population of monocytes called intermediate monocytes [120]. This subset of monocytes is defined by CD14 and CD16 expression and produces IL-6 in COVID-19 patients [121].
Monocytes are the main source of several cells such as dendritic cells and especially macrophages when they migrate into the tissues. Based on their function and activation mode, macrophages are divided into M1 and M2 types.
M1 macrophages present antigens to T cells, inhibit tumor growth, and produce pro-inflammatory cytokines such as IL-6, IL-12, and TNF-α, while M2 macrophages participate in tissue repair and wound healing and produce anti-inflammatory cytokines (IL-10 and TGF-β), and hence, restrict inflammation [122].
In patients with COVID-19, monocytes and macrophages detect SARS-CoV-2 using a variety of PRRs, which sense viral PAMPs such as viral RNA. During SARS-CoV-2 infection, the virus attacks tissues, especially the lung, and induces active immune responses through recruiting monocytes and macrophages [123]. These cells restrict the spread of the virus by phagocytosis or release cytokines to stimulate T cells and other adaptive immune responses [123]. However, a dysregulated activation of monocytes and macrophages contributes to hyper-inflammation and SARS-associated pathology [124].
A study on patients with COVID-19 showed that the serum level of pro-inflammatory cytokines, including IP-10, MCP-1, MIP-1α, and TNF-α was higher in ICU patients than those receiving non-ICU care [125]. Moreover, BALF samples from SARS-CoV-2 patients showed a high expression of CCL-2 and CCL-7 as the main chemokines at monocyte recruitment [126].
In addition, a higher percentage of monocytes and macrophages in BALF was observed in patients with a severe form of COVID-19 compared with patients with mild disease or healthy patients [127]. The studies also indicated that the BALF from severe COVID-19 patients contained higher frequencies of M1 macrophages, while in patients with moderate disease a higher percentage of M2 type was observed [127].
However, the research on an animal model of respiratory viruses such as respiratory syncytial virus has shown that the control of infection depends on M1 macrophages [128]. Despite this, in other respiratory viruses including SARS and influenza, the number of M1 macrophages decreases through apoptosis and necrosis which leads to the spread of virus [128]. On the other hand, the polarization of macrophages toward the M2 type may contribute to the viral spread through inhibiting effective antiviral immune responses. Moreover, the over-activation of M1 macrophages and production of the pro-inflammatory cytokines or cytokine storm can lead to severe disease [128].
Researches have also revealed that cytokine storm, particularly macrophage activation syndrome (MAS), is the most important complication in COVID-19 infection [129]. MAS results in high expression of inflammatory cytokines such as TNF-α and IL-6 and subsequently these mediators promote inflammatory pathways [130]. This over-activation of immune responses from MAS leads to acute respiratory distress syndrome (ARDS) in COVID-19 patients [131].
Recent researches suggest that monocyte responses might be dysregulated in COVID-19 patients. These dysregulations may be due to the involvement of different subsets of monocytes in the disease. Classical monocytes, whose major function is phagocytosis, uptake pathogens and produce pro-inflammatory cytokines and activate the other immune cells to modulate inflammation. Intermediate monocytes produce various types of inflammatory cytokines such as IL-1β, IL-6, IL-8, and TNF-α and participate in cytokine storm.
In COVID-19 patients, the numbers of CD14++CD16− cells, as classical monocytes, decrease, while the numbers of CD14++CD16+ (intermediate) and CD14+CD16++ (non-classical) monocytes increase [113].
Furthermore, patients with a severe form of COVID-19 display a high increase in the number of intermediate monocytes than a mild form of the disease [132]. In addition, these subsets of monocytes also showed the capability to secrete GM-CSF, TNF-α, and IL-6 especially in ICU patients, which led to inflammatory cytokine storm.
In contrast, the results in the case of CD14+ CD16++ non-classical monocytes are controversial. A study by Gatti on patients infected with SARS-CoV-2 revealed a decrease of peripheral non-classical monocytes in the severe form of disease [133]. Other work also reported depletion of non-classical monocytes in COVID-19 patients [134]. However, non-classical monocytes consist of two subsets, including type 1 and type 2 (non-classical monocyte 1 and 2) based on the expression of SLAN (a glycosylated form of P-selectin glycoprotein-1 (PSGL-1). The research found that only SLAN+ non-classical monocytes (type 2) were absent in the moderate and severe COVID-19 patients, while type 1 non-classical monocytes were increased [132].
In addition, in numerous inflammatory diseases such as inflammatory bowel disease, cardiovascular disease, and stroke, the number of CD16 + monocytes (intermediate and non-classical) was correlated to disease severity [135,136,137]. This provides insight into the pro-inflammatory properties of non-classical and intermediate monocytes, while classical monocytes exhibit scavenging and phagocytic properties.
Therefore, it seems likely that in patients infected with SARS-CoV-2, the severity of pulmonary immune injury correlates with intermediate and non-classical monocytes, especially the first one; since intermediate monocytes seldom exist in healthy individuals, it is believed that they are a transitional stage between classical and non-classical monocytes.
Accordingly, it seems that although monocytes can limit the virus through phagocytosis or promoting adaptive immune responses, the plasticity of classical monocytes toward intermediate or non-classical types determines the outcome of disease. When monocytes are biased towards intermediate and non-classical types, it can drive severe lung injury during infection with human coronaviruses, but when the monocytes shift to phagocytic type (classical type), the outcome will be a decrease in viral spreading and a mild form of the disease (Table 1).
It is well known that in addition to the role of monocytes/macrophages in the pathogenesis of COVID-19, a different expression of ACE-2 (as a receptor for SARS-CoV-2) by these cells is involved in determining the disease severity.
Evidence revealed that SARS-CoV-2 utilizes this receptor to attach and enter into the host cells; hence, the high expression of ACE2 is considered in the pathogenesis of SARS-CoV-2. This receptor is widely distributed on macrophages and the findings demonstrated that not all macrophages have this receptor [121]. The findings of a research on samples obtained from patients who died from SARS-CoV-2 revealed the high expression of SARS-CoV-2 nucleoprotein (NP) and high level of ACE-2 in macrophages from lymph node and spleen [138]. Moreover, pulmonary macrophages express a high level of ACE2; therefore, pulmonary macrophages may contribute to pulmonary infection during SARS-CoV-2 invasion [63].
Recently, the heterogeneity of ACE-2 expression on monocyte subsets has also been reported in patients with rheumatoid arthritis (RA). In RA patients the results revealed the frequency of non-classical and intermediate monocytes were related to disease severity and also the expression of ACE-2 was increased in CD16 + monocyte subsets than classical monocytes [139].
In patients with chronic kidney disease, the CD14 + CD16 + monocytes with high expression of ACE-2 were associated with increased mortality in comparison to other patients [140].
However, the expression of ACE-2 on monocyte subsets in COVID-19 patients has not yet been fully clarified; therefore, further studies are warranted.
As mentioned in the previous sections, the life and death of COVID-19 patients are highly dependent on monocytes/macrophages.
Collectively, the factors that drive severe lung pathology during infection with human coronaviruses include (i) increased production of proinflammatory cytokines or cytokine storm by monocytes and macrophages, (ii) high expression of ACE-2 by monocytes and macrophages, especially pulmonary macrophages, and (iii) high frequency of pro-inflammatory subsets of monocytes such as intermediate/non-classical monocytes (Table 1).
Therefore, although manipulation of monocyte/macrophages seems to be a promising option for the treatment of COVID-19 patients, it should be noted that various types of monocytes/macrophages are involved in the different stages of disease.
Thus, a better understanding of which subsets of monocytes/macrophages drive disease pathology is important for the development of proper therapeutic interventions.
CONCLUDING REMARKS
Based on the information presented here, it appears that innate immunity plays significant roles to determine the outcome of COVID-19 infection. Based on the fact that the young infected human has stronger innate immunity than older ones, they eradicate the virus faster. Additionally, most hospitalized patients suffer from pro-inflammatory responses which may be related to the uncontrolled innate immune responses. Furthermore, innate immunity may participate in the distribution of COVID-19, especially by attachment of COVID-19 to DC-SIGN on the dendritic cells and migration of the cells to the lymph nodes. Monocyte/macrophages are also the main cells that participate in the defense against COVID-19; however, they are also the main source of pro-inflammatory cytokines, including IL-6 that play key roles in the induction of pro-inflammatory-related complications in infected patients.
Data Availability
All the data are provided on request to the corresponding author.
References
Ruggieri, A., L. Di Trani, I. Gatto, M. Franco, E. Vignolo, B. Bedini, G. Elia, and C. Buonavoglia. 2007. Canine coronavirus induces apoptosis in cultured cells. Veterinary microbiology 121 (1–2): 64–72.
Drexler, Jan Felix, Florian Gloza-Rausch, Jörg. Glende, Victor Max Corman, Doreen Muth, Matthias Goettsche, Antje Seebens, Matthias Niedrig, Susanne Pfefferle, and Stoian Yordanov. 2010. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. Journal of virology 84 (21): 11336–11349.
Singhal, Tanu. 2020. A review of coronavirus disease-2019 (COVID-19). The Indian Journal of Pediatrics:1–6.
Ksiazek, Thomas G., Dean Erdman, Cynthia S. Goldsmith, Sherif R. Zaki, Teresa Peret, Shannon Emery, Suxiang Tong, Carlo Urbani, James A. Comer, and Wilina Lim. 2003. A novel coronavirus associated with severe acute respiratory syndrome. New England journal of medicine 348 (20): 1953–1966.
Memish, Ziad A., Nischay Mishra, Kevin J. Olival, Shamsudeen F. Fagbo, Vishal Kapoor, Jonathan H. Epstein, Rafat AlHakeem, Abdulkareem Durosinloun, Mushabab Al Asmari, and Ariful Islam. 2013. Middle East respiratory syndrome coronavirus in bats. Saudi Arabia. Emerging infectious diseases 19 (11): 1819.
Sahin, Ahmet Riza, Aysegul Erdogan, Pelin Mutlu Agaoglu, Yeliz Dineri, Ahmet Yusuf Cakirci, Mahmut Egemen Senel, Ramazan Azim Okyay, and Ali Muhittin Tasdogan. 2020. 2019 novel coronavirus (COVID-19) outbreak: A review of the current literature. EJMO 4 (1): 1–7.
Yi, Ye., Philip NP. Lagniton, Sen Ye, Enqin Li, and Xu. Ren-He. 2020. COVID-19: What has been learned and to be learned about the novel coronavirus disease. International journal of biological sciences 16 (10): 1753.
Wang, Li-sheng, Yi-ru Wang, Da-wei Ye, and Qing-quan Liu. 2020. A review of the 2019 Novel coronavirus (COVID-19) based on current evidence. International journal of antimicrobial agents:105948.
Prompetchara, Eakachai, Chutitorn Ketloy, and Tanapat Palaga. 2020. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific Journal of Allergy and Immunology 38 (1): 1–9.
Cong, Yingying, Pauline Verlhac, and Fulvio Reggiori. 2017. The interaction between nidovirales and autophagy components. Viruses 9 (7): 182.
Li, Jing, and Wenjun Liu. 2020. Puzzle of highly pathogenic human coronaviruses (2019-nCoV). Protein & Cell 11 (4): 235–238.
Wang, Ning, Jian Shang, Shibo Jiang, and Du. Lanying. 2020. Subunit vaccines against emerging pathogenic human coronaviruses. Frontiers in microbiology 11: 298.
Dhama, Kuldeep, Khan Sharun, Ruchi Tiwari, Shubhankar Sircar, Sudipta Bhat, Yashpal Singh Malik, Karam Pal Singh, Wanpen Chaicumpa, D Katterine Bonilla-Aldana, and Alfonso J Rodriguez-Morales. 2020. Coronavirus disease 2019–COVID-19.
Mohammadi, Mahtab, Seyed Ali Mohammad Arabzadeh, Hamid Reza Mollaei, Seyed Hamidreza Monavari, and Najmeh Nikpour. 2020. Frequency of coronavirus NL63 infection in children with upper respiratory infection by real-time PCR. Iranian Journal of Pediatrics 30(3).
Rothan, Hussin A, and Siddappa N Byrareddy. 2020. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. Journal of autoimmunity:102433.
Ahmed, Syed Faraz, Ahmed A. Quadeer, and Matthew R. McKay. 2020. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 12 (3): 254.
Lu, Roujian, Xiang Zhao, Juan Li, Peihua Niu, Bo. Yang, Wu. Honglong, Wenling Wang, Hao Song, Baoying Huang, and Na. Zhu. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. The Lancet 395 (10224): 565–574.
Liang, Yanwen, Mong-Lien. Wang, Chian-Shiu. Chien, Aliaksandr A. Yarmishyn, Yi-Ping. Yang, Wei-Yi. Lai, Yung-Hung. Luo, Yi-Tsung. Lin, Yann-Jang. Chen, and Pei-Ching. Chang. 2020. Highlight of immune pathogenic response and hematopathologic effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 infection. Frontiers in Immunology 11: 1022.
Tai, Wanbo, Lei He, Xiujuan Zhang, Pu. Jing, Denis Voronin, Shibo Jiang, Yusen Zhou, and Du. Lanying. 2020. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cellular & molecular immunology 17 (6): 613–620.
Kruse, Robert L. 2020. Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Research 9.
ul Qamar, Muhammad Tahir, Safar M Alqahtani, Mubarak A Alamri, and Ling-Ling Chen. 2020. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. Journal of pharmaceutical analysis.
Vabret, Nicolas, Graham J Britton, Conor Gruber, Samarth Hegde, Joel Kim, Maria Kuksin, Rachel Levantovsky, Louise Malle, Alvaro Moreira, and Matthew D Park. 2020. Immunology of COVID-19: current state of the science. Immunity.
Zhai, Yanfang, Chenguang Wang, and Zhengfan Jiang. 2018. Cross-talk between bacterial PAMPs and host PRRs. National Science Review 5 (6): 791–792.
Amarante-Mendes, Gustavo P., Sandy Adjemian, Laura Migliari Branco, Larissa C. Zanetti, Ricardo Weinlich, and Karina R. Bortoluci. 2018. Pattern recognition receptors and the host cell death molecular machinery. Frontiers in Immunology 9: 2379.
Brubaker, Sky W., Kevin S. Bonham, Ivan Zanoni, and Jonathan C. Kagan. 2015. Innate immune pattern recognition: A cell biological perspective. Annual review of immunology 33: 257–290.
Nosratabadi, Reza, Seyed Moayed Alavian, Mohammad Zare-Bidaki, Vahid Mohammadi Shahrokhi, and Mohammad Kazemi Arababadi. 2017. Innate immunity related pathogen recognition receptors and chronic hepatitis B infection. Molecular immunology 90: 64–73.
Abbas, Abul K, Andrew H Lichtman, and Shiv Pillai. 2014. Cellular and molecular immunology E-book. Elsevier Health Sciences.
Arababadi, Mohammad Kazemi, Reza Nosratabadi, and Gholamreza Asadikaram. 2018. Vitamin D and toll like receptors. Life sciences 203: 105–111.
Said, Elias A, Nicolas Tremblay, Mohammed S Al-Balushi, Ali A Al-Jabri, and Daniel Lamarre. 2018. Viruses seen by our cells: the role of viral RNA sensors. Journal of immunology research 2018.
Shahrakyvahed, Aziz, Javad Sanchooli, Nima Sanadgol, Mohammad Kazemi Arababadi, and Derek Kennedy. 2014. TLR9: An important molecule in the fight against hepatitis B virus. Postgraduate medical journal 90 (1065): 396–401.
Totura, Allison L., and Ralph S. Baric. 2012. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Current opinion in virology 2 (3): 264–275.
Pillai, Balakrishna, Cleetus Cherupanakkal Agieshkumar, Jeffrey Immanuel, Elanthiraiyan Saravanan, Vignewswari Eswar Kumar, Tamilarasu Kadhiravan, and Soundravally Rajendiran. 2020. Expression pattern of selected Toll-like receptors (TLR’s) in the PBMC’s of severe and non-severe dengue cases. Immunological investigations 49 (4): 443–452.
Kawasaki, Takumi, and Taro Kawai. 2014. Toll-like receptor signaling pathways. Frontiers in Immunology 5: 461.
Rubio, Daniel, Xu. Ren-Huan, Sanda Remakus, Tracy E. Krouse, Mary Ellen Truckenmiller, Roshan J. Thapa, Siddharth Balachandran, Antonio Alcamí, Christopher C. Norbury, and Luis J. Sigal. 2013. Crosstalk between the type 1 interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell host & microbe 13 (6): 701–710.
Felsenstein, Susanna, Jenny A Herbert, Paul S McNamara, and Christian M Hedrich. 2020. COVID-19: immunology and treatment options. Clinical Immunology:108448.
Totura, Allison L, Alan Whitmore, Sudhakar Agnihothram, Alexandra Schäfer, Michael G Katze, Mark T Heise, and Ralph S Baric. 2015. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio 6(3).
Sohn, Kyung Mok, Sung-Gwon Lee, Hyeon Ji Kim, Shinhyea Cheon, Hyeongseok Jeong, Jooyeon Lee, In Soo Kim, Prashanta Silwal, Young Jae Kim, and Seungwha Paik. 2020. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial sepsis. Journal of Korean medical science 35(38).
Zhou, Shujun, Gui Wang, and Wenbin Zhang. 2018. Effect of TLR4/MyD88 signaling pathway on sepsis-associated acute respiratory distress syndrome in rats, via regulation of macrophage activation and inflammatory response. Experimental and therapeutic medicine 15 (4): 3376–3384.
Imai, Yumiko, G. Keiji Kuba, Greg Neely, Rubina Yaghubian-Malhami, Thomas Perkmann, Geert van Loo, Maria Ermolaeva, Y.H. Ruud Veldhuizen, Connie Leung, and Hongliang Wang. 2008. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133 (2): 235–249.
Moreno-Eutimio, Mario Adán, Constantino Lopez-Macias, and Rodolfo Pastelin-Palacios. 2020. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes and Infection.
Der Made, Van, I. Caspar, Annet Simons, Janneke Schuurs-Hoeijmakers, Guus Van Den Heuvel, Tuomo Mantere, Simone Kersten, Rosanne C. Van Deuren, Marloes Steehouwer, Simon V. Van Reijmersdal, and Martin Jaeger. 2020. Presence of genetic variants among young men with severe COVID-19. JAMA 324 (7): 663–673.
Angelopoulou, Athina, Nikos Alexandris, Evangelia Konstantinou, Konstantinos Mesiakaris, Charilaos Zanidis, Konstantinos Farsalinos, and Konstantinos Poulas. 2020. Imiquimod-A toll like receptor 7 agonist-Is an ideal option for management of COVID 19. Environmental Research 188:109858.
Wang, Yi, and Li Liu. 2016. The membrane protein of severe acute respiratory syndrome coronavirus functions as a novel cytosolic pathogen-associated molecular pattern to promote beta interferon induction via a Toll-like-receptor-related TRAF3-independent mechanism. MBio 7(1).
Yueh-Ming, L., Michael Gale Jr. 2011. Immune signaling by RIG-I-like receptors. Immunity 34(5): 680-692.
Khodarev, Nikolai N. 2019. Intracellular RNA sensing in mammalian cells: role in stress response and cancer therapies. In International review of cell and molecular biology, 31–89. Elsevier.
Ebrahim, Maryam, Vahid Mirzaei, Reza Bidaki, Ziba Shabani, Hamid Daneshvar, Masoud Karimi-Googheri, Mehdi Khaleghinia, Mohammad Reza Afrooz, Yaser Yousefpoor, and Mohammad Kazemi Arababadi. 2015. Are RIG-1 and MDA5 expressions associated with chronic HBV infection? Viral immunology 28 (9): 504–508.
Rehwinkel, Jan, and Michaela U Gack. 2020. RIG-I-like receptors: their regulation and roles in RNA sensing. Nature Reviews Immunology:1–15.
Brielle, Esther S., Dina Schneidman-Duhovny, and Michal Linial. 2020. The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses 12 (5): 497.
Ren, Xiaoming, Melissa M Linehan, Akiko Iwasaki, and Anna Marie Pyle. 2019. RIG-I recognition of RNA targets: the influence of terminal base pair sequence and overhangs on affinity and signaling. Cell Reports 29 (12):3807–3815. e3803.
Uchikawa, Emiko, Mathilde Lethier, Hélène. Malet, Joanna Brunel, Denis Gerlier, and Stephen Cusack. 2016. Structural analysis of dsRNA binding to anti-viral pattern recognition receptors LGP2 and MDA5. Molecular cell 62 (4): 586–602.
Fang, Run, Qifei Jiang, Xiang Zhou, Chenguang Wang, Yukun Guan, Jianli Tao, Jianzhong Xi, Ji-Ming Feng, and Zhengfan Jiang. 2017. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS pathogens 13 (11):e1006720.
Hüsser, Linda, Marco P. Alves, Nicolas Ruggli, and Artur Summerfield. 2011. Identification of the role of RIG-I, MDA-5 and TLR3 in sensing RNA viruses in porcine epithelial cells using lentivirus-driven RNA interference. Virus research 159 (1): 9–16.
Zalinger, Zachary B., Ruth Elliott, Kristine M. Rose, and Susan R. Weiss. 2015. MDA5 is critical to host defense during infection with murine coronavirus. Journal of virology 89 (24): 12330–12340.
Zhao, Xiaoyu, Hin Chu, Bosco Ho-Yin. Wong, Man Chun Chiu, Dong Wang, Cun Li, Xiaojuan Liu, Dong Yang, Vincent Kwok-Man. Poon, and Jianpiao Cai. 2020. Activation of C-type lectin receptor and (RIG)-I-like receptors contributes to proinflammatory response in Middle east respiratory syndrome coronavirus-infected macrophages. The Journal of Infectious Diseases 221 (4): 647–659.
Li, Jianfeng, Yin Liu, and Xuming Zhang. 2010. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. Journal of virology 84 (13): 6472–6482.
Slater, Louise, Nathan W Bartlett, Jennifer J Haas, Jie Zhu, Ross P Walton, Annemarie Sykes, Samer Dahdaleh, Deborah L Clarke, Maria G Belvisi, and Onn M Kon. 2010. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog 6(11):e1001178.
Wang, Jennifer P., Anna Cerny, Damon R. Asher, Evelyn A. Kurt-Jones, Roderick T. Bronson, and Robert W. Finberg. 2010. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. Journal of virology 84 (1): 254–260.
Josset, Laurence, Vineet D Menachery, Lisa E Gralinski, Sudhakar Agnihothram, Pavel Sova, Victoria S Carter, Boyd L Yount, Rachel L Graham, Ralph S Baric, and Michael G Katze. 2013. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio 4(3).
Kindler, Eveline, Hulda R Jónsdóttir, Doreen Muth, Ole J Hamming, Rune Hartmann, Regulo Rodriguez, Robert Geffers, Ron AM Fouchier, Christian Drosten, and Marcel A Müller. 2013. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. MBio 4(1).
Lee, Jeong Yoon, Sojung Bae, and Jinjong Myoung. 2019. Middle East respiratory syndrome coronavirus-encoded accessory proteins impair MDA5-and TBK1-mediated activation of NF-κB. Journal of Microbiology and Biotechnology 29 (8): 1316–1323.
Liu, GuanQun, Jung-Hyun Lee, Zachary M Parker, Dhiraj Acharya, Jessica J Chiang, Michiel van Gent, William Riedl, Meredith E Davis-Gardner, Effi Wies, and Cindy Chiang. 2020. ISG15-dependent activation of the RNA sensor MDA5 and its antagonism by the SARS-CoV-2 papain-like protease. bioRxiv.
Yang, Ziwei, Xiaolin Zhang, Fan Wang, Peihui Wang, Xiaojuan Li, and Ersheng Kuang. 2020. Suppression of MDA5-mediated antiviral immune responses by NSP8 of SARS-CoV-2. bioRxiv.
Fu, Yu-Zhi, Su-Yun Wang, Zhou-Qin Zheng, Yi Huang, Wei-Wei Li, Zhi-Sheng Xu, and Yan-Yi Wang. 2020. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cellular & molecular immunology:1–8.
Geurtsen, Jeroen, Nicole N Driessen, and Ben J Appelmelk. 2010. Mannose–fucose recognition by DC-SIGN. In Microbial Glycobiology, 673–695. Elsevier.
da Silva, Ronaldo, Ludovica Segat Celerino, and Sergio Crovella. 2011. Role of DC-SIGN and L-SIGN receptors in HIV-1 vertical transmission. Human immunology 72 (4): 305–311.
Gramberg, Thomas, Heike Hofmann, Peggy Möller, Patricia F. Lalor, Andrea Marzi, Martina Geier, Mandy Krumbiegel, Thomas Winkler, Frank Kirchhoff, and David H. Adams. 2005. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 340 (2): 224–236.
Lugo-Villarino, Geanncarlo, Anthony Troegeler, Luciana Balboa, Claire Lastrucci, Carine Duval, Ingrid Mercier, Alan Bénard, Florence Capilla, Talal Al Saati, and Renaud Poincloux. 2018. The C-type lectin receptor DC-SIGN has an anti-inflammatory role in human M (IL-4) macrophages in response to Mycobacterium tuberculosis. Frontiers in Immunology 9: 1123.
van Kooyk, Yvette, Wendy WJ. Unger, Cynthia M. Fehres, Hakan Kalay, and Juan J. García-Vallejo. 2013. Glycan-based DC-SIGN targeting vaccines to enhance antigen cross-presentation. Molecular immunology 55 (2): 143–145.
Zhang, Yueting, Elizabeth Buckles, and Gary R. Whittaker. 2012. Expression of the C-type lectins DC-SIGN or L-SIGN alters host cell susceptibility for the avian coronavirus, infectious bronchitis virus. Veterinary microbiology 157 (3–4): 285–293.
Devaux, Christian A, Jean-Marc Rolain, and Didier Raoult. 2020. ACE2 receptor polymorphism: susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. Journal of Microbiology, Immunology and Infection.
Amraei, Razie, Marc Napoleon, Wenqing Yin, Jacob Berrigan, Ellen Suder, Grace Zhao, Judith Olejnik, Suryaram Gummuluru, Elke Muhlberger, and Vipul Chitalia. 2020. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. bioRxiv.
Han, Dong P, Motashim Lohani, and Michael W Cho. 2007. Specific asparagine-linked glycosylation sites are critical for DC-SIGN-and L-SIGN-mediated SARS-CoV entry. Journal of virology.
Regan, Andrew D., and Gary R. Whittaker. 2008. Utilization of DC-SIGN for entry of feline coronaviruses into host cells. Journal of virology 82 (23): 11992–11996.
Jeffers, Scott A., Sonia M. Tusell, Laura Gillim-Ross, Erin M. Hemmila, Jenna E. Achenbach, Gregory J. Babcock, William D. Thomas, Larissa B. Thackray, Mark D. Young, and Robert J. Mason. 2004. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proceedings of the National Academy of Sciences 101 (44): 15748–15753.
Hofmann, Heike, Graham Simmons, Andrew J. Rennekamp, Chawaree Chaipan, Thomas Gramberg, Elke Heck, Martina Geier, Anja Wegele, Andrea Marzi, and Paul Bates. 2006. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. Journal of virology 80 (17): 8639–8652.
Jeffers, Scott A, Erin M Hemmila, and Kathryn V Holmes. 2006. Human coronavirus 229E can use CD209L (L-SIGN) to enter cells. In The Nidoviruses, 265–269. Springer.
Han, Dong P., Motashim Lohani, and Michael W. Cho. 2007. Specific asparagine-linked glycosylation sites are critical for DC-SIGN-and L-SIGN-mediated severe acute respiratory syndrome coronavirus entry. Journal of virology 81 (21): 12029–12039.
Zhou, Jian-hua, Yi.-ning Wang, Qiu-yan Chang, Peng Ma, Hu. Yonghao, and Xin Cao. 2018. Type III interferons in viral infection and antiviral immunity. Cellular Physiology and Biochemistry 51 (1): 173–185.
Teijaro, John R. 2016. Type I interferons in viral control and immune regulation. Current opinion in virology 16: 31–40.
Acosta, Patricio L, Alana B Byrne, Diego R Hijano, and Laura B Talarico. 2020. Human type I interferon antiviral effects in respiratory and reemerging viral infections. Journal of immunology research 2020.
Zhou, Zhuo, Lili Ren, Li Zhang, Jiaxin Zhong, Yan Xiao, Zhilong Jia, Li Guo, Jing Yang, Chun Wang, and Shuai Jiang. 2020. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell host & microbe.
Wilk, Aaron J, Arjun Rustagi, Nancy Q Zhao, Jonasel Roque, Giovanny J Martínez-Colón, Julia L McKechnie, Geoffrey T Ivison, Thanmayi Ranganath, Rosemary Vergara, and Taylor Hollis. 2020. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nature Medicine:1–7.
Gao, Hong, LL Zhang, Qiang Wei, ZJ Duan, XM Tu, ZA Yu, Wei Deng, LP Zhang, LL Bao, and Bin Zhang. 2005. Preventive and therapeutic effects of recombinant IFN-alpha2b nasal spray on SARS-CoV infection in Macaca mulata. Zhonghua shi yan he lin chuang bing du xue za zhi= Zhonghua shiyan he linchuang bingduxue zazhi= Chinese journal of experimental and clinical virology 19 (3):207–210.
Falzarano, Darryl, Emmie De Wit, Angela L. Rasmussen, Friederike Feldmann, Atsushi Okumura, Dana P. Scott, Doug Brining, Trenton Bushmaker, Cynthia Martellaro, and Laura Baseler. 2013. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV–infected rhesus macaques. Nature Medicine 19 (10): 1313–1317.
Sallard, Erwan, François-Xavier Lescure, Yazdan Yazdanpanah, France Mentre, Nathan Peiffer-Smadja, ADER Florence, Yazdan YAZDANPANAH, France MENTRE, François-Xavier LESCURE, and Nathan PEIFFER-SMADJA. 2020. Type 1 interferons as a potential treatment against COVID-19. Antiviral Research:104791.
Omrani, Ali S., Mustafa M. Saad, Kamran Baig, Abdelkarim Bahloul, Mohammed Abdul-Matin, Amal Y. Alaidaroos, Ghaleb A. Almakhlafi, Mohammed M. Albarrak, Ziad A. Memish, and Ali M. Albarrak. 2014. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: A retrospective cohort study. The Lancet Infectious Diseases 14 (11): 1090–1095.
Hecker, Michael, Christiane Hartmann, Ole Kandulski, Brigitte Katrin Paap, Dirk Koczan, Hans-Juergen. Thiesen, and Uwe Klaus Zettl. 2013. Interferon-beta therapy in multiple sclerosis: The short-term and long-term effects on the patients’ individual gene expression in peripheral blood. Molecular neurobiology 48 (3): 737–756.
Spiegel, Martin, Andreas Pichlmair, Luis Martínez-Sobrido, Jerome Cros, Adolfo García-Sastre, Otto Haller, and Friedemann Weber. 2005. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. Journal of virology 79 (4): 2079–2086.
Tang, Bone SF., Kwok-hung Chan, Vincent CC. Cheng, Patrick CY. Woo, Susanna KP. Lau, Clarence CK. Lam, Tsun-leung Chan, Alan KL. Wu, Ivan FN. Hung, and Suet-yi Leung. 2005. Comparative host gene transcription by microarray analysis early after infection of the Huh7 cell line by severe acute respiratory syndrome coronavirus and human coronavirus 229E. Journal of virology 79 (10): 6180–6193.
Jones, B.M., E.S.K. Ma, J.S.M. Peiris, P.C. Wong, J.C.M. Ho, B. Lam, K.N. Lai, and K.W.T. Tsang. 2004. Prolonged disturbances of in vitro cytokine production in patients with severe acute respiratory syndrome (SARS) treated with ribavirin and steroids. Clinical & Experimental Immunology 135 (3): 467–473.
Frieman, Matthew, Mark Heise, and Ralph Baric. 2008. SARS coronavirus and innate immunity. Virus research 133 (1): 101–112.
De Lang, Anna, Tracey Baas, Saskia L Smits, Michael G Katze, Albert DME Osterhaus, and Bart L Haagmans. 2009. Unraveling the complexities of the interferon response during SARS-CoV infection.
Spiegel, Martin, Andreas Pichlmair, Elke Mühlberger, Otto Haller, and Friedemann Weber. 2004. The antiviral effect of interferon-beta against SARS-coronavirus is not mediated by MxA protein. Journal of clinical virology 30 (3): 211–213.
Wang, Nan, Yan Zhan, Linyu Zhu, Zhibing Hou, Feng Liu, Pinhong Song, Feng Qiu, Xiaolin Wang, Xiafei Zou, and Deyun Wan. 2020. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID-19 patients. Cell host & microbe 28(3):455–464. e452.
Lokugamage, K. 2020. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. bioRxiv Prepr. Serv. Biol.
Lopez, LeAnn, Peter C. Sang, Yun Tian, and Yongming Sang. 2020. Dysregulated interferon response underlying severe COVID-19. Viruses 12 (12): 1433.
Channappanavar, Rudragouda, Anthony R. Fehr, Rahul Vijay, Matthias Mack, Jincun Zhao, David K. Meyerholz, and Stanley Perlman. 2016. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell host & microbe 19 (2): 181–193.
Jamilloux, Yvan, Thomas Henry, Alexandre Belot, Sébastien Viel, Maxime Fauter, Thomas El Jammal, Thierry Walzer, Bruno François, and Pascal Sève. 2020. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmunity Reviews:102567.
O’Connell, Patrick, and Yasser A Aldhamen. 2020. Systemic innate and adaptive immune responses to SARS-CoV-2 as it relates to other coronaviruses. Human Vaccines & Immunotherapeutics:1–12.
Zipfel, Cyril, and Silke Robatzek. 2010. Pathogen-associated molecular pattern-triggered immunity: Veni, vidi…? Plant physiology 154 (2): 551–554.
García-Sastre, Adolfo, and Christine A. Biron. 2006. Type 1 interferons and the virus-host relationship: A lesson in detente. Science 312 (5775): 879–882.
Gasteiger, Georg, and Andrea D’osualdo, David A Schubert, Alexander Weber, Emanuela M Bruscia, and Dominik Hartl. . 2017. Cellular innate immunity: An old game with new players. Journal of innate immunity 9 (2): 111–125.
Olukitibi, Titus Abiola, Zhujun Ao, Mona Mahmoudi, Gary A. Kobinger, and Xiaojian Yao. 2019. Dendritic cells/macrophages-targeting feature of Ebola glycoprotein and its potential as immunological facilitator for antiviral vaccine approach. Microorganisms 7 (10): 402.
Worbs, Tim, Swantje I. Hammerschmidt, and Reinhold Förster. 2017. Dendritic cell migration in health and disease. Nature Reviews Immunology 17 (1): 30.
Sato, Katsuaki, and Shigeharu Fujita. 2007. Dendritic cells-nature and classification. Allergology International 56 (3): 183–191.
Asselin-Paturel, Carine, and Giorgio Trinchieri. 2005. Production of type I interferons: Plasmacytoid dendritic cells and beyond. The Journal of experimental medicine 202 (4): 461–465.
Mueller, Scott N. 2017. Spreading the load: Antigen transfer between migratory and lymph node-resident dendritic cells promotes T-cell priming. European Journal of Immunology 47 (10): 1798–1801.
Collin, Matthew, and Venetia Bigley. 2018. Human dendritic cell subsets: An update. Immunology 154 (1): 3–20.
Chu, Hin, Jie Zhou, Bosco Ho-Yin. Wong, Cun Li, Zhong-Shan. Cheng, Xiang Lin, Vincent Kwok-Man. Poon, Tianhao Sun, Candy Choi-Yi. Lau, and Jasper Fuk-Woo. Chan. 2014. Productive replication of Middle East respiratory syndrome coronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology 454: 197–205.
Zhou, Jie, Hin Chu, Cun Li, Bosco Ho-Yin. Wong, Zhong-Shan. Cheng, Vincent Kwok-Man. Poon, Tianhao Sun, Candy Choi-Yi. Lau, Kenneth Kak-Yuen. Wong, and Jimmy Yu-Wai. Chan. 2014. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: Implications for pathogenesis. The Journal of Infectious Diseases 209 (9): 1331–1342.
Law, Helen KW., Chung Yan Cheung, Hoi Yee Ng, Sin Fun Sia, Yuk On Chan, Winsie Luk, John M. Nicholls, J.S. Peiris, and Yu Lung Lau. 2005. Chemokine up-regulation in sars-coronavirus–infected, monocyte-derived human dendritic cells. Blood 106 (7): 2366–2374.
Tseng, Chien-Te K., Lucy A. Perrone, Hongbing Zhu, Shinji Makino, and Clarence J. Peters. 2005. Severe acute respiratory syndrome and the innate immune responses: Modulation of effector cell function without productive infection. The Journal of Immunology 174 (12): 7977–7985.
Yang, Dong, Hin Chu, Yuxin Hou, Yue Chai, Huiping Shuai, Andrew Chak-Yiu. Lee, Xi. Zhang, Yixin Wang, Hu. Bingjie, and Xiner Huang. 2020. Attenuated interferon and proinflammatory response in SARS-CoV-2–infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. The Journal of Infectious Diseases 222 (5): 734–745.
Ziegler, Thedi, Sampsa Matikainen, Esa Rönkkö, Pamela Österlund, Maarit Sillanpää, Jukka Sirén, Riku Fagerlund, Milla Immonen, Krister Melén, and Ilkka Julkunen. 2005. Severe acute respiratory syndrome coronavirus fails to activate cytokine-mediated innate immune responses in cultured human monocyte-derived dendritic cells. Journal of virology 79 (21): 13800–13805.
Lau, Yu-Lung., J.S. Peiris, and H.K. Law. 2012. Role of dendritic cells in SARS coronavirus infection. Hong Kong Medical Journal 18 (Suppl 3): 28–30.
Law, Helen KW., Chung Yan Cheung, Sin Fun Sia, Yuk On Chan, J.S. Malik Peiris, and Yu Lung Lau. 2009. Toll-like receptors, chemokine receptors and death receptor ligands responses in SARS coronavirus infected human monocyte derived dendritic cells. BMC immunology 10 (1): 35.
Yang, Dong, Hin Chu, Yuxin Hou, Yue Chai, Huiping Shuai, Andrew Chak-Yiu Lee, Xi Zhang, Yixin Wang, Bingjie Hu, and Xiner Huang. 2020. Attenuated interferon and pro-inflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. The Journal of Infectious Diseases.
Franken, Lars, Marzena Schiwon, and Christian Kurts. 2016. Macrophages: Sentinels and regulators of the immune system. Cellular microbiology 18 (4): 475–487.
Strauss-Ayali, Dalit, Sean M. Conrad, and David M. Mosser. 2007. Monocyte subpopulations and their differentiation patterns during infection. Journal of leukocyte biology 82 (2): 244–252.
Merah-Mourah, F., S.O. Cohen, N. Dominique Charron, and Mooney, and Alain Haziot. . 2020. Identification of novel human monocyte subsets and evidence for phenotypic groups defined by interindividual variations of expression of adhesion molecules. Scientific reports 10 (1): 1–16.
Merad, Miriam, and Jerome C Martin. 2020. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nature Reviews Immunology:1–8.
Wang, Youhan, Wanli Smith, Dingjun Hao, Baorong He, and Lingbo Kong. 2019. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. International immunopharmacology 70: 459–466.
Guan, Wei-jie, Zheng-yi Ni, Yu Hu, Wen-hua Liang, Chun-quan Ou, Jian-xing He, Lei Liu, Hong Shan, Chun-liang Lei, and David SC Hui. 2020. Clinical characteristics of 2019 novel coronavirus infection in China. MedRxiv.
Blanco-Melo, Daniel, Benjamin E Nilsson-Payant, Wen-Chun Liu, Skyler Uhl, Daisy Hoagland, Rasmus Møller, Tristan X Jordan, Kohei Oishi, Maryline Panis, and David Sachs. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell.
Catanzaro, Michele, Francesca Fagiani, Marco Racchi, Emanuela Corsini, Stefano Govoni, and Cristina Lanni. 2020. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduction and Targeted Therapy 5 (1): 1–10.
Zhou, Zhuo, Lili Ren, Li Zhang, Jiaxin Zhong, Yan Xiao, Zhilong Jia, Li Guo, Jing Yang, Chun Wang, and Shuai Jiang. 2020. Overly exuberant innate immune response to SARS-CoV-2 infection.
Liao, Mingfeng, Yang Liu, Jing Yuan, Yanling Wen, Gang Xu, Juanjuan Zhao, Lin Cheng, Jinxiu Li, Xin Wang, and Fuxiang Wang. 2020. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nature Medicine:1–3.
Sang, Yongming, Laura C Miller, and Frank Blecha. 2015. Macrophage polarization in virus-host interactions. Journal of clinical & cellular immunology 6(2).
Napoli, Claudio, Giuditta Benincasa, Clelia Criscuolo, Mario Faenza, Cinzia Liberato, and Mariangela Rusciano. 2021. Immune reactivity during COVID-19: Implications for treatment. Immunology letters.
McGonagle, Dennis, Kassem Sharif, Anthony O'Regan, and Charlie Bridgewood. 2020. Interleukin-6 use in COVID-19 pneumonia related macrophage activation syndrome. Autoimmunity Reviews:102537.
Gómez-Rial, Jose, Irene Rivero-Calle, Antonio Salas, and Federico Martinón-Torres. 2020. Role of monocytes/macrophages in Covid-19 pathogenesis: Implications for therapy. Infection and Drug Resistance 13: 2485.
Padgett, Lindsey E, Huy Q Dinh, Serena J Chee, Claire E Olingy, Runpei Wu, Daniel J Araujo, Pandurangan Vijayanand, Christian H Ottensmeier, and Catherine C Hedrick. 2020. Interplay of monocytes and T lymphocytes in COVID-19 Severity. bioRxiv.
Gatti, Arianna, Danilo Radrizzani, Paolo Viganò, Antonino Mazzone, and Bruno Brando. 2020. Decrease of non-classical and intermediate monocyte subsets in severe acute SARS-CoV-2 infection. Cytometry Part A 97 (9): 887–890.
Schulte-Schrepping, Jonas, Nico Reusch, Daniela Paclik, Kevin Baßler, Stephan Schlickeiser, Bowen Zhang, Benjamin Krämer, Tobias Krammer, Sophia Brumhard, and Lorenzo Bonaguro. 2020. Suppressive myeloid cells are a hallmark of severe COVID-19. MedRxiv.
Hilgendorf, Ingo, and Filip K. Swirski. 2012. Making a difference: Monocyte heterogeneity in cardiovascular disease. Current atherosclerosis reports 14 (5): 450–459.
Koch, Stefan, T. Kucharzik, J. Heidemann, A. Nusrat, and A. Luegering. 2010. Investigating the role of proinflammatory CD16+ monocytes in the pathogenesis of inflammatory bowel disease. Clinical & Experimental Immunology 161 (2): 332–341.
Urra, Xabier, Neus Villamor, Sergio Amaro, Manuel Gomez-Choco, Víctor. Obach, Laura Oleaga, Anna M. Planas, and Angel Chamorro. 2009. Monocyte subtypes predict clinical course and prognosis in human stroke. Journal of Cerebral Blood Flow & Metabolism 29 (5): 994–1002.
Feng, Zeqing, Bo Diao, Rongshuai Wang, Gang Wang, Chenhui Wang, Yingjun Tan, Liang Liu, Changsong Wang, Ying Liu, and Yueping Liu. 2020. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. MedRxiv.
Rodríguez-Carrio, Javier, Mercedes Alperi-López, Patricia López, Ángel Pérez-Álvarez, Lorena Benavente, Francisco Javier Ballina-García, and Ana Suárez. 2019. SAT0064 expansion of CD16+ monocyte subsets and ace expression are associated with arterial thickening and vascular function in very early rheumatoid arthritis: BMJ Publishing Group Ltd.
Ulrich, Christof, Gunnar H. Heine, Eric Seibert, Danilo Fliser, and Matthias Girndt. 2010. Circulating monocyte subpopulations with high expression of angiotensin-converting enzyme predict mortality in patients with end-stage renal disease. Nephrology Dialysis Transplantation 25 (7): 2265–2272.
Acknowledgements
The authors would like to thank the authorities in the Research Council of Kerman University of Medical Sciences (KMU).
Author information
Authors and Affiliations
Contributions
AA and HRM performed the literature search and drafted the manuscript. RN and MKA provided suggestions and edited the manuscript. All the authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
This article does not contain any experiments with human participants or animals performed by any of the authors.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Anaeigoudari, A., Mollaei, H.R., Arababadi, M.K. et al. Severe Acute Respiratory Syndrome Coronavirus 2: The Role of the Main Components of the Innate Immune System. Inflammation 44, 2151–2169 (2021). https://doi.org/10.1007/s10753-021-01519-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-021-01519-7