
Type I Interferon Induction and Exhaustion during Viral Infection: Plasmacytoid Dendritic Cells and Emerging COVID-19 Findings

Division of Biological Sciences, University of California, San Diego, CA 92093, USA
*
Author to whom correspondence should be addressed.
Viruses 2021, 13(9), 1839; https://doi.org/10.3390/v13091839
Submission received: 27 June 2021
/
Revised: 1 September 2021
/
Accepted: 1 September 2021
/
Published: 15 September 2021
(This article belongs to the Special Issue In Memory of Stefan Kunz)
{kind=link}
Abstract
:Type I Interferons (IFN-I) are a family of potent antiviral cytokines that act through the direct restriction of viral replication and by enhancing antiviral immunity. However, these powerful cytokines are a caged lion, as excessive and sustained IFN-I production can drive immunopathology during infection, and aberrant IFN-I production is a feature of several types of autoimmunity. As specialized producers of IFN-I plasmacytoid (p), dendritic cells (DCs) can secrete superb quantities and a wide breadth of IFN-I isoforms immediately after infection or stimulation, and are the focus of this review. Notably, a few days after viral infection pDCs tune down their capacity for IFN-I production, producing less cytokines in response to both the ongoing infection and unrelated secondary stimulations. This process, hereby referred to as “pDC exhaustion”, favors viral persistence and associates with reduced innate responses and increased susceptibility to secondary opportunistic infections. On the other hand, pDC exhaustion may be a compromise to avoid IFN-I driven immunopathology. In this review we reflect on the mechanisms that initially induce IFN-I and subsequently silence their production by pDCs during a viral infection. While these processes have been long studied across numerous viral infection models, the 2019 coronavirus disease (COVID-19) pandemic has brought their discussion back to the fore, and so we also discuss emerging results related to pDC-IFN-I production in the context of COVID-19.
Keywords:
IFN-I; plasmacytoid dendritic cells; viral infection; LCMV; influenza; HIV-1; HCV; HBV; COVID-19; SARS-CoV-21. Introduction
The Type I interferon (IFN-I) family are antiviral and antineoplastic cytokines critical for the control of most types of viral infection [1,2]. This family includes 13 subtypes of IFN-α in humans (14 in mice) one IFN-β as well as a handful of other gene products (reviewed in [2]). While all cell types can produce IFN-I, plasmacytoid (p) dendritic cells (DCs) produce IFN-I and other interferons at exceptional levels, including all 13 subtypes of IFN-α, IFN-β, and 3 subtypes of IFN-λ, and IFN-τ (reviewed in [3,4,5] and described in [6]). As such, pDCs promote control of multiple types of viral infection (reviewed in [4]). However, despite the critical importance of IFN-I to control viral infections the consequences of IFN-I are not exclusively beneficial. Excessive and/or late IFN-I is associated with increased mortality in several animal models of viral infection (e.g., Influenza [7], arenavirus mediated hemorrhagic fever [8], MERS and SARS-CoV-1 infections in mice [9,10]). Furthermore, persistent IFN-I signaling in a mouse model of chronic viral infection promotes T cell exhaustion and compromises viral control [11,12]. Although it is notable that the addition of recombinant IFN-I early in the same infection promotes viral clearance [13]. Finally, aberrant IFN-I signaling is a signature of many autoimmune disorders several of which have been associated with activation of pDCs (reviewed in [14]).
Despite the importance of pDC derived IFN-I for the control of many viruses, after their initial activation pDCs rapidly lose their capacity to produce these antiviral mediators, a state that we refer to as “pDC exhaustion” (Reviewed in [15]). While this pDC exhaustion can favor prolonged viral replication and opportunistic secondary infections [16,17,18], it may have evolved as a “default” behavior to avoid the potentially harmful effects of sustained IFN-I production. Additionally, this adaptation may also help protect against interferonopathies in the absence of infection.
Here we provide a review of the available literature on pDC IFN-I induction and the mechanisms that support and oppose this important function of pDCs. First, we describe the pathways that promote or suppress pDC IFN-I production on a per-cell basis including modulators of signaling, inhibitory receptors, cytokines, and metabolism. Afterwards, we describe mechanisms that reduce or enhance pDC numbers. Together both types of regulation help to determine the availability of pDC derived IFN-I upon a viral infection.
The COVID-19 pandemic caused by SARS Coronavirus-2 (SARS-CoV-2) has led to rapidly evolving investigation of this disease. These studies have revealed that many of the features of other acute and potentially deadly infections are conserved in COVID-19, including critical roles for IFN-I in both protection and disease. Furthermore, COVID-19 has been associated directly with suppression of pDC IFN-I on a per cell basis [19], as well as with reduced pDC numbers [19,20,21,22]. At the end of this review, we describe emerging literature on pDCs, IFN-I, and COVID-19 and discuss how this relates to the established framework in the field. We will avoid a full review of pDC development, the roles of pDCs in tolerogenic responses, and the role of pDC IFN-I production in cancer. We suggest the reader consults these recent reviews as resources for those topics [5,15,23,24].
2. pDC IFN-I Production after Viral Infections
2.1. Uptake of Nucleic Acids
The first step in recognition of a viral infection by a pDC is the uptake of nucleic acids, and their delivery to toll-like receptor (TLR) containing endosomes. While the uptake of stimulatory oligonucleotides used to study pDC function in vitro primarily occurs through clathrin mediated endocytosis [25], viral material uptake can reach the TLR-containing endosomes through endocytosis or autophagy [26,27]. Furthermore, the viral material sensed can be functional or defective viral particles [28,29], exosomes [30], and even productive infection and replication within the pDC themselves [26]. Additionally, Fc receptors can facilitate uptake of immunocomplexed nucleic acids into pDCs [31] and this can trigger their IFN-I production [32]. Indeed, this process has been implicated in abnormal IFN-I elevation in systemic lupus erythematosus (SLE) [32,33]. In contrast, FcγIIB-based internalization of immune complexes has been shown to oppose IFN-I production in pDCs, and this is associated with reduced IFN-I production in response to Sendai virus (SV) infection when virus-specific-antibodies are present, suggesting this route could exist as a way to quell IFN-I responses [31]. Recently, it has also become well-established that, in some cases, pDCs require cell-cell contact with infected cells to robustly produce IFN-I [34,35,36,37,38,39,40,41], and recent observations in vitro support the idea that an “interferogenic synapse” can develop between pDCs and infected cells [42]. How stimulatory material is exchanged in this synapse, and its potential function in vivo still needs further study. Ultimately, the precise route for nucleic acid uptake depends on the type of viral infection and context, and this route can influence the magnitude of pDC IFN-I responses [31,34].
2.2. Signaling by TLR7/TLR9
The primary mechanism by which pDCs are known to recognize exogenous and endogenous RNA and DNA is through TLRs 7 and 9, respectively. These TLRs are highly expressed in pDCs and stimulation of either can lead to high levels of IFN-I production (Reviewed in ref [43]). TLR7 and TLR9 signal through a combination of MyD88-NF-kB and MyD88-IRF7 pathways (Reviewed in refs [44,45]). The relative use of these signaling pathways depends on the subcellular compartments in which the specific TLR is located and varies with the stimulus [46,47,48]. For example, the multimeric oligonucleotide family of CpG-A localizes to early endosomes where they preferentially activate the MYD88-IRF7 pathway inducing high levels of IFN-I [46,47]. In contrast the monomeric oligonucleotide family of CpG-B localizes to an endolysosomal compartment [46,47]. In the last-mentioned case the MYD88-NF-kB pathway is preferentially activated and leads to production of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNFα) [46]. In pDCs, but not in conventional (c) DCs, the trafficking of CpG-A to early endosomes and their cytokine production relies on signaling through Src kinases Lyn and Fyn [49]. This connection between differential trafficking and signaling has been used to explain the long-standing observation that CpG-A induces higher levels of IFN-I, while CpG-B induces more pro-inflammatory cytokines [50,51]. It is important to note that neither of these cases is absolute as CpG-B induces some production of IFN-I while CpG-A also induces pro-inflammatory cytokines.
2.3. TLR2/12
While most of the work in pDCs focuses on TLR7 and TLR9, pDCs can also express TLR2 and TLR12 [52,53]. As with TLR7 and TLR9, recognition of Toxoplasma gondii profilin by TLR12 is sufficient to stimulate pDCs to produce IL-12. These observations, along with the observation that TLR12 deficient mice are more susceptible to T. gondii infection than wild type animals, suggest a potential role for pDCs in host resistance to this pathogen [52]. However, not all TLRs expressed in pDC stimulate the production of IFN-I or pro-inflammatory cytokines. Indeed, the commensal associated molecule polysaccharide A (PSA), which is sensed via TLR2 [54], has been shown to drive an immunoregulatory phenotype in pDCs that protects mice against 2,4,6-trinitrobenzenesulfonic acid (TNBS) induced colitis [53]. Further research into the roles for these and other TLRs expressed by pDCs are an exciting avenue of future work and will provide deeper insight into how pDC responses are regulated in a variety of contexts.
2.4. Cytosolic Nucleic Acid Sensors
In addition to TLRs pDCs express several cytosolic sensors of nucleic acids, some of which have been established to induce pDC activation and IFN-I production. The cytosolic nucleic acid receptors DHX36 and DHX9 have been shown to bind CpG-A and CpG-B and induce IFN-I and pro-inflammatory cytokine production, respectively [55]. On the other hand, it has been proposed that the cytosolic sensor retinoic acid-inducible gene I (RIG-I) does not induce IFN-I production in pDCs. This assertion is based on IFN-I levels in response to Newcastle disease virus (NDV), which did not change in RIG-I deficient pDCs [56]. However, it has since been shown that in some cases RIG-I can contribute to IFN-I production in pDCs. Specifically, adaptors of RIG-I help drive IFN-I production in pDCs in response to NDV in the absence of IFN-I restriction of viral growth [57]. Additionally, RIG-I may drive pDC responses to the cell-free yellow fever live vaccine (YF-17D) [34]. Though notably, the amount of IFN-I produced as the result of RIG-I signaling is less than that produced in response to TLR7 signaling [34].
Recently the cyclic GMP-AMP synthase (cGAS)-stimulator of IFN genes (STING) pathway has also emerged as a method by which pDCs can recognize and respond to insult. In this pathway the enzyme cGAS recognizes cytosolic dsDNA and makes cyclic GMP-AMP (cGAMP). Cyclic dinucleotides can then be recognized by STING which induces IFN-I expression (Reviewed in ref [58]). Human pDCs express both cGAS and STING and can produce IFN-I in response to both cytosolic dsDNA as well as the STING ligands cGAMP, dAMP, and diGMP [59]. STING stimulation of IFN-I in human pDCs associates primarily with nuclear translocation of IRF3 (in contrast to IRF7 for TLR7 and TLR9) [59,60]. Importantly, potentially as a result of these differences, stimulation with cGAMP induces significantly lower levels of IFN-α, IFN-β, and IFN-λ, that is also produced with slower kinetics, as compared to CpG-A stimulation of TLR9 [60]. Furthermore, STING pre-activation reduces pDC IFN-I production in response to TLR9 ligation [60].
3. Suppression of pDC IFN-I Production after Viral Infections
3.1. pDC Functional Exhaustion
Within a few days after an in vivo viral infection the capacity of pDCs to produce IFN-I is severely reduced [16,61], a phenotype that we now refer to as pDC exhaustion. This was first described after acute and chronic LCMV infection in mice, and coincided with a dramatic attenuation of systemic IFN-I early after its peak at day one post infection [16]. pDC exhaustion was also shown to associate with compromised Natural Killer cell activation, Interferon-γ production and cytotoxicity in response to an unrelated secondary virus [16]. Lee et al. confirmed these findings, and additionally showed pDC exhaustion associated with deficient cross-priming of T cells to an unrelated antigen [61]. Importantly, the pDC exhaustion phenotype described in vivo is consistent with other studies showing that pDCs from patients infected with HIV [62,63,64,65], HCV [66,67,68,69,70] and HBV [71,72,73] as well as macaques infected with SIV [74,75] exhibited reduced per-cell production of IFN-I when re-stimulated ex vivo. Additionally, in HIV-infected humans systemic IFN-I is attenuated early after infection [76], and reduced pDC numbers and/or loss of their ex vivo IFN-I production are correlated with opportunistic infections [18,77,78,79]. Remarkably, this is true even when considered independently of CD4 T cell numbers [77]. It is additionally important to note that IFN-I exhaustion in pDCs is sometimes associated with reduced capacity to produce other cytokines such as TNFα [17]. pDC exhaustion, however, is primarily studied in the context of IFN-I and it is unclear whether other interferons that can also be produced by pDCs (i.e., IFN-λ, and IFN-τ) are likewise suppressed in this condition.
More recently, it has been established that, analogous to T cell receptor (TCR) induction of CD8 T cell exhaustion, in vivo pDC exhaustion in LCMV-infected mice is dependent on TLR7, which reduces the expression of E2-2, a transcription factor essential for pDC development and function [80], via cell-intrinsic signaling in pDCs [17]. Evidence from HIV infection also suggests that persistent stimulation may promote pDC exhaustion as ex vivo IFN-I production by pDCs is reduced in patients with high viral load [62,63], recovers during the administration of successful antiretroviral therapy [65], but becomes suppressed again upon interruption of antiretroviral treatment [63]. Additionally, E2-2 expression is reduced in pDCs from HIV viremic patients [17], and in a human pDC cell line when stimulated with TLR ligands for 2 days [81]. Taken together, these data suggest that persistent TLR engagement may promote pDC IFN-I exhaustion in mice and human pDCs, analogous to what has been described for CD8 T cell exhaustion after chronic stimulation through the TCR [82].
3.2. Inhibitory Receptors
IFN-I production in pDCs can be modified by receptors expressed on their surface. Diverse families of receptors such as BDCA2 [83], Siglec-H [84], ILT7 [85], FcεRI [86], DCIR [87], Tim-3 [64], CD28 [88], E-cadherin [89], and Nkp44 [90] all have established rolls in modulating pDC cytokine production downstream of ligation. Much of this evidence exists ex vivo and the role of most of these receptors in pDC function during in vivo viral infection is not yet established. Even for the few that have been investigated in vivo little is known. Mice deficient in CD28 have enhanced serum IFN-I and increased IFN-I transcript levels in pDCs after either LCMV or MCMV infection, and this associates with improved control of MCMV [88]. Similarly, Siglec-H deficient mice also have increased systemic IFN-I after MCMV infection [91]. In addition, in HIV patients a functionally distinct subset of pDC expressing Tim-3 has been identified, correlating expression of this marker to distinctly reduced IFN-I production capacity in human infection [64].
As blockade of inhibitory receptors has been an effective treatment strategy for the modulation of immune cell functions, it is tempting to speculate that surface inhibitory receptors could be promising targets for enhancing pDC function. For example, the asthma drug Omiluzimab, which acts by sequestering free IgE, drives downregulation of FcεRI (a receptor for IgE) on mast cells and basophils [92]. However, as a side-effect Omiluzimab treatment also enhances IFN-α production capacity in pDCs [93]. This enhancement associated with reduced frequency of rhinovirus detection in a cohort of asthma patients treated with Omiluzimab [94], suggesting that targeting pDC inhibitory receptors has the potential to improve human anti-viral responses in vivo. However, pDC inhibitory receptors may also provide pathogens routes to modulate pDC function. For example, HIV protein Vpu has been shown to subvert anti-viral pDC functions by redistributing BST2 on infected cells. This permits viral exit while still engaging the inhibitory receptor ILT7 on pDCs to suppress IFN-I production [95]. Thus, the multiple inhibitory receptors expressed by pDCs may offer mechanisms by which both hosts, and pathogens can modulate pDC-derived IFN-I in the context of infections.
3.3. Cytokines and Hormones
Many soluble factors can modulate pDC function. Negative regulators of pDC IFN-I production include Transforming Growth Factor Beta (TGFβ), Interleukin (IL)-10, prostaglandin E2 (PGE2), and TNFα [96,97,98,99,100]. While positive regulators include IL-4 [97], IL-7 [97], IL-15 [97], Estrogen [101], and IFN-I itself [97,102,103,104]. As with inhibitory receptors the functions of most of these soluble molecules are primarily established in vitro or ex vivo and the roles these factors play and the mechanisms by which they modulate pDC function during viral infection in vivo are not fully elucidated.
The fact that IFN-I itself enhances pDC IFN-I production is worth noting as availability of autocrine or paracrine IFN-I signaling can enhance the percentage of IFN-I producing pDCs [97,102,103,104], although IFN-I also induces pDC death in HSV-1 infected mice [105]. The role of TNFα is also notable as pDCs also produce TNFα upon TLR [106] or FCεR1 engagement [86,107] and thus this may represent another autocrine and/or paracrine negative feedback loop. Integrating the potential autocrine and/or paracrine effects of IFN-I and TNFα may offer a partial explanation of differences between CpG-B and CpG-A stimulation. For example, it would be predicted that a pDC which starts producing IFN-I would receive autocrine signals that drive it toward further production of IFN-I. In contrast, a pDC producing primarily TNFα would receive feedback that opposes IFN-I production. This has been partially proposed before although these studies considered only the impacts of IFN-I positive feedback [104] and TNFα negative feedback [108,109] in isolation. Further study, and perhaps mathematical modeling may shed light on how to disentangle these feedback loops, as well as the established intrinsic differences in cell signaling, which support this phenomenon.
It is also worth noting the positive impact of Estrogen on pDC function [101]. This is one part of a larger convergence of factors that drive increased pDC responses in people with more than one X chromosome [101,110,111,112,113]. A phenomenon also partially driven by TLR7, which is expressed on the X chromosome but resistant to X chromosome inactivation [114]. Indeed, both X chromosome content and sex hormone levels contribute to increased IFN-I production in mice [111]. Furthermore, a study inclusive of both cis and transgender male and female humans recently demonstrated that both chromosome number and sex hormone levels associate independently with pDC functional capacity in humans as well [115].
3.4. Metabolic Requirements in pDCs
IFN-I responses in pDC require large-scale and rapid synthesis of IFN-I mRNA/protein. The metabolic requirements for this process are just beginning to be uncovered. It has been established that pDCs require both Oxidative metabolism (OxPhos) as well as glycolysis to maintain their function [116,117,118]. Furthermore, mammalian target of rapamycin (mTOR), a central regulator of metabolism, is essential for pDC IFN-I production [119]. While multiple studies agree that pDCs adjust their metabolism after stimulation the details of these changes are still being established, Bajwa et al. show that at 24 h post stimulation with influenza, lactate terminal glycolysis is enhanced in pDCs while OxPhos is slightly suppressed [116]. In contrast Hurley et al. show that 6 hr of influenza or HSV exposure drives a slight increase in oxygen consumption rate (OCR) indicating increased OxPhos [118]. It is likely that the choice of distinct time-points in these studies is responsible for the observed differences. Metabolic changes after stimulation may also provide feedback loops that help control and tune down IFN-I production. Indeed, as high levels of lactate can inhibit pDC IFN-I production [120], then it might be expected that over time enhanced levels of lactate terminal glycolysis after stimulation as described in Bajwa et al. [116] could inhibit pDC IFN-I production. Furthermore, Wu et al. show that IFN-I signaling can enhance basal OCR in both pDCs and epithelial cells, and show oxidative metabolism is essential to pDC function [117]. Thus, this could be part of the mechanism by which IFN-I enhances its own production. Further studies will be necessary to work out the precise details explaining how changes in pDC metabolism in response to environmental cues may tune their function. Furthermore, it will be especially important to assess how pDCs adapt their metabolism after in vivo infection.
4. Reduction of pDC Numbers after Viral Infections
In addition to the direct suppression of pDC IFN-I production by the above mechanisms pDC-derived-IFN-I is also compromised in many infections by a reduction in pDC numbers. This has been well established for HIV-1 [18,62,63,64,65,121,122], HCV [66,67,68,69,70,123,124,125] and HBV [71,72,73] infections, in humans. Reduced numbers of pDCs have also been reported during SIV infection in macaques [74,75,126,127], and LCMV, CMV, HSV, and VSV in mice [13,16,17,61]. The mechanisms by which pDC numbers may be altered include reductions in development from bone marrow (BM) progenitors, increased apoptosis, changes in pDC proliferative capacity, and conversion of pDCs into conventional (c)DC-like cells, as discussed below.
4.1. Compromised pDC Development from Bone Marrow Progenitors
As pDCs are typically short-lived (persisting for only ~3 days in the periphery of C57Bl/6 mice) [128,129] it is expected the entirety of the pDC population will turn over in the course of any infection lasting longer than a few days. However, in non-resolving infections pDC exhaustion and reduced numbers of pDCs are maintained long-term [16]. One possibility is that pDC progenitors are modulated by the infection in a way that they generate fewer and functionally deficient pDCs. Indeed, pDC generation from BM is reduced for long term in mice infected with a persistent LCMV variant, and transiently decreased after infection with an acute LCMV strain [17]. This associates with a numerical reduction in pDC progenitors in the BM, but also extends to a reduced capacity of these progenitors to produce pDCs and reduced expression of E2-2 [17]. Furthermore, the few pDCs generated from BM progenitors from infected mice develop to produce less IFN-I when stimulated ex vivo [17]. Reduced E2-2 in both the progenitors and matured DCs from these cultures raises the possibility that a transcriptional state inherited from progenitors may contribute to the exhaustion phenotype in their pDC progeny. It should, however, be considered as a caveat that virus does persist in these BM cultures and thus may provide persistent TLR7 stimulation to newly generated pDCs driving the observed exhaustion phenotype. Additionally, as advances are made in understanding the development of pDCs, it has become apparent that the BM pDC progenitors analyzed in this study were heterogeneous [130]. Thus, it will now be necessary to distinguish whether a specific depletion of E2-2 high pDC committed progenitors or a general suppression of E2-2 expression across multiple progenitor populations is responsible for these observations.
4.2. Apoptosis
Increased levels of apoptosis in pDCs have been observed in HIV and SIV infections in humans and macaques respectively and HSV infection in mice [75,105,126,131]. For HSV-1 infection in mice IFN-I signaling is at least partially responsible for this effect [105], suggesting that IFN-I driven apoptosis is part of a negative feedback loop limiting pDC IFN-I production after infection. It is also worth considering a role for glucocorticoids (GCs), which can be enhanced after viral infection [132,133] and can also promote pDC apoptosis [134,135,136]. However, as TLR stimulation opposes GC induced apoptosis, it is unclear whether increased GC levels during infection would ultimately result in increased pDC apoptosis in vivo and so further study will be needed to evaluate this.
4.3. Proliferation
Although during persistent LCMV infection pDC development is compromised, and apoptosis is increased, pDC numbers in peripheral lymphoid tissues are not universally decreased after infection. Indeed, at day 10 after LCMV infection there is an increase in pDC numbers in the spleens of infected mice [17]. This corresponded to an expansion of CD4−CCR9− and CD4−CCR9+ pDC subsets with increased proliferative capacity [17]. While CD4−CCR9− and CD4−CCR9+ pDCs have been described in the BM [128,137,138], where they do exhibit proliferative potential as precursors of CD4+CCR9+ pDCs, splenic pDCs are predominantly CCR9+ at steady state and do not proliferate [128,137,138]. In LCMV infection this gain in proliferative subsets is dependent on both IFN-I and TLR7 signaling, in contrast to functional exhaustion which requires only the latter [17]. Notably, the cell-cycle marker Ki67 has also been reported to be enhanced in pDCs from SIV infected macaques [75,126,127]. As development from the BM is compromised at this time, this pool of proliferating pDC in the spleen (and potentially other lymphoid tissues) may perpetuate, at least partially, the pool of functionally exhausted pDCs during infection.
4.4. pDC Conversion during Viral Infection
Both acute and chronic LCMV infection as well as the direct administration of PolyI:C or IFN-I drives pDCs to differentiate into cells resembling CD11b+ cDCs (cDC2s). These pDC-derived-cDCs exhibit phenotypic and functional features of cDC2s both in vitro [139] and in vivo [140]. While it was first observed in viral infection, pDC conversion has also been reported after stimulation with GM-CSF or intestinal epithelium supernatant [137], in Ly49Q− pDC from PolyI:C injected mice [141], and even after pDC transfer in steady-state conditions [138], suggesting this phenomenon may also regulate pDC numbers at steady state.
Recent subdivision of DC subsets has identified a potential intermediate stage in the transition between pDC and cDC2. High dimensional analysis of the DC compartments of humans and mice identified a “bridge” population termed transitional (t)DCs that occupy a spectrum between pDC and cDC2 [142,143]. This population is formed as an intermediate between pDC and cDC2 like fates in in vitro culture [144], shows intermediate E2-2 expression and IFN-I production when compared to pure pDCs and cDC2, and maps inside the recently described populations of “non-canonical DCs” [5] (i.e., Axl+ DC in humans [142,145] and CX3CR1+CD8+ cDC in mice [146,147]) that exhibit features of both pDCs and cDCs. Importantly, tDCs accumulate in the lungs of influenza virus infected mice [143]. This is in line with a model where there is increased differentiation from pDC, through tDC, to cDC2 after infection. While these results are in line with a role for pDC differentiation into tDC/cDC2 as a mechanism for reduced pDC numbers during infection, further work will be necessary to definitively show whether the transition between pDC and cDC2 like cells, that was first reported during in vivo viral infection [139,140], uses tDC as an intermediate, and whether disrupting this phenomenon can restore pDC numbers.
5. pDCs, IFN-I, SARS-CoV-2, and COVID-19
The COVID-19 pandemic caused by SARS-CoV-2 has led to rapidly evolving investigation of this disease and the virus that causes it. Although pDC have been shown to be protective against beta coronavirus infection in mice [148], the direct protective impact of pDC in SARS-CoV-2 infection has not yet been assessed. Still, much work has been done in a short amount of time to discover how IFN-I regulates SARS-CoV-2 infection and associated COVID-19, how this novel virus is sensed by pDCs, and how infection with SARS-CoV-2 modulates pDC population dynamics and function. Here we summarize the most recent work on these subjects as of the writing of this review.
5.1. The Complex Role of IFN-I in COVID-19
The precise role of IFN-I in COVID-19 patients is complex and still emerging. COVID-19 patients that have deficiencies in IFN-I responses as a result of germline mutations or anti-IFN-I autoantibodies are more susceptible to severe disease [149,150,151,152,153]. This is in line with the beneficial role of IFN-I in the control of a variety of viral infections. Furthermore, male identified patients (which typically have low levels of estrogen and only one X chromosome encoding TLR7), are significantly more likely to be hospitalized and develop severe COVID-19 [154], and this could be partly a result of reduced IFN-I production by their pDCs. Notably, use of recombinant IFN-β as a therapeutic for COVID-19 has shown promise in some trials, but not others [155,156,157], and a retrospective study described that early treatment of COVID-19 patients with IFN-α decreased mortality while late treatment resulted in the opposite outcome [158]. In addition, several reports have associated low levels of systemic IFN and reduced IFN-I signatures with severe disease [22,159] while other studies have found positive correlations among IFN-I and severe disease [160,161], although the later may be a consequence of more sustained viral replication that induces more IFN-I in severe cases.
5.2. Sensing of SARS-CoV-2 by pDC
It has been established that pDCs can produce IFN-I in response to stimulation with SARS-CoV-2 in vitro [162]. Much work has been done triangulating the PRR used by pDCs to sense SARS-CoV-2. For example, it has been shown that SARS-CoV-2 possesses more ssRNA fragments capable of being recognized by TLR7 and TLR8 when compared to SARS-CoV [163]. Furthermore, a study looking at humans carrying loss of function mutations in UNC93b or IRAK4, which are respectively essential for TLR endosomal localization and signaling [164,165], has shown that their pDCs fail to produce cytokines in response to SARS-CoV-2 exposure [162]. In the same line, SARS-CoV-2, like other coronaviruses, is an RNA virus and TLR7 is essential for pDC sensing of beta-coronavirus in mice [148]. A definitive role for TLR7 in SARS-CoV-2 was recently demonstrated in patients with familial deficiency for TLR7 function. These patients show little to no production of IFN-α2 in their pDCs after stimulation with SARS-CoV-2, though interestingly production of several other inflammatory cytokines was not significantly impacted in this case [153]. Together these studies provide compelling evidence that pDCs sense SARS-CoV2 via TLR7. Consistent with this, familial deficiency in TLR7 leads to increased susceptibility to severe COVID-19 [151,166]. It is important to note that pDC sensing of SARS-CoV-2 is likely to be independent of SARS-CoV-2 infection of pDCs, since human pDCs do not express the SARS-CoV-2 entry receptor ACE2 or support productive replication [162].
5.3. Reduced pDC Numbers in SARS-CoV-2 Infection
It is also now well established that, as in other human infections, pDCs are reduced in the peripheral blood of COVID-19 patients [19,20,21,22]. The mechanism underlying this phenomenon has not been fully established, but one study has shown that circulating pDCs from COVID-19 patients have an increased apoptosis signature [22]. As expected, increased pDC apoptosis signatures are negatively correlated with pDC numbers, suggesting increased apoptosis may be responsible for decreased pDC numbers in COVID-19 patients [22]. Intriguingly, increased apoptosis in pDCs also positively correlated with severe disease. Given that pDC numbers also correlate with IFN-I signatures in the same patients, this data is suggestive of a model in which pDC are protective against severe COVID-19, but increased apoptosis limits pDC numbers and IFN-I availability in severe disease [22].
5.4. Decreased pDC Function in SARS-CoV-2 Infection
It has been recently shown that pDC from the peripheral blood of COVID-19 patients show reduced IFN-I production in a per-cell basis [19]. Many plausible models exist that might explain this. Several cytokines, including IL-10 and TNFα (discussed above for their potential to suppress pDC function) are elevated in COVID-19 patients [160,167]. Additionally, in COVID-19 patients pDCs show reduced phosphorylation of S6 protein [19], suggesting that mTOR signaling, which supports IFN-I production in pDCs [119], may be compromised in this context. Furthermore, reduction of E2-2 expression upon TLR7 stimulation, as mentioned above for pDC exhaustion in other viral infections [17], is a likely mechanism driving pDC hypo-functionality in COVID-19 patients, although E2-2 levels have not been studied in pDC from SARS-CoV-2 infected cohorts.
5.5. Proposed Model for the Biology and Role of pDCs in SARS-CoV-2 Infection
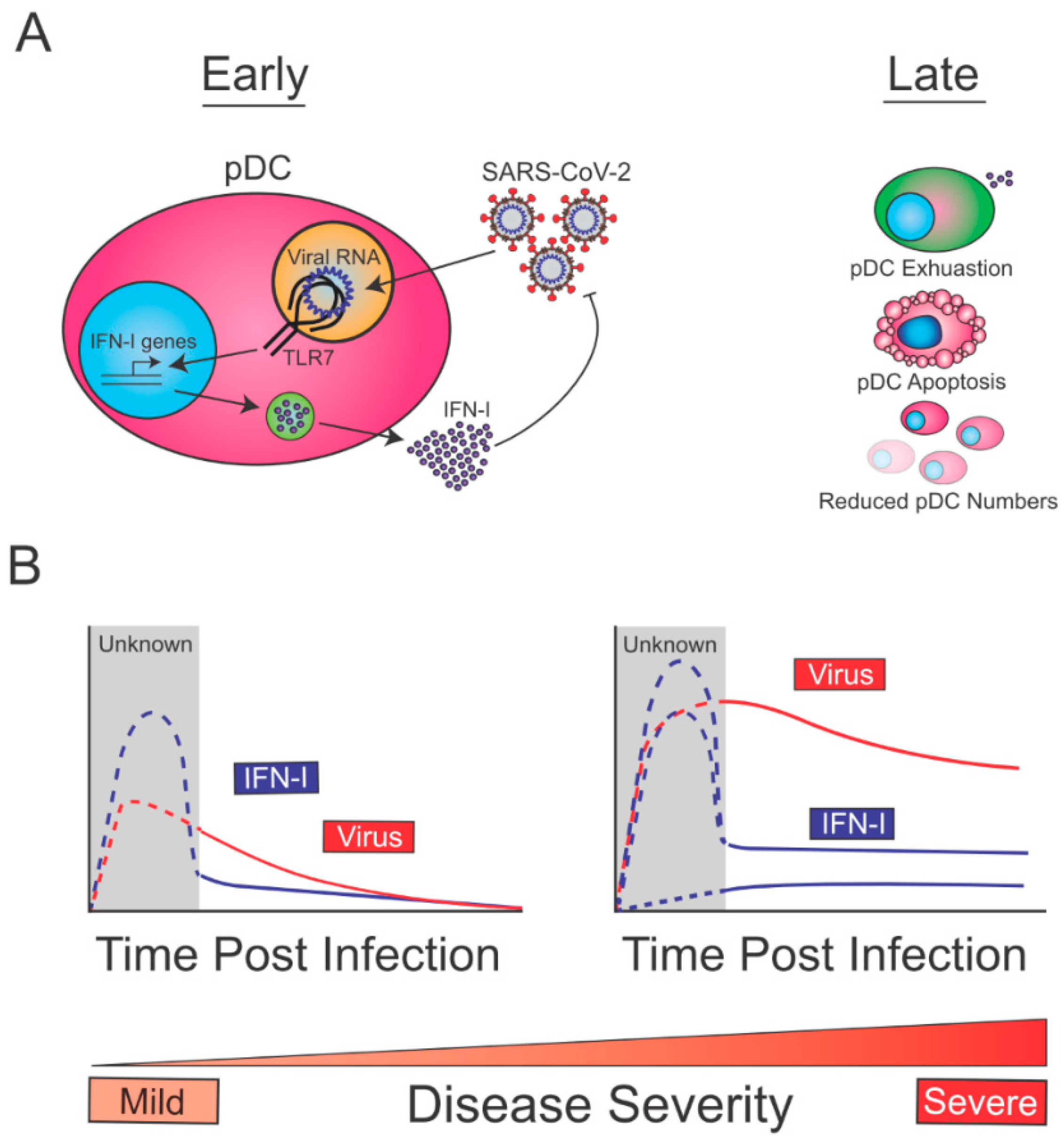
Overall, the aforementioned studies are compatible with a model in which, at early times post-infection, pDCs are sensing SARS-CoV-2 through TLR7, which drives the production of IFN-I, and contains SARS-CoV-2 replication (Figure 1A, left). At later time points post-infection, reduction in pDC numbers (exacerbated in severe cases due to increased apoptosis) and decreased pDC function (exhaustion), may lead to reduced pDC-derived IFN-I (Figure 1A, right). Given that early pDC-derived IFN-I is expected to be beneficial for the host to control SARS-CoV-2, this immune function is likely intact in patients with mild disease (Figure 1B, left). On the other hand, COVID-19 severe cases may show diverse profiles of pDC-derived-IFN-I (Figure 1B, right). This may range from defective pDC IFN-I production (e.g., patients with TLR7 loss of function mutations) to intact or even enhanced pDC responses in patients infected with high viral inoculum or fast-spreading variants, or in individuals with deficiencies in other innate defense mechanisms. In the latter patients, sustained viral replication would perpetuate IFN-I levels from a variety of infected cells thereby leading to increased IFN-I signatures. While Arunachalam et al. did not observe any correlation between severity of disease and pDC intrinsic loss of function in COVID-19 patients (albeit with a limited cohort) [19], it is still likely that enhancing or sustaining pDC-derived-IFN-I during the early phase of SARS-CoV-2 infection would help contain the viral spread and favor a rapid resolution of the disease. Future work aiming at better defining the mechanisms by which pDC numbers are reduced and how pDC function is suppressed in COVID-19 patients, may allow us to design therapies to restore/enhance pDC-derived IFN-I early enough to quickly contain viral spread.
6. Discussion
IFN-I are powerful cytokines for the control of viruses, but their power also has the potential to harm their host. The diverse mechanisms of IFN-I control in pDCs likely represent an evolved abundance of caution surrounding the power of pDCs to produce IFN-I. Therefore, these suppressive systems are analogous to PD1 expression in CD8 T cells, which prevents autoimmunity and immunopathology, but also favors sustained infections or tumors [168]. They likely represent a natural mechanism of immune regulation more than direct viral strategies to suppress immune responses. This is emphasized most recently in COVID-19 infection where IFN-I can protect the host when administered early [158], and IFN-I deficiencies are associated with severe disease [149,150,151], but, in contrast, late IFN-I treatment may be detrimental [158].
The commonalities that COVID-19 shows with previously-studied infections reemphasizes the importance of the knife’s-edge control that evolution has maintained with interferon. These commonalities also emphasize that the work that has been done understanding IFN-I regulation in other systems offers opportunities to quickly develop strategies to combat novel viral diseases. Beyond viral infection, our understanding of these mechanisms also has implications for the treatment of cancer as pDCs also become exhausted within the tumor microenvironment (Reviewed in [15]). Similarly, for autoimmunity and chronic inflammatory diseases where harnessing of the IFN-I natural braking systems may provide novel therapies. Altogether, the power of pDCs to produce IFN-I is paired by evolution with multiple pathways by which to contain it. By working toward a deeper understanding of those pathways we can harness pDCs for the treatment of a wide spectrum of human illnesses.
Author Contributions
Conceptualization T.T.G. and E.I.Z.; writing—original draft preparation, T.T.G. and E.I.Z.; writing—review and editing, T.T.G. and E.I.Z.; funding acquisition, E.I.Z. Both authors have read and agreed to the published version of the manuscript.
Funding
Research in the Zuniga lab is supported by NIH grants AI145314, AI081923, AI113923 and AI132122.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank members of the Zuniga lab for fruitful discussions. As space was limited, we were unable to cite all available studies, and so apologize to any authors who feel their studies were not adequately represented in our accounting.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Fitzgerald-Bocarsly, P.; Feng, D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007, 89, 843–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Kanzler, H.; Duramad, O.; Cao, W.; Liu, Y.-J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 2006, 107, 2423–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldstone, M.B.A.; Ware, B.C.; Horton, L.E.; Welch, M.J.; Aiolfi, R.; Zarpellon, A.; Ruggeri, Z.M.; Sullivan, B.M. Lymphocytic choriomeningitis virus Clone 13 infection causes either persistence or acute death dependent on IFN-1, cytotoxic T lymphocytes (CTLs), and host genetics. Proc. Natl. Acad. Sci. USA 2018, 115, E7814–E7823. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.F.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B.A. Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and Magnitude of Type I Interferon Responses by Distinct Sensors Impact CD8 T Cell Exhaustion and Chronic Viral Infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Lee-Kirsch, M.A. The Type I Interferonopathies. Annu. Rev. Med. 2017, 68, 297–315. [Google Scholar] [CrossRef]
- Greene, T.T.; Jo, Y.R.; Zuniga, E.I. Infection and cancer suppress pDC derived IFN-I. Curr. Opin. Immunol. 2020, 66, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.I.; Liou, L.-Y.; Mack, L.; Mendoza, M.; Oldstone, M.B.A. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 2008, 4, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macal, M.; Jo, Y.; Dallari, S.; Chang, A.Y.; Dai, J.; Swaminathan, S.; Wehrens, E.J.; Fitzgerald-Bocarsly, P.; Zúñiga, E.I. Self-Renewal and Toll-like Receptor Signaling Sustain Exhausted Plasmacytoid Dendritic Cells during Chronic Viral Infection. Immunity 2018, 48, 730–744.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soumelis, V.; Scott, I.; Gheyas, F.; Bouhour, D.; Cozon, G.; Cotte, L.; Huang, L.; Levy, J.A.; Liu, Y.-J. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood 2001, 98, 906–912. [Google Scholar] [CrossRef] [Green Version]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.A.P.M.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tsang, O.T.Y.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef]
- Peruzzi, B.; Bencini, S.; Capone, M.; Mazzoni, A.; Maggi, L.; Salvati, L.; Vanni, A.; Orazzini, C.; Nozzoli, C.; Morettini, A.; et al. Quantitative and qualitative alterations of circulating myeloid cells and plasmacytoid DC in SARS-CoV-2 infection. Immunology 2020, 161, 345–353. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; del Barrio, I.D.M.; Das, A.; Fish, M.; Monin, L.; Muñoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Martins, A.J.; Lau, W.W.; Rachmaninoff, N.; Chen, J.; Imberti, L.; Mostaghimi, D.; Fink, D.L.; Burbelo, P.D.; Dobbs, K.; et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 2021, 184, 1836–1857.e22. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Su, L. A pathogenic role of plasmacytoid dendritic cells in autoimmunity and chronic viral infection. J. Exp. Med. 2019, 216, 1974–1985. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, S.; Herfs, M.; Delvenne, P.; Hubert, P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: Insight into the molecular mechanisms. J. Leukoc. Biol. 2013, 93, 343–352. [Google Scholar] [CrossRef]
- Latz, E.; Schoenemeyer, A.; Visintin, A.; Fitzgerald, K.A.; Monks, B.G.; Knetter, C.F.; Lien, E.; Nilsen, N.J.; Espevik, T.; Golenbock, D.T. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 2004, 5, 190–198. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [Green Version]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like Receptor 9–mediated Recognition of Herpes Simplex Virus-2 by Plasmacytoid Dendritic Cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Feldman, S.B.; Ferraro, M.; Zheng, H.M.; Patel, N.; Gould-Fogerite, S.; Fitzgerald-Bocarsly, P. Viral Induction of Low Frequency Interferon-α Producing Cells. Virology 1994, 204, 1–7. [Google Scholar] [CrossRef]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Flores, M.; Chew, C.; Tyan, K.; Huang, W.Q.; Salem, A.; Clynes, R. FcγRIIB Prevents Inflammatory Type I IFN Production from Plasmacytoid Dendritic Cells during a Viral Memory Response. J. Immunol. 2015, 194, 4240–4250. [Google Scholar] [CrossRef] [Green Version]
- Båve, U.; Magnusson, M.; Eloranta, M.-L.; Perers, A.; Alm, G.V.; Rönnblom, L. FcγRIIa Is Expressed on Natural IFN-α-Producing Cells (Plasmacytoid Dendritic Cells) and Is Required for the IFN-α Production Induced by Apoptotic Cells Combined with Lupus IgG. J. Immunol. 2003, 171, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Bruni, D.; Chazal, M.; Sinigaglia, L.; Chauveau, L.; Schwartz, O.; Desprès, P.; Jouvenet, N. Viral entry route determines how human plasmacytoid dendritic cells produce type i interferons. Sci. Signal. 2015, 8, ra25. [Google Scholar] [CrossRef]
- Décembre, E.; Assil, S.; Hillaire, M.L.B.; Dejnirattisai, W.; Mongkolsapaya, J.; Screaton, G.R.; Davidson, A.D.; Dreux, M. Sensing of Immature Particles Produced by Dengue Virus Infected Cells Induces an Antiviral Response by Plasmacytoid Dendritic Cells. PLoS Pathog. 2014, 10, e1004434. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Li, Y.; McKnight, K.L.; Hensley, L.; Lanford, R.E.; Walker, C.M.; Lemon, S.M. Human pDCs preferentially sense enveloped hepatitis A virions. J. Clin. Investig. 2015, 125, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Nicolás, O.; Auray, G.; Sautter, C.A.; Rappe, J.C.F.; McCullough, K.C.; Ruggli, N.; Summerfield, A. Sensing of Porcine Reproductive and Respiratory Syndrome Virus-Infected Macrophages by Plasmacytoid Dendritic Cells. Front. Microbiol. 2016, 7, 771. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.W.; Pothlichet, J.; Schilte, C.; Chaperot, L.; Plumas, J.; Randall, R.E.; Si-Tahar, M.; et al. Innate Sensing of HIV-Infected Cells. PLoS Pathog. 2011, 7, e1001284. [Google Scholar] [CrossRef] [Green Version]
- Wieland, S.F.; Takahashi, K.; Boyd, B.; Whitten-Bauer, C.; Ngo, N.; de la Torre, J.-C.; Chisari, F.V. Human Plasmacytoid Dendritic Cells Sense Lymphocytic Choriomeningitis Virus-Infected Cells In Vitro. J. Virol. 2014, 88, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Yun, T.J.; Igarashi, S.; Zhao, H.; Perez, O.A.; Pereira, M.R.; Zorn, E.; Shen, Y.; Goodrum, F.; Rahman, A.; Sims, P.A.; et al. Human plasmacytoid dendritic cells mount a distinct antiviral response to virus-infected cells. Sci. Immunol. 2021, 6, eabc7302. [Google Scholar] [CrossRef]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assil, S.; Coléon, S.; Dong, C.; Décembre, E.; Sherry, L.; Allatif, O.; Webster, B.; Dreux, M. Plasmacytoid Dendritic Cells and Infected Cells Form an Interferogenic Synapse Required for Antiviral Responses. Cell Host Microbe 2019, 25, 730–745.e6. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Cao, W.; Liu, Y.-J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef]
- Blasius, A.L.; Beutler, B. Intracellular Toll-like Receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Ohba, Y.; Yanai, H.; Hegishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef]
- Guiducci, C.; Ott, G.; Chan, J.H.; Damon, E.; Calacsan, C.; Matray, T.; Lee, K.D.; Coffman, R.L.; Barrat, F.J. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J. Exp. Med. 2006, 203, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Sasai, M.; Linehan, M.M.; Iwasaki, A. Bifurcation of toll-like receptor 9 signaling by adaptor protein 3. Science 2010, 329, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Dallari, S.; Macal, M.; Loureiro, M.E.; Jo, Y.; Swanson, L.; Hesser, C.; Ghosh, P.; Zuniga, E.I. Src family kinases Fyn and Lyn are constitutively activated and mediate plasmacytoid dendritic cell responses. Nat. Commun. 2017, 8, 14830. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, N.; Antonenko, S.; Liu, Y.-J. Distinct CpG DNA and Polyinosinic-Polycytidylic Acid Double-Stranded RNA, Respectively, Stimulate CD11c − Type 2 Dendritic Cell Precursors and CD11c + Dendritic Cells to Produce Type I IFN. J. Immunol. 2001, 166, 2291–2295. [Google Scholar] [CrossRef] [Green Version]
- Kerkmann, M.; Rothenfusser, S.; Hornung, V.; Towarowski, A.; Wagner, M.; Sarris, A.; Giese, T.; Endres, S.; Hartmann, G. Activation with CpG-A and CpG-B Oligonucleotides Reveals Two Distinct Regulatory Pathways of Type I IFN Synthesis in Human Plasmacytoid Dendritic Cells. J. Immunol. 2003, 170, 4465–4474. [Google Scholar] [CrossRef] [Green Version]
- Koblansky, A.A.; Jankovic, D.; Oh, H.; Hieny, S.; Sungnak, W.; Mathur, R.; Hayden, M.S.; Akira, S.; Sher, A.; Ghosh, S. Recognition of Profilin by Toll-like Receptor 12 Is Critical for Host Resistance to Toxoplasma gondii. Immunity 2013, 38, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.; Erturk-Hasdemir, D.; Ochoa-Reparaz, J.; Reinecker, H.C.; Kasper, D.L. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe 2014, 15, 413–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-like receptor pathway establishes commensal gut colonization. Science 2011, 332, 974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Pazhoor, S.; Bao, M.; Zhang, Z.; Hanabuchi, S.; Facchinetti, V.; Bover, L.; Plumas, J.; Chaperot, L.; Qin, J.; et al. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase a helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15181–15186. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, Y.; Kumar, H.; Koyama, S.; Kawai, T.; Takeuchi, O.; Akira, S. Cutting Edge: TLR-Dependent Viral Recognition Along with Type I IFN Positive Feedback Signaling Masks the Requirement of Viral Replication for IFN-α Production in Plasmacytoid Dendritic Cells. J. Immunol. 2009, 182, 3960–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Bode, C.; Fox, M.; Tewary, P.; Steinhagen, A.; Ellerkmann, R.K.; Klinman, D.; Baumgarten, G.; Hornung, V.; Steinhagen, F. Human plasmacytoid dendritic cells elicit a Type I Interferon response by sensing DNA via the cGAS-STING signaling pathway. Eur. J. Immunol. 2016, 46, 1615–1621. [Google Scholar] [CrossRef]
- Deb, P.; Dai, J.; Singh, S.; Kalyoussef, E.; Fitzgerald-Bocarsly, P. Triggering of the cGAS–STING Pathway in Human Plasmacytoid Dendritic Cells Inhibits TLR9-Mediated IFN Production. J. Immunol. 2020, 205, 223–236. [Google Scholar] [CrossRef]
- Lee, L.N.; Burke, S.; Montoya, M.; Borrow, P. Multiple Mechanisms Contribute to Impairment of Type 1 Interferon Production during Chronic Lymphocytic Choriomeningitis Virus Infection of Mice. J. Immunol. 2009, 182, 7178–7189. [Google Scholar] [CrossRef] [Green Version]
- Feldman, S.; Stein, D.; Amrute, S.; Denny, T.; Garcia, Z.; Kloser, P.; Sun, Y.; Megjugorac, N.; Fitzgerald-Bocarsly, P. Decreased Interferon-α Production in HIV-Infected Patients Correlates with Numerical and Functional Deficiencies in Circulating Type 2 Dendritic Cell Precursors. Clin. Immunol. 2001, 101, 201–210. [Google Scholar] [CrossRef]
- Tilton, J.C.; Manion, M.M.; Luskin, M.R.; Johnson, A.J.; Patamawenu, A.A.; Hallahan, C.W.; Cogliano-Shutta, N.A.; Mican, J.M.; Davey, R.T.; Kottilil, S.; et al. Human Immunodeficiency Virus Viremia Induces Plasmacytoid Dendritic Cell Activation In Vivo and Diminished Alpha Interferon Production In Vitro. J. Virol. 2008, 82, 3997–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.A.; Clayton, K.L.; Mujib, S.; Zhang, H.; Rahman, A.K.M.N.; Liu, J.; Yue, F.Y.; Benko, E.; Kovacs, C.; Ostrowski, M.A. Tim-3 is a Marker of Plasmacytoid Dendritic Cell Dysfunction during HIV Infection and Is Associated with the Recruitment of IRF7 and p85 into Lysosomes and with the Submembrane Displacement of TLR9. J. Immunol. 2017, 198, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fu, J.; Zhao, Q.; He, Y.; Jin, L.; Zhang, H.; Yao, J.; Zhang, L.; Wang, F.-S. Differential Restoration of Myeloid and Plasmacytoid Dendritic Cells in HIV-1-Infected Children after Treatment with Highly Active Antiretroviral Therapy. J. Immunol. 2006, 176, 5644–5651. [Google Scholar] [CrossRef]
- Kanto, T.; Inoue, M.; Miyatake, H.; Sato, A.; Sakakibara, M.; Yakushijin, T.; Oki, C.; Itose, I.; Hiramatsu, N.; Takehara, T.; et al. Reduced Numbers and Impaired Ability of Myeloid and Plasmacytoid Dendritic Cells to Polarize T Helper Cells in Chronic Hepatitis C Virus Infection. J. Infect. Dis. 2004, 190, 1919–1926. [Google Scholar] [CrossRef] [Green Version]
- Ulsenheimer, A.; Gerlach, J.T.; Jung, M.-C.; Gruener, N.; Wächtler, M.; Backmund, M.; Santantonio, T.; Schraut, W.; Heeg, M.H.J.; Schirren, C.A.; et al. Plasmacytoid dendritic cells in acute and chronic hepatitis C virus infection. Hepatology 2005, 41, 643–651. [Google Scholar] [CrossRef]
- Goutagny, N.; Vieux, C.; Decullier, E.; Ligeoix, B.; Epstein, A.; Trépo, C.; Couzigou, P.; Inchauspé, G.; Bain, C. Quantification and Functional Analysis of Plasmacytoid Dendritic Cells in Patients with Chronic Hepatitis C Virus Infection. J. Infect. Dis. 2004, 189, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, W.K.; Curbishley, S.M.; Goddard, S.; Alabraba, E.; Shaw, J.; Youster, J.; McKeating, J.; Adams, D.H. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J. Hepatol. 2007, 47, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Chang, S.; Kodys, K.; Mandrekar, P.; Bakis, G.; Cormier, M.; Szabo, G. Hepatitis C Virus (HCV) Core Protein-Induced, Monocyte-Mediated Mechanisms of Reduced IFN-α and Plasmacytoid Dendritic Cell Loss in Chronic HCV Infection. J. Immunol. 2006, 177, 6758–6768. [Google Scholar] [CrossRef] [Green Version]
- van der Molen, R.G.; Sprengers, D.; Binda, R.S.; de Jong, E.C.; Niesters, H.G.M.; Kusters, J.G.; Kwekkeboom, J.; Janssen, H.L.A. Functional impairment of myeloid and plasmacytoid dendritic cells of patients with chronic hepatitis B. Hepatology 2004, 40, 738–746. [Google Scholar] [CrossRef]
- Xie, Q.; Shen, H.-C.; Jia, N.-N.; Wang, H.; Lin, L.-Y.; An, B.-Y.; Gui, H.-L.; Guo, S.-M.; Cai, W.; Yu, H.; et al. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect. 2009, 11, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-Z.; Wang, M.; Li, H.-W.; Zhuang, H.; Xu, D.; Wang, F.-S. Decreased Frequency and Function of Circulating Plasmocytoid Dendritic Cells (pDC) in Hepatitis B Virus Infected Humans. J. Clin. Immunol. 2004, 24, 637–646. [Google Scholar] [CrossRef]
- Malleret, B.; Manéglier, B.; Karlsson, I.; Lebon, P.; Nascimbeni, M.; Perié, L.; Brochard, P.; Delache, B.; Calvo, J.; Andrieu, T.; et al. Primary infection with simian immunodeficiency virus: Plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood 2008, 112, 4598–4608. [Google Scholar] [CrossRef]
- Bruel, T.; Dupuy, S.; Démoulins, T.; Rogez-Kreuz, C.; Dutrieux, J.; Corneau, A.; Cosma, A.; Cheynier, R.; Dereuddre-Bosquet, N.; Le Grand, R.; et al. Plasmacytoid Dendritic Cell Dynamics Tune Interferon-Alfa Production in SIV-Infected Cynomolgus Macaques. PLoS Pathog. 2014, 10, e1003915. [Google Scholar] [CrossRef] [Green Version]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a Striking Systemic Cytokine Cascade prior to Peak Viremia in Acute Human Immunodeficiency Virus Type 1 Infection, in Contrast to More Modest and Delayed Responses in Acute Hepatitis B and C Virus Infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegal, F.P.; Fitzgerald-Bocarsly, P.; Holland, B.K.; Shodell, M. Interferon-α generation and immune reconstitution during antiretroviral therapy for human immunodeficiency virus infection. AIDS 2001, 15, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Fitzgerald, P.A.; Siegal, F.P. Severe Acquired Immune Deficiency Syndrome in Male Homosexuals: Diminished Capacity to Make Interferon-a in Vitro Associated with Severe Opportunistic Infections. J. Infect. Dis. 1983, 148, 962–966. [Google Scholar] [CrossRef]
- Siegal, F.P.; Lopez, C.; Fitzgerald, P.A.; Shah, K.; Baron, P.; Leiderman, I.Z.; Imperato, D.; Landesman, S. Opportunistic infections in acquired immune deficiency syndrome result from synergistic defects of both the natural and adaptive components of cellular immunity. J. Clin. Investig. 1986, 78, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisse, B.; Caton, M.L.; Lehner, M.; Maeda, T.; Scheu, S.; Locksley, R.; Holmberg, D.; Zweier, C.; den Hollander, N.S.; Kant, S.G.; et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 2008, 135, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, H.S.; Cisse, B.; Bunin, A.; Lewis, K.L.; Reizis, B. Continuous Expression of the Transcription Factor E2-2 Maintains the Cell Fate of Mature Plasmacytoid Dendritic Cells. Immunity 2010, 33, 905–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Günther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a Novel Plasmacytoid Dendritic Cell–specific Type II C-type Lectin, Mediates Antigen Capture and Is a Potent Inhibitor of Interferon α/β Induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Cella, M.; Maldonado, J.; Takai, T.; Colonna, M. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood 2006, 107, 2474–2476. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Rosen, D.B.; Ito, T.; Bover, L.; Bao, M.; Watanabe, G.; Yao, Z.; Zhang, L.; Lanier, L.L.; Liu, Y.-J. Plasmacytoid dendritic cell–specific receptor ILT7–FcεRIγ inhibits Toll-like receptor–induced interferon production. J. Exp. Med. 2006, 203, 1399–1405. [Google Scholar] [CrossRef] [Green Version]
- Novak, N.; Allam, J.-P.; Hagemann, T.; Jenneck, C.; Laffer, S.; Valenta, R.; Kochan, J.; Bieber, T. Characterization of FcεRI-bearing CD123+ blood dendritic cell antigen-2+ plasmacytoid dendritic cells in atopic dermatitis. J. Allergy Clin. Immunol. 2004, 114, 364–370. [Google Scholar] [CrossRef]
- Meyer-Wentrup, F.; Benitez-Ribas, D.; Tacken, P.J.; Punt, C.J.A.; Figdor, C.G.; de Vries, I.J.M.; Adema, G.J. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-α production. Blood 2008, 111, 4245–4253. [Google Scholar] [CrossRef] [Green Version]
- Macal, M.; Tam, M.A.; Hesser, C.; Di Domizio, J.; Leger, P.; Gilliet, M.; Zuniga, E.I. CD28 Deficiency Enhances Type I IFN Production by Murine Plasmacytoid Dendritic Cells. J. Immunol. 2016, 196, 1900–1909. [Google Scholar] [CrossRef]
- Bi, E.; Li, R.; Bover, L.C.; Li, H.; Su, P.; Ma, X.; Huang, C.; Wang, Q.; Liu, L.; Yang, M.; et al. E-cadherin expression on multiple myeloma cells activates tumor-promoting properties in plasmacytoid DCs. J. Clin. Investig. 2018, 128, 4821–4831. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, A.; Cella, M.; Kondo, T.; Colonna, M. Paradoxic inhibition of human natural interferon-producing cells by the activating receptor NKp44. Blood 2005, 106, 2076–2082. [Google Scholar] [CrossRef] [Green Version]
- Puttur, F.; Arnold-Schrauf, C.; Lahl, K.; Solmaz, G.; Lindenberg, M.; Mayer, C.T.; Gohmert, M.; Swallow, M.; van Helt, C.; Schmitt, H.; et al. Absence of Siglec-H in MCMV Infection Elevates Interferon Alpha Production but Does Not Enhance Viral Clearance. PLoS Pathog. 2013, 9, e1003648. [Google Scholar] [CrossRef] [Green Version]
- Thomson, N.C.; Chaudhuri, R. Omalizumab: Clinical use for the management of asthma. Clin. Med. Insights Circ. Respir. Pulm. Med. 2011, 6, 27–40. [Google Scholar] [CrossRef]
- Gill, M.A.; Liu, A.H.; Calatroni, A.; Krouse, R.Z.; Shao, B.; Schiltz, A.; Gern, J.E.; Togias, A.; Busse, W.W. Enhanced plasmacytoid dendritic cell antiviral responses after omalizumab. J. Allergy Clin. Immunol. 2018, 141, 1735–1743.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquivel, A.; Busse, W.W.; Calatroni, A.; Togias, A.G.; Grindle, K.G.; Bochkov, Y.A.; Gruchalla, R.S.; Kattan, M.; Kercsmar, C.M.; Hershey, G.K.; et al. Effects of Omalizumab on Rhinovirus Infections, Illnesses, and exacerbations of asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Bego, M.G.; Côté, É.; Aschman, N.; Mercier, J.; Weissenhorn, W.; Cohen, É.A. Vpu Exploits the Cross-Talk between BST2 and the ILT7 Receptor to Suppress Anti-HIV-1 Responses by Plasmacytoid Dendritic Cells. PLoS Pathog. 2015, 11, e1005024. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.-D.; Faget, J.; Mithieux, F.; Cassignol, A.; et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Gary-Gouy, H.; Lebon, P.; Dalloul, A.H. Type I Interferon Production by Plasmacytoid Dendritic Cells and Monocytes Is Triggered by Viruses, but the Level of Production Is Controlled by Distinct Cytokines. J. Interf. Cytokine Res. 2002, 22, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian-Ding, I.; Schäfer, M.; Hartmann, E.; Pries, R.; Parcina, M.; Schneider, P.; Giese, T.; Endres, S.; Wollenberg, B.; Hartmann, G. Tumour-derived prostaglandin E 2 and transforming growth factor-β synergize to inhibit plasmacytoid dendritic cell-derived interferon-α. Immunology 2009, 128, 439–450. [Google Scholar] [CrossRef]
- Li, L.; Liu, S.; Zhang, T.; Pan, W.; Yang, X.; Cao, X. Splenic Stromal Microenvironment Negatively Regulates Virus-Activated Plasmacytoid Dendritic Cells through TGF-β. J. Immunol. 2008, 180, 2951–2956. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Ito, T.; Ozaki, Y.; Tanijiri, T.; Yokoi, T.; Nakamura, K.; Takebayashi, M.; Amakawa, R.; Fukuhara, S. Prostaglandin E2 is a negative regulator on human plasmacytoid dendritic cells. Immunology 2006, 119, 36–42. [Google Scholar] [CrossRef]
- Seillet, C.; Laffont, S.; Trémollières, F.; Rouquié, N.; Ribot, C.; Arnal, J.F.; Douin-Echinard, V.; Gourdy, P.; Guéry, J.C. The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor α signaling. Blood 2012, 119, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asselin-Paturel, C.; Brizard, G.; Chemin, K.; Boonstra, A.; O’Garra, A.; Vicari, A.; Trinchieri, G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J. Exp. Med. 2005, 201, 1157–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimmers, F.; Subedi, N.; van Buuringen, N.; Heister, D.; Vivié, J.; Beeren-Reinieren, I.; Woestenenk, R.; Dolstra, H.; Piruska, A.; Jacobs, J.F.M.; et al. Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-alpha production by plasmacytoid dendritic cells. Nat. Commun. 2018, 9, 3317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kaiser, V.; Beier, E.; Bechheim, M.; Guenthner-Biller, M.; Ablasser, A.; Berger, M.; Endres, S.; Hartmann, G.; Hornung, V. Self-priming determines high type I IFN production by plasmacytoid dendritic cells. Eur. J. Immunol. 2014, 44, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Wang, Y.; Vermi, W.; Gilfillan, S.; Schreiber, R.D.; Colonna, M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 2011, 208, 2367–2374. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, N.; Antonenko, S.; Lau, J.Y.-N.; Liu, Y.-J. Natural Interferon α/β–Producing Cells Link Innate and Adaptive Immunity. J. Exp. Med. 2000, 192, 219–226. [Google Scholar] [CrossRef]
- Schroeder, J.T.; Bieneman, A.P.; Xiao, H.; Chichester, K.L.; Vasagar, K.; Saini, S.; Liu, M.C. TLR9- and FcεRI-Mediated Responses Oppose One Another in Plasmacytoid Dendritic Cells by Down-Regulating Receptor Expression. J. Immunol. 2005, 175, 5724–5731. [Google Scholar] [CrossRef] [Green Version]
- Palucka, A.K.; Blanck, J.P.; Bennett, L.; Pascual, V.; Banchereau, J. Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc. Natl. Acad. Sci. USA 2005, 102, 3372–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psarras, A.; Antanaviciute, A.; Alase, A.; Carr, I.; Wittmann, M.; Emery, P.; Tsokos, G.C.; Vital, E.M. TNF-α Regulates Human Plasmacytoid Dendritic Cells by Suppressing IFN-α Production and Enhancing T Cell Activation. J. Immunol. 2021, 206, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Chang, J.J.; Chan, E.S.; Pollard, R.B.; Sidhu, H.K.; Kulkarni, S.; Wen, T.F.; Lindsay, R.J.; Orellana, L.; Mildvan, D.; et al. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat. Med. 2009, 15, 955–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffont, S.; Rouquié, N.; Azar, P.; Seillet, C.; Plumas, J.; Aspord, C.; Guéry, J.-C. X-Chromosome Complement and Estrogen Receptor Signaling Independently Contribute to the Enhanced TLR7-Mediated IFN-α Production of Plasmacytoid Dendritic Cells from Women. J. Immunol. 2014, 193, 5444–5452. [Google Scholar] [CrossRef] [Green Version]
- Berghöfer, B.; Frommer, T.; Haley, G.; Fink, L.; Bein, G.; Hackstein, H. TLR7 Ligands Induce Higher IFN-α Production in Females. J. Immunol. 2006, 177, 2088–2096. [Google Scholar] [CrossRef] [Green Version]
- Griesbeck, M.; Ziegler, S.; Laffont, S.; Smith, N.; Chauveau, L.; Tomezsko, P.; Sharei, A.; Kourjian, G.; Porichis, F.; Hart, M.; et al. Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN- Production in Women. J. Immunol. 2015, 195, 5327–5336. [Google Scholar] [CrossRef] [Green Version]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, K.; Peckham, H.; Radziszewska, A.; Menon, M.; Oliveri, P.; Simpson, F.; Deakin, C.T.; Lee, S.; Ciurtin, C.; Butler, G.; et al. Sex and pubertal differences in the type 1 interferon pathway associate with both X chromosome number and serum sex hormone concentration. Front. Immunol. 2019, 10, 3167. [Google Scholar] [CrossRef]
- Bajwa, G.; DeBerardinis, R.J.; Shao, B.; Hall, B.; Farrar, J.D.; Gill, M.A. Cutting Edge: Critical Role of Glycolysis in Human Plasmacytoid Dendritic Cell Antiviral Responses. J. Immunol. 2016, 196, 2004–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.-H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Hurley, H.J.; Dewald, H.; Rothkopf, Z.S.; Singh, S.; Jenkins, F.; Deb, P.; De, S.; Barnes, B.J.; Fitzgerald-Bocarsly, P. Frontline Science: AMPK regulates metabolic reprogramming necessary for interferon production in human plasmacytoid dendritic cells. J. Leukoc. Biol. 2021, 109, 299–308. [Google Scholar] [CrossRef]
- Cao, W.; Manicassamy, S.; Tang, H.; Kasturi, S.P.; Pirani, A.; Murthy, N.; Pulendran, B. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat. Immunol. 2008, 9, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; Bandopadhyay, P.; Sarif, J.; D’Rozario, R.; Paul, S.; et al. Lactate Induces Pro-tumor Reprogramming in Intratumoral Plasmacytoid Dendritic Cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chehimi, J.; Campbell, D.E.; Azzoni, L.; Bacheller, D.; Papasavvas, E.; Jerandi, G.; Mounzer, K.; Kostman, J.; Trinchieri, G.; Montaner, L.J. Persistent Decreases in Blood Plasmacytoid Dendritic Cell Number and Function Despite Effective Highly Active Antiretroviral Therapy and Increased Blood Myeloid Dendritic Cells in HIV-Infected Individuals. J. Immunol. 2002, 168, 4796–4801. [Google Scholar] [CrossRef]
- Meyers, J.H.; Justement, J.S.; Hallahan, C.W.; Blair, E.T.; Sun, Y.A.; O’Shea, M.A.; Roby, G.; Kottilil, S.; Moir, S.; Kovacs, C.M.; et al. Impact of HIV on Cell Survival and Antiviral Activity of Plasmacytoid Dendritic Cells. PLoS ONE 2007, 2, e458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, D.D.; Yonkers, N.L.; Post, A.B.; Asaad, R.; Heinzel, F.P.; Lederman, M.M.; Lehmann, P.V.; Valdez, H. Selective Impairments in Dendritic Cell-Associated Function Distinguish Hepatitis C Virus and HIV Infection. J. Immunol. 2004, 172, 4907–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertheimer, A.M.; Bakke, A.; Rosen, H.R. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology 2004, 40, 335–345. [Google Scholar] [CrossRef]
- Szabo, G.; Dolganiuc, A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology 2005, 210, 237–247. [Google Scholar] [CrossRef]
- Brown, K.N.; Wijewardana, V.; Liu, X.; Barratt-Boyes, S.M. Rapid Influx and Death of Plasmacytoid Dendritic Cells in Lymph Nodes Mediate Depletion in Acute Simian Immunodeficiency Virus Infection. PLoS Pathog. 2009, 5, e1000413. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Evans, T.I.; Gillis, J.; Connole, M.; Reeves, R.K. Bone Marrow–Imprinted Gut-Homing of Plasmacytoid Dendritic Cells (pDCs) in Acute Simian Immunodeficiency Virus Infection Results in Massive Accumulation of Hyperfunctional CD4 + pDCs in the Mucosae. J. Infect. Dis. 2015, 211, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Chow, K.V.; Soo, P.; Xu, Z.; Brady, J.L.; Lawlor, K.E.; Masters, S.L.; O’keeffe, M.; Shortman, K.; Zhang, J.-G.; et al. Plasmacytoid dendritic cells are short-lived: Reappraising the influence of migration, genetic factors and activation on estimation of lifespan. Sci. Rep. 2016, 6, 25060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Waskow, C.; Liu, X.; Yao, K.; Hoh, J.; Nussenzweig, M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat. Immunol. 2007, 8, 578–583. [Google Scholar] [CrossRef]
- Dress, R.J.; Dutertre, C.-A.; Giladi, A.; Schlitzer, A.; Low, I.; Shadan, N.B.; Tay, A.; Lum, J.; Kairi, M.F.B.M.; Hwang, Y.Y.; et al. Plasmacytoid dendritic cells develop from Ly6D+ lymphoid progenitors distinct from the myeloid lineage. Nat. Immunol. 2019, 20, 852–864. [Google Scholar] [CrossRef]
- Lehmann, C.; Lafferty, M.; Garzino-Demo, A.; Jung, N.; Hartmann, P.; Fätkenheuer, G.; Wolf, J.S.; van Lunzen, J.; Romerio, F. Plasmacytoid Dendritic Cells Accumulate and Secrete Interferon Alpha in Lymph Nodes of HIV-1 Patients. PLoS ONE 2010, 5, e11110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, M.M.; Bhangoo, A. Human immune deficiency virus (HIV) infection and the hypothalamic pituitary adrenal axis. Rev. Endocr. Metab. Disord. 2013, 14, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Spencer, R.L.; Pearce, B.D.; Pisell, T.L.; Tanapat, P.; Leung, J.J.; Dhabhar, F.S.; McEwen, B.S.; Biron, C.A. Effects of viral infection on corticosterone secretion and glucocorticoid receptor binding in immune tissues. Psychoneuroendocrinology 1997, 22, 455–474. [Google Scholar] [CrossRef]
- Boor, P.P.C.; Metselaar, H.J.; Mancham, S.; Tilanus, H.W.; Kusters, J.G.; Kwekkeboom, J. Prednisolone Suppresses the Function and Promotes Apoptosis of Plasmacytoid Dendritic Cells. Am. J. Transplant. 2006, 6, 2332–2341. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Gong, M.; Xu, Z.; Gill, M.; Chaussabel, D.; Meeker, T.; Chan, J.H.; Wright, T.; Punaro, M.; Bolland, S.; et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 2010, 465, 937–941. [Google Scholar] [CrossRef] [Green Version]
- Lepelletier, Y.; Zollinger, R.; Ghirelli, C.; Raynaud, F.; Hadj-Slimane, R.; Cappuccio, A.; Hermine, O.; Liu, Y.-J.; Soumelis, V. Toll-like receptor control of glucocorticoid-induced apoptosis in human plasmacytoid predendritic cells (pDCs). Blood 2010, 116, 3389–3397. [Google Scholar] [CrossRef]
- Schlitzer, A.; Heiseke, A.F.; Einwächter, H.; Reindl, W.; Schiemann, M.; Manta, C.-P.; See, P.; Niess, J.-H.; Suter, T.; Ginhoux, F.; et al. Tissue-specific differentiation of a circulating CCR9− pDC-like common dendritic cell precursor. Blood 2012, 119, 6063–6071. [Google Scholar] [CrossRef] [Green Version]
- Schlitzer, A.; Loschko, J.; Mair, K.; Vogelmann, R.; Henkel, L.; Einwächter, H.; Schiemann, M.; Niess, J.-H.; Reindl, W.; Krug, A. Identification of CCR9− murine plasmacytoid DC precursors with plasticity to differentiate into conventional DCs. Blood 2011, 117, 6562–6570. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, E.I.; McGavern, D.B.; Pruneda-Paz, J.L.; Teng, C.; Oldstone, M.B.A. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat. Immunol. 2004, 5, 1227–1234. [Google Scholar] [CrossRef]
- Liou, L.-Y.; Blasius, A.L.; Welch, M.J.; Colonna, M.; Oldstone, M.B.A.; Zuniga, E.I. In vivo conversion of BM plasmacytoid DC into CD11b+ conventional DC during virus infection. Eur. J. Immunol. 2008, 38, 3388–3394. [Google Scholar] [CrossRef]
- Toma-Hirano, M.; Namiki, S.; Miyatake, S.; Arai, K.; Kamogawa-Schifter, Y. Type I interferon regulates pDC maturation and Ly49Q expression. Eur. J. Immunol. 2007, 37, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Alcántara-Hernández, M.; Leylek, R.; Wagar, L.E.; Engleman, E.G.; Keler, T.; Marinkovich, M.P.; Davis, M.M.; Nolan, G.P.; Idoyaga, J. High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 2017, 47, 1037–1050.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leylek, R.; Alcántara-Hernández, M.; Lanzar, Z.; Lüdtke, A.; Perez, O.A.; Reizis, B.; Idoyaga, J. Integrated Cross-Species Analysis Identifies a Conserved Transitional Dendritic Cell Population. Cell Rep. 2019, 29, 3736–3750.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leylek, R.; Alcántara-Hernández, M.; Granja, J.M.; Chavez, M.; Perez, K.; Diaz, O.R.; Li, R.; Satpathy, A.T.; Chang, H.Y.; Idoyaga, J. Chromatin Landscape Underpinning Human Dendritic Cell Heterogeneity. Cell Rep. 2020, 32, 108180. [Google Scholar] [CrossRef]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [Green Version]
- Bar-On, L.; Birnberg, T.; Lewis, K.L.; Edelson, B.T.; Bruder, D.; Hildner, K.; Buer, J.; Murphy, K.M.; Reizis, B.; Jung, S. CX3CR1+ CD8α+ dendritic cells are a steady-state population related to plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 14745–14750. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.M.; Nish, S.A.; Yogev, N.; Waisman, A.; Reiner, S.L.; Reizis, B. Leukemia-associated activating mutation of Flt3 expands dendritic cells and alters T cell responses. J. Exp. Med. 2016, 213, 415–431. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Barragan, L.; Züst, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell- derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Z.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Bigio, B.; Yang, R.; Arias, A.A.; Zhou, Q.; Han, J.E.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Van Den Heuvel, G.; Mantere, T.; Kersten, S.; Van Deuren, R.C.; Steehouwer, M.; Van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants among Young Men with Severe COVID-19. JAMA—J. Am. Med. Assoc. 2020, 324, 663–673. [Google Scholar] [CrossRef]
- Bastard, P.; Gervais, A.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Manry, J.; Michailidis, E.; Hoffmann, H.-H.; Eto, S.; Garcia-Prat, M.; et al. Autoantibodies neutralizing type I IFNs are present in ~4% of uninfected individuals over 70 years old and account for ~20% of COVID-19 deaths. Sci. Immunol. 2021, 6, eabl4340. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Renkilaraj, M.R.L.M.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef]
- Klein, S.L.; Dhakal, S.; Ursin, R.L.; Deshpande, S.; Sandberg, K.; Mauvais-Jarvis, F. Biological sex impacts COVID-19 outcomes. PLOS Pathog. 2020, 16, e1008570. [Google Scholar] [CrossRef] [PubMed]
- Consortium, W.S.T. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020, 384, 497–511. [Google Scholar] [CrossRef]
- Monk, P.D.; Marsden, R.J.; Tear, V.J.; Brookes, J.; Batten, T.N.; Mankowski, M.; Gabbay, F.J.; Davies, D.E.; Holgate, S.T.; Ho, L.P.; et al. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021, 9, 196–206. [Google Scholar] [CrossRef]
- Hung, I.F.N.; Lung, K.C.; Tso, E.Y.K.; Liu, R.; Chung, T.W.H.; Chu, M.Y.; Ng, Y.Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D.; et al. Retrospective Multicenter Cohort Study Shows Early Interferon Therapy Is Associated with Favorable Clinical Responses in COVID-19 Patients. Cell Host Microbe 2020, 28, 455–464.e2. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Onodi, F.; Bonnet-Madin, L.; Meertens, L.; Karpf, L.; Poirot, J.; Zhang, S.-Y.; Picard, C.; Puel, A.; Jouanguy, E.; Zhang, Q.; et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J. Exp. Med. 2021, 218, e20201387. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Eutimio, M.A.; López-Macías, C.; Pastelin-Palacios, R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020, 22, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Puel, A.; Bonnet, M.; Ku, C.-L.; Bustamante, J.; Yang, K.; Soudais, C.; Dupuis, S.; Feinberg, J.; Fieschi, C.; et al. Pyogenic Bacterial Infections in Humans with IRAK-4 Deficiency. Science. 2003, 299, 2076–2079. [Google Scholar] [CrossRef]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef]
- Fallerini, C.; Daga, S.; Mantovani, S.; Benetti, E.; Picchiotti, N.; Francisci, D.; Paciosi, F.; Schiaroli, E.; Baldassarri, M.; Fava, F.; et al. Association of toll-like receptor 7 variants with life-threatening COVID-19 disease in males: Findings from a nested case-control study. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Biology of pDCs during SARS-CoV-2 Infection. (A, left) pDC are likely to uptake viral material independent of cell-intrinsic infection, and to sense SARS-CoV-2 RNA via TLR7. This first step stimulates the production of IFN-I which promotes early viral control. (A, right) Late after infection blood pDCs become hypofunctional (exhausted), undergo apoptosis and show reduced numbers, and all this may limit pDC derived IFN-I. (B, left) pDC from patients with mild disease are expected to produce high IFN-I levels promoting the control of SARS-CoV-2 early after infection. (B, right) On the other hand, individuals with severe disease course may exhibit varying degrees of pDC-IFN-I production. The latter may range from low pDC-derived IFN-I in patients with defective TLR7 signaling to normal or even enhanced pDC-derived IFN-I in patients with defects in other immune pathways, or infected with a high viral inoculum or a fast-replicating variant. In such individuals the initial IFN-I wave is expected to be present but insufficient to contain the virus, which may result in sustained stimulation of IFN-I production by a variety of cell types. Importantly, the viral loads or IFN-I levels have not been measured in patients at early times post infection, prior to symptom development, and so the levels of this initial IFN-I response are hypothetical (dotted lines).
Figure 1.
Biology of pDCs during SARS-CoV-2 Infection. (A, left) pDC are likely to uptake viral material independent of cell-intrinsic infection, and to sense SARS-CoV-2 RNA via TLR7. This first step stimulates the production of IFN-I which promotes early viral control. (A, right) Late after infection blood pDCs become hypofunctional (exhausted), undergo apoptosis and show reduced numbers, and all this may limit pDC derived IFN-I. (B, left) pDC from patients with mild disease are expected to produce high IFN-I levels promoting the control of SARS-CoV-2 early after infection. (B, right) On the other hand, individuals with severe disease course may exhibit varying degrees of pDC-IFN-I production. The latter may range from low pDC-derived IFN-I in patients with defective TLR7 signaling to normal or even enhanced pDC-derived IFN-I in patients with defects in other immune pathways, or infected with a high viral inoculum or a fast-replicating variant. In such individuals the initial IFN-I wave is expected to be present but insufficient to contain the virus, which may result in sustained stimulation of IFN-I production by a variety of cell types. Importantly, the viral loads or IFN-I levels have not been measured in patients at early times post infection, prior to symptom development, and so the levels of this initial IFN-I response are hypothetical (dotted lines).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Greene, T.T.; Zuniga, E.I. Type I Interferon Induction and Exhaustion during Viral Infection: Plasmacytoid Dendritic Cells and Emerging COVID-19 Findings. Viruses 2021, 13, 1839. https://doi.org/10.3390/v13091839
AMA Style
Greene TT, Zuniga EI. Type I Interferon Induction and Exhaustion during Viral Infection: Plasmacytoid Dendritic Cells and Emerging COVID-19 Findings. Viruses. 2021; 13(9):1839. https://doi.org/10.3390/v13091839
Chicago/Turabian StyleGreene, Trever T., and Elina I. Zuniga. 2021. "Type I Interferon Induction and Exhaustion during Viral Infection: Plasmacytoid Dendritic Cells and Emerging COVID-19 Findings" Viruses 13, no. 9: 1839. https://doi.org/10.3390/v13091839
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.