
Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models

 , ,
, ,
Abstract
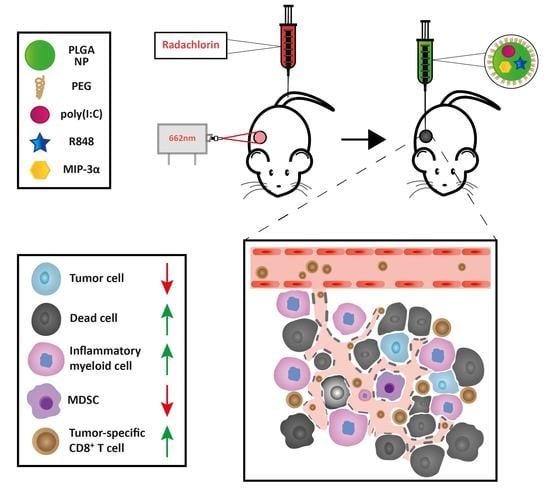
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Preparation of PLGA-NPs
2.3. Size Distribution and Surface Charge of the NPs
2.4. Cell Lines
2.5. Animal Models
2.6. Photosensitizer Uptake and Retention Experiments
2.7. PDT In Vitro Cytotoxicity
2.8. Maturation of D1DCs after Incubation with NPs
2.9. Toxicity of the NPs
2.10. Maturation of D1DCs after Incubation with PDT-Treated Tumor Cells
2.11. PDT and NP Tumor Treatments In Vivo
2.12. Detection of Blood Tetramers
2.13. Depletion of CD8+ Cells
2.14. Analysis of the Tumor Microenvironment, Draining Lymph Node and Spleen
2.15. Intracellular Cytokine Staining
2.16. Statistics
3. Results
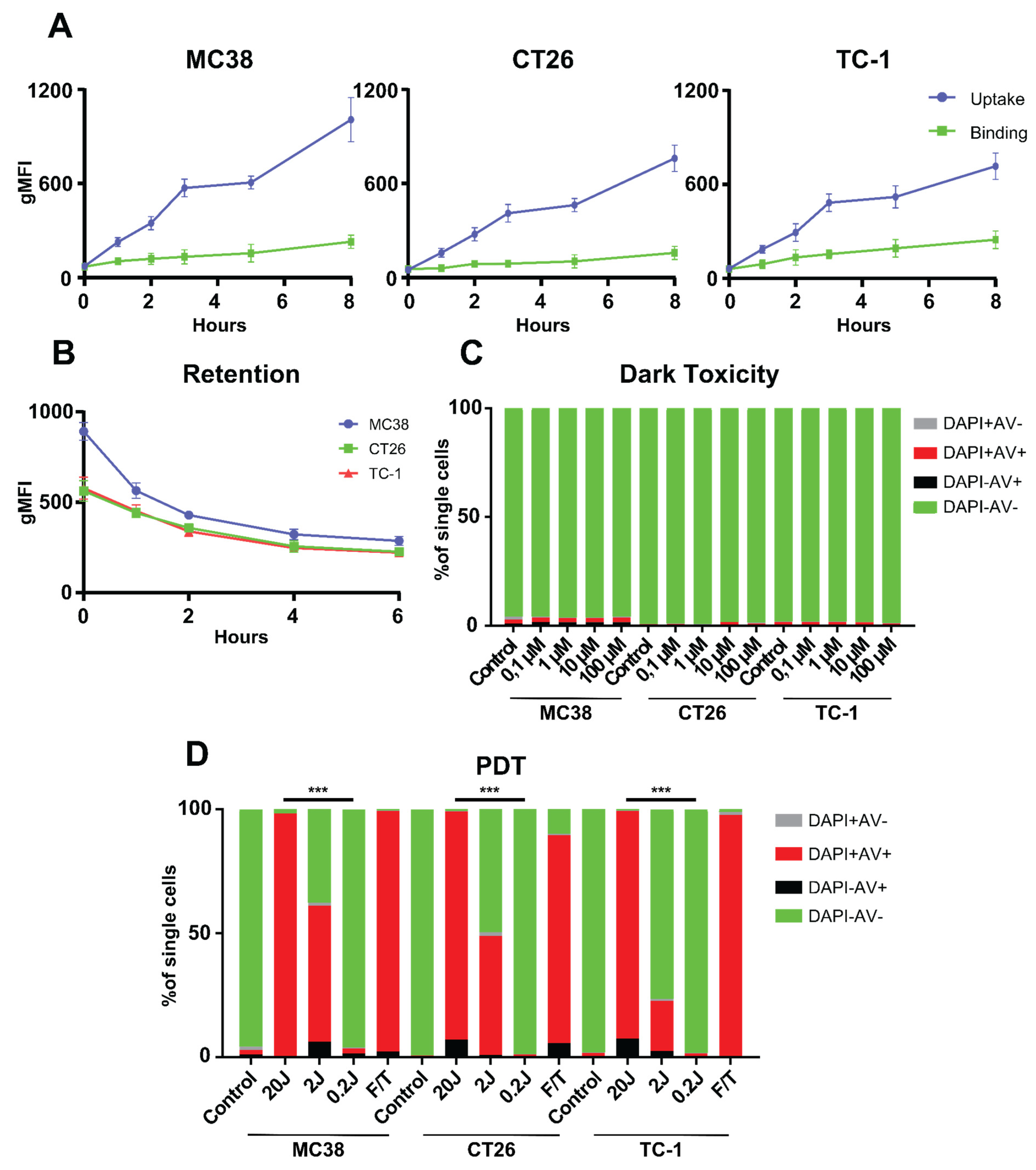
3.1. PDT In Vitro
3.2. Radachlorin PDT Induces Immunogenic Cell Death
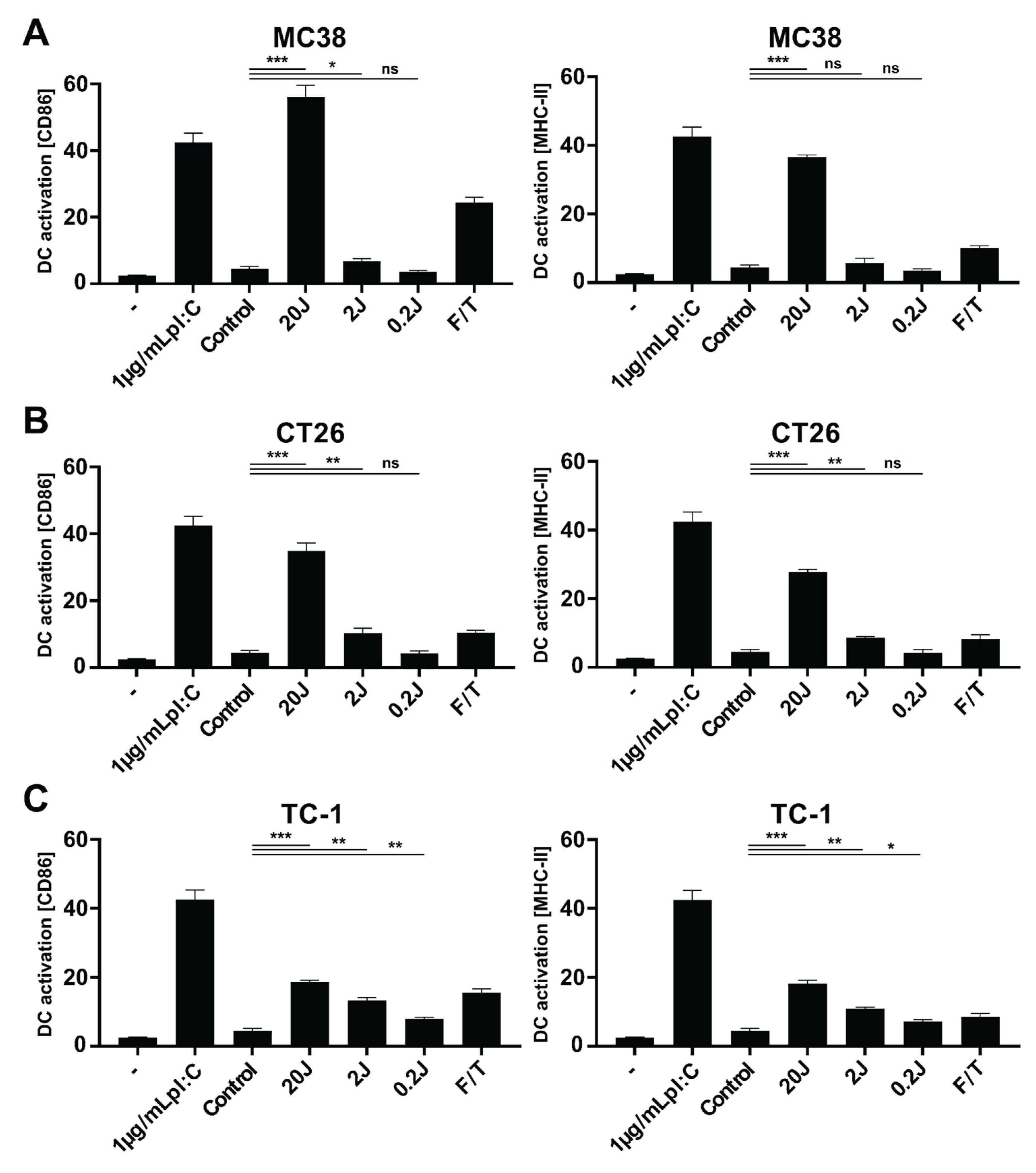
3.3. Physicochemical Characterization and Biological Activity of PLGA-PEG (poly(I:C), R848, MIP3α) NPs
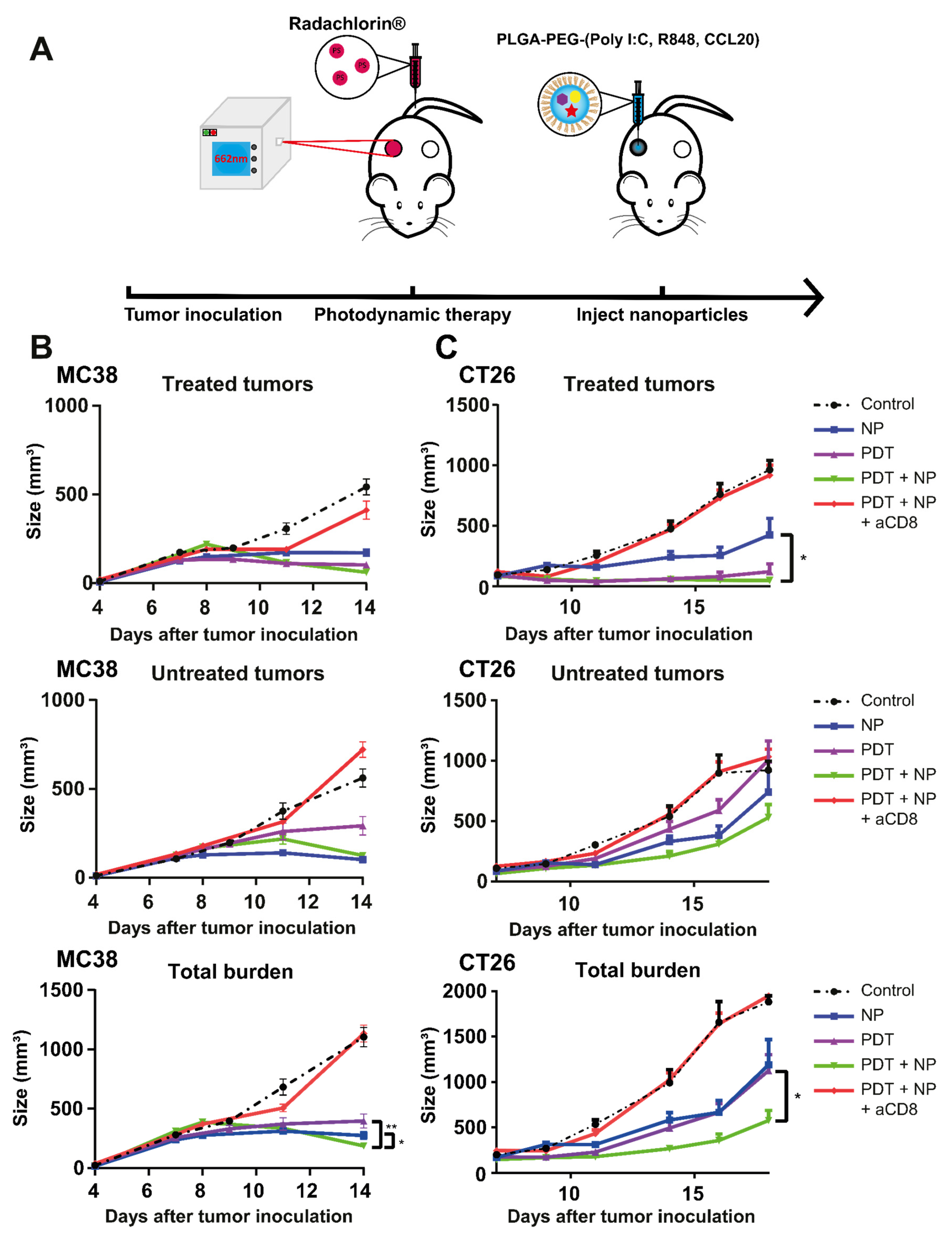
3.4. The Combination of PDT and NP Elicits a CD8+ T cell-Dependent Abscopal Effect in Mice Bearing Bilateral MC38 or CT26 Tumors
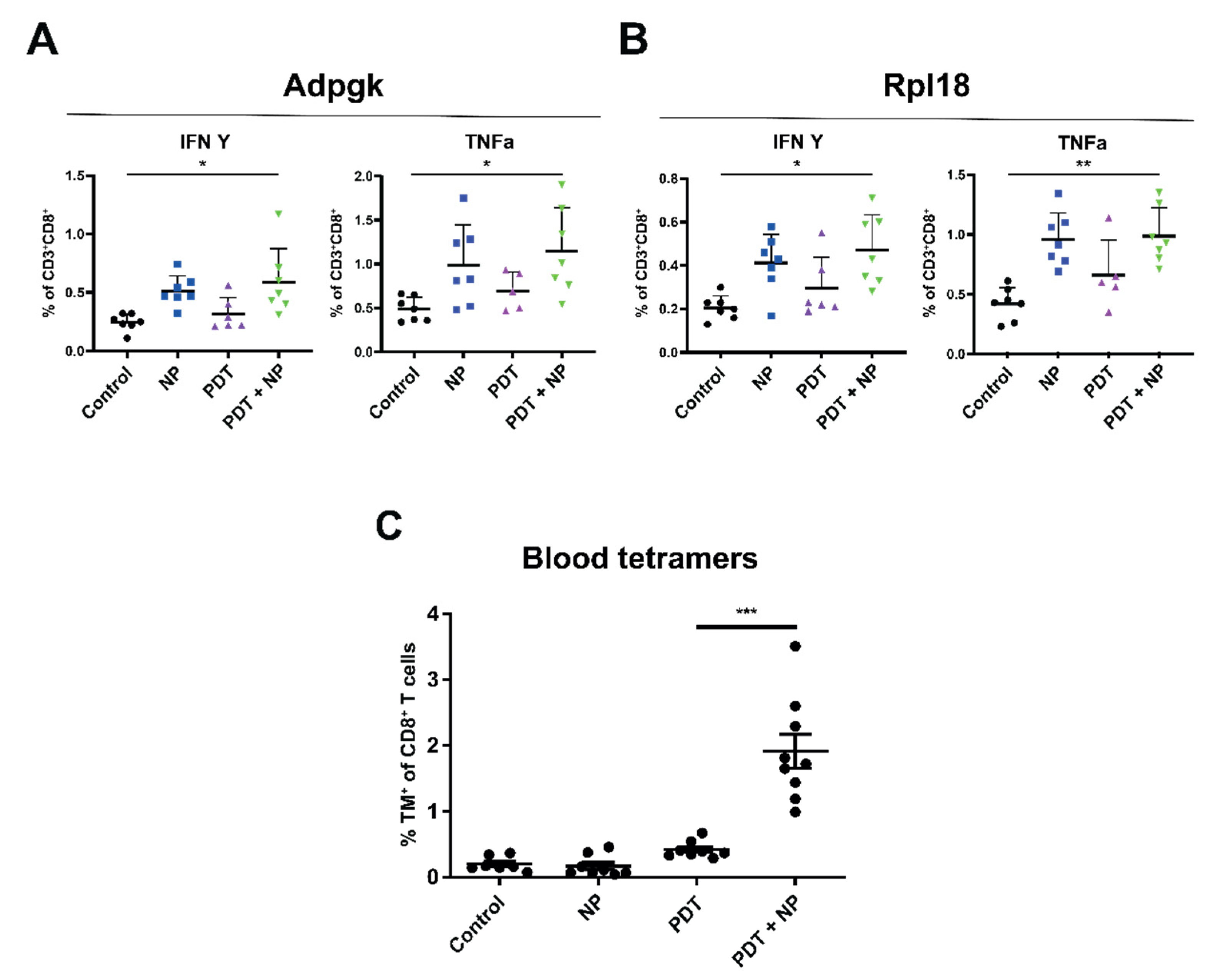
3.5. The Combination of PDT and Immunostimulatory NPs Provides Enhanced, Tumor-Specific Immune Responses in Mice Bearing Bilateral MC38 or TC-1 Tumors
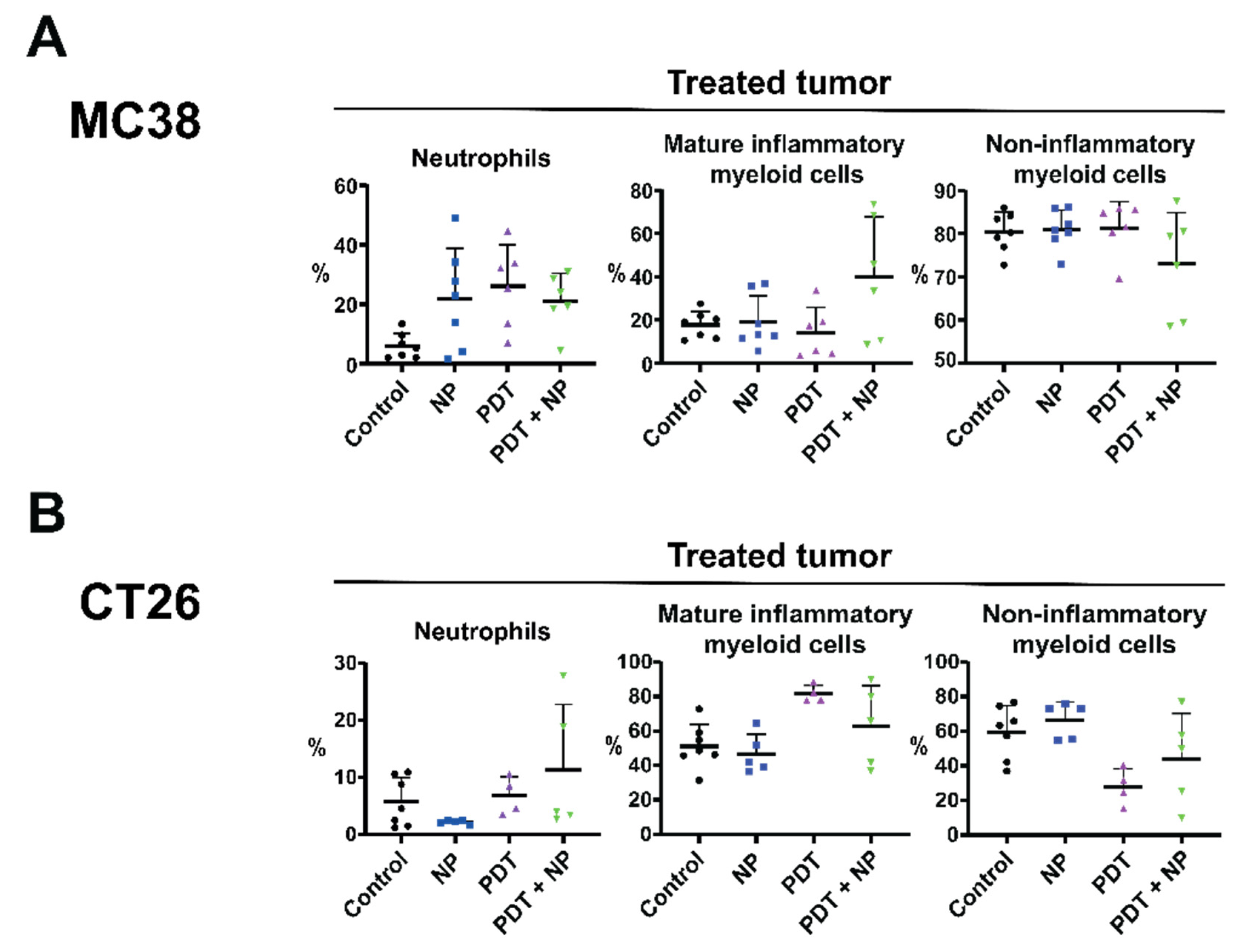
3.6. The Combination Treatment Induces an Inflammatory State in Colon Tumors in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Straten, D.; Mashayekhi, V.; De Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef]
- Hernández, I.B.; Yu, Y.; Ossendorp, F.; Korbelik, M.; Oliveira, S. Preclinical and Clinical Evidence of Immune Responses Triggered in Oncologic Photodynamic Therapy: Clinical Recommendations. J. Clin. Med. 2020, 9, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.; Obaid, G.; Hasan, T. The Course of Immune Stimulation by Photodynamic Therapy: Bridging Fundamentals of Photochemically Induced Immunogenic Cell Death to the Enrichment of T-Cell Repertoire. Photochem. Photobiol. 2019, 95, 1288–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yang, J.; Luo, L.; Jiang, M.; Qin, B.; Yin, H.; Zhu, C.; Yuan, X.; Zhang, J.; Luo, Z.; et al. Targeting photodynamic and photothermal therapy to the endoplasmic reticulum enhances immunogenic cancer cell death. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nat. Cell Biol. 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef]
- Korbelik, M.; Sun, J.; Cecic, I. Photodynamic therapy-induced cell surface expression and release of heat shock proteins: Relevance for tumor response. Cancer Res. 2005, 65, 1018–1026. [Google Scholar] [PubMed]
- Krysko, D.; Garg, A.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Panzarini, E.; Inguscio, V.; Fimia, G.M.; Dini, L. Rose Bengal Acetate PhotoDynamic Therapy (RBAc-PDT) Induces Exposure and Release of Damage-Associated Molecular Patterns (DAMPs) in Human HeLa Cells. PLoS ONE 2014, 9, e105778. [Google Scholar] [CrossRef]
- Vabulas, R.M.; Wagner, H.; Schild, H. Heat Shock Proteins as Ligands of Toll-Like Receptors. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2002; Volume 270, pp. 169–184. [Google Scholar] [CrossRef]
- Flechtner, J.B.; Cohane, K.P.; Mehta, S.; Slusarewicz, P.; Leonard, A.K.; Barber, B.H.; Levey, D.L.; Andjelic, S. High-affinity interactions between peptides and heat shock protein 70 augment CD8+ T lymphocyte immune responses. J. Immunol. 2006, 177, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Salimu, J.; Spary, L.; Al-Taei, S.; Clayton, A.; Mason, M.D.; Staffurth, J.; Tabi, Z. Cross-Presentation of the Oncofetal Tumor Antigen 5T4 from Irradiated Prostate Cancer Cells—A Key Role for Heat-Shock Protein 70 and Receptor CD91. Cancer Immunol. Res. 2015, 3, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Krysko, D.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovitch, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nat. Cell Biol. 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β–dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Kleinovink, J.W.; Fransen, M.F.; Löwik, C.W.; Ossendorp, F. Photodynamic-Immune Checkpoint Therapy Eradicates Local and Distant Tumors by CD8+ T Cells. Cancer Immunol. Res. 2017, 5, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Locy, H.; DE Mey, S.L.; De Mey, W.; De Ridder, M.; Thielemans, K.; Maenhout, S.K. Immunomodulation of the Tumor Microenvironment: Turn Foe into Friend. Front. Immunol. 2018, 9, 2909. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.G.; Camps, M.G.; Li, T.M.; Zerrillo, L.; Löwik, C.W.; Ossendorp, F.; Cruz, L.J. Effective chemoimmunotherapy by co-delivery of doxorubicin and immune adjuvants in biodegradable nanoparticles. Theranostics 2019, 9, 6485–6500. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.; Camps, M.; Li, T.; Chan, A.; Ossendorp, F.; Cruz, L. Co-delivery of immunomodulators in biodegradable nanoparticles improves therapeutic efficacy of cancer vaccines. Biomaterials 2019, 220, 119417. [Google Scholar] [CrossRef]
- Smith, M.; García-Martínez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. OncoImmunology 2018, 7, e1526250. [Google Scholar] [CrossRef]
- Cheng, Y.-S.; Xu, F. Anticancer function of polyinosinic-polycytidylic acid. Cancer Biol. Ther. 2010, 10, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Shime, H.; Matsumoto, M.; Seya, T. Double-stranded RNA promotes CTL-independent tumor cytolysis mediated by CD11b+Ly6G+ intratumor myeloid cells through the TICAM-1 signaling pathway. Cell Death Differ. 2017, 24, 385–396. [Google Scholar] [CrossRef]
- Shime, H.; Matsumoto, M.; Oshiumi, H.; Tanaka, S.; Nakane, A.; Iwakura, Y.; Tahara, H.; Inoue, N.; Seya, T. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc. Natl. Acad. Sci. USA 2012, 109, 2066–2071. [Google Scholar] [CrossRef] [Green Version]
- Takemura, R.; Takaki, H.; Okada, S.; Shime, H.; Akazawa, T.; Oshiumi, H.; Matsumoto, M.; Teshima, T.; Seya, T. PolyI: C–Induced, TLR3/RIP3-Dependent Necroptosis Backs Up Immune Effector–Mediated Tumor Elimination In Vivo. Cancer Immunol. Res. 2015, 3, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Friboulet, L.; Pioche-Durieu, C.; Rodriguez, S.; Valent, A.; Souquère, S.; Ripoche, H.; Khabir, A.; Tsao, S.W.; Bosq, J.; Wai v, K.; et al. Recurrent Overexpression of c-IAP2 in EBV-Associated Nasopharyngeal Carcinomas: Critical Role in Resistance to Toll-like Receptor 3-Mediated Apoptosis. Neoplasia 2008, 10, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, A.; Starace, D.; Galli, R.; Padula, F.; De Cesaris, P.; Filippini, A.; Ziparo, E.; Riccioli, A. Toll-like receptor 3 triggers apoptosis of human prostate cancer cells through a PKC- -dependent mechanism. Carcinogenesis 2008, 29, 1334–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.P.M.; Kirejczyk, Z.; Besch, R.; Potthoff, S.; Leverkus, M.; Hacker, G.W. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. 2009, 17, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estornes, Y.; Toscano, F.; Virard, F.; Jacquemin, G.; Pierrot, A.; Vanbervliet, B.; Bonnin, M.; Lalaoui, N.; Mercier-Gouy, P.; Pachéco, Y.; et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012, 19, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-X.; Sun, L. Immune effects of R848: Evidences that suggest an essential role of TLR7/8-induced, Myd88- and NF-κB-dependent signaling in the antiviral immunity of Japanese flounder (Paralichthys olivaceus). Dev. Comp. Immunol. 2015, 49, 113–120. [Google Scholar] [CrossRef]
- Yin, T.; He, S.; Wang, Y. Toll-like receptor 7/8 agonist, R848, exhibits antitumoral effects in a breast cancer model. Mol. Med. Rep. 2015, 12, 3515–3520. [Google Scholar] [CrossRef] [Green Version]
- Spinetti, T.; Spagnuolo, L.; Mottas, I.; Secondini, C.; Treinies, M.; Rüegg, C.; Hotz, C.; Bourquin, C. TLR7-based cancer immunotherapy decreases intratumoral myeloid-derived suppressor cells and blocks their immunosuppressive function. OncoImmunology 2016, 5, e1230578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30345161 (accessed on 30 April 2019). [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR 8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef]
- Hieshima, K.; Imai, T.; Opdenakker, G.; Van Damme, J.; Kusuda, J.; Tei, H.; Sakaki, Y.; Takatsuki, K.; Miura, R.; Yoshie, O.; et al. Molecular Cloning of a Novel Human CC Chemokine Liver and Activation-regulated Chemokine (LARC) Expressed in Liver. J. Biol. Chem. 1997, 272, 5846–5853. [Google Scholar] [CrossRef] [Green Version]
- McLean, M.; Murray, G.I.; Stewart, K.N.; Norrie, G.; Mayer, C.; Hold, G.L.; Thomson, J.; Fyfe, N.; Hope, M.; Mowat, N.A.G.; et al. The Inflammatory Microenvironment in Colorectal Neoplasia. PLoS ONE 2011, 6, e15366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frick, V.O.; Rubie, C.; Keilholz, U.; Ghadjar, P. Chemokine/chemokine receptor pair CCL20/CCR6 in human colorectal malignancy: An overview. World J. Gastroenterol. 2016, 22, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Kwong, B.; Liu, H.; Irvine, D.J. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 2011, 32, 5134–5147. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, C.; Rueda, F.; Löwik, C.; Ossendorp, F.; Cruz, L.J. Combinatorial prospects of nano-targeted chemoimmunotherapy. Biomaterials 2016, 83, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) As biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Lavan, D.; McGuire, T.; Langer, R. Small-scale systems for in vivo drug delivery. Nat. Biotechnol. 2003, 21, 1184–1191. [Google Scholar] [CrossRef]
- In’t Veld, R.V.H.; Ritsma, L.; Kleinovink, J.W.; Que, I.; Ossendorp, F.; Cruz, L.J. Photodynamic cancer therapy enhances accumulation of nanoparticles in tumor-associated myeloid cells. J. Control. Release 2020, 320, 19–31. [Google Scholar] [CrossRef]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, N.G.R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Xu, L.; Wang, C.; Yang, R.; Zhuang, Q.; Han, X.; Dong, Z.; Zhu, W.; Peng, R.; Liu, Z. Near-Infrared-Triggered Photodynamic Therapy with Multitasking Upconversion Nanoparticles in Combination with Checkpoint Blockade for Immunotherapy of Colorectal Cancer. ACS Nano 2017, 11, 4463–4474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Hu, C.; Cai, L.; Pang, M. A covalent organic framework as a nanocarrier for synergistic phototherapy and immunotherapy. J. Mater. Chem. B 2020, 8, 5451–5459. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wei, G.; Zeng, Z.; Huang, Y.; Huang, L.; Shen, Y.; Sun, X.; Xu, C.; Zhao, C. Enhanced cancer therapy through synergetic photodynamic/immune checkpoint blockade mediated by a liposomal conjugate comprised of porphyrin and IDO inhibitor. Theranostics 2019, 9, 5542–5557. [Google Scholar] [CrossRef]
- Barr, H.; Krasner, N.; Boulos, P.B.; Chatlani, P.; Bown, S.G. Photodynamic therapy for colorectal cancer: A quantitative pilot study. J. Br. Surg. 2005, 77, 93–96. [Google Scholar] [CrossRef] [PubMed]
- McCaughan, J.S., Jr.; Hawley, P.C.; Bethel, B.H.; Walker, J. Photodynamic therapy of endobronchial malignancies. Cancer 1988, 62, 691–701. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Fokkink, R.; Joosten, B.; Stuart, M.C.; Albericio, F.; Torensma, R.; Figdor, C.G. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J. Control. Release 2010, 144, 118–126. [Google Scholar] [CrossRef]
- Cruz, L.J.; Tacken, P.J.; Bonetto, F.; Buschow, S.I.; Croes, H.J.; Wijers, M.; de Vries, I.J.; Figdor, C.G. Multimodal Imaging of Nanovaccine Carriers Targeted to Human Dendritic Cells. Mol. Pharm. 2011, 8, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.J.; Tacken, P.J.; Rueda, F.; Domingo, J.C.; Albericio, F.; Figdor, C.G. Targeting Nanoparticles to Dendritic Cells for Immunotherapy. Methods Enzymol. 2012, 509, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.J.; Stammes, M.A.; Que, I.; van Beek, E.R.; Knol-Blankevoort, V.T.; Snoeks, T.J.; Chan, A.; Kaijzel, E.L.; Löwik, C.W. Effect of PLGA NP size on efficiency to target traumatic brain injury. J. Control. Release 2016, 223, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Lambeck, A.J.A.; Cruz, L.J.; Tacken, P.J.; De Vries, I.J.M.; Figdor, C.G.; De Vries, I.J.M. Human Plasmacytoid Dendritic Cells Phagocytose, Process, and Present Exogenous Particulate Antigen. J. Immunol. 2010, 184, 4276–4283. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.C. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar]
- Ossendorp, F.; Fu, N.; Camps, M.; Granucci, F.; Gobin, S.J.P.; Elsen, P.V.D.; Schuurhuis, D.; Adema, G.J.; Lipford, G.B.; Chiba, T.; et al. Differential Expression Regulation of the α and β Subunits of the PA28 Proteasome Activator in Mature Dendritic Cells. J. Immunol. 2005, 174, 7815–7822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zom, G.G.; Khan, S.; Britten, C.M.; Sommandas, V.; Camps, M.G.; Loof, N.M.; Budden, C.F.; Meeuwenoord, N.J.; Filippov, D.V.; Van Der Marel, G.A.; et al. Efficient Induction of Antitumor Immunity by Synthetic Toll-like Receptor Ligand–Peptide Conjugates. Cancer Immunol. Res. 2014, 2, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinovink, J.W.; Van Driel, P.B.; Snoeks, T.J.; Prokopi, A.; Fransen, M.F.; Cruz, L.J.; Mezzanotte, L.; Chan, A.; Löwik, C.W.; Ossendorp, F. Combination of Photodynamic Therapy and Specific Immunotherapy Efficiently Eradicates Established Tumors. Clin. Cancer Res. 2016, 22, 1459–1468. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.J.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Hos, B.J.; Camps, M.G.; Bulk, J.V.D.; Tondini, E.; Ende, T.C.V.D.; Ruano, D.; Franken, K.; Janssen, G.M.; de Ru, A.H.; Filippov, D.V.; et al. Identification of a neo-epitope dominating endogenous CD8 T cell responses to MC-38 colorectal cancer. OncoImmunology 2020, 9, 1673125. [Google Scholar] [CrossRef] [Green Version]
- Cecic, I.; Parkins, C.S.; Korbelik, M. Induction of Systemic Neutrophil Response in Mice by Photodynamic Therapy of Solid Tumors. Photochem. Photobiol. 2001, 74, 712. [Google Scholar] [CrossRef]
- Nejad, E.B.; Labrie, C.; Abdulrahman, Z.; Van Elsas, M.J.; Rademaker, E.; Kleinovink, J.W.; Van Der Sluis, T.C.; Van Duikeren, S.; Teunisse, A.F.A.; Jochemsen, A.G.; et al. Lack of myeloid cell infiltration as an acquired resistance strategy to immunotherapy. J. Immunother. Cancer 2020, 8, e001326. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Fan, Z.; Zhou, F.; Wang, X.; Shi, L.; Zhang, H.; Wang, P.; Yang, D.; Zhang, L.; Chen, W.R.; et al. Improvement of DC vaccine with ALA-PDT induced immunogenic apoptotic cells for skin squamous cell carcinoma. Oncotarget 2015, 6, 17135–17146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, N.-C.; Kim, H.J.; Kang, M.-S.; Lee, J.-H.; Song, J.-Y.; Seo, H.G.; Bae, Y.-S.; Lim, D.-S. Photodynamic therapy-mediated DC immunotherapy is highly effective for the inhibition of established solid tumors. Cancer Lett. 2012, 324, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xin, F.; Yao, C.; Liu, X.; Wu, M.; Lin, X.; Du, X.; Chen, G.; Zhang, D.; Zhang, Z.; et al. Photodynamic Therapy Combined with Antihypoxic Signaling and CpG Adjuvant as an In Situ Tumor Vaccine Based on Metal–Organic Framework Nanoparticles to Boost Cancer Immunotherapy. Adv. Healthcare Mater. 2020, 9, 1900996. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, L.; Wang, C.; Han, Y.; Lu, Y.; Liu, J.; Hu, X.; Yao, T.; Lin, Y.; Liang, S.; et al. Tumor-Targeted Drug and CpG Delivery System for Phototherapy and Docetaxel-Enhanced Immunotherapy with Polarization toward M1-Type Macrophages on Triple Negative Breast Cancers. Adv. Mater. 2019, 31, e1904997. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, F.; Deng, H.; Lin, L.; Wang, S.; Kang, F.; Yu, G.; Lau, J.; Tian, R.; Zhang, M.; et al. Smart Nanovesicle-Mediated Immunogenic Cell Death through Tumor Microenvironment Modulation for Effective Photodynamic Immunotherapy. ACS Nano 2019, 14, 620–631. [Google Scholar] [CrossRef]
- Bao, R.; Wang, Y.; Lai, J.; Zhu, H.; Zhao, Y.; Li, S.; Li, N.; Huang, J.; Yang, Z.; Wang, F.; et al. Enhancing Anti-PD-1/PD-L1 Immune Checkpoint Inhibitory Cancer Therapy by CD276-Targeted Photodynamic Ablation of Tumor Cells and Tumor Vasculature. Mol. Pharm. 2019, 16, 339–348. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, F.; Shao, D.; Chang, Z.; Wang, L.; Hu, H.; Zheng, X.; Li, X.; Chen, F.; Tu, Z.; et al. Janus Nanobullets Combine Photodynamic Therapy and Magnetic Hyperthermia to Potentiate Synergetic Anti-Metastatic Immunotherapy. Adv. Sci. 2019, 6, 1901690. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hou, M.; Sun, W.; Wu, Q.; Xu, J.; Xiong, L.; Chai, Y.; Liu, Y.; Yu, M.; Wang, H.; et al. Sequential PDT and PTT Using Dual-Modal Single-Walled Carbon Nanohorns Synergistically Promote Systemic Immune Responses against Tumor Metastasis and Relapse. Adv. Sci. 2020, 7, 2001088. [Google Scholar] [CrossRef]
- Yang, W.; Zhu, G.; Wang, S.; Yu, G.; Yang, Z.; Lin, L.; Zhou, Z.; Liu, Y.; Dai, Y.; Zhang, F.; et al. In Situ Dendritic Cell Vaccine for Effective Cancer Immunotherapy. ACS Nano 2019, 13, 3083–3094. [Google Scholar] [CrossRef]
- Wu, X.; Yang, H.; Chena, X.; Gaoa, J.; Duana, Y.; Weia, D.; Zhangc, J.; Gec, K.; Liangde, X.-J.; Huangf, Y.; et al. Nano-herb medicine and PDT induced synergistic immunotherapy for colon cancer treatment. Biomaterials 2021, 269, 120654. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Diameter | ζ Potential (mV) | PDI | Encapsulation Efficiency (% w/w) | |||
|---|---|---|---|---|---|---|---|
| NIR | poly(I:C) | R848 | MIP3α | ||||
| NP(NIR + pIC + R848 + MIP3α)-PEG | 249.6 ± 85.4 | −21.4 ± 4.75 | 0.178 ± 0.042 | 62.4 ± 6.9 | 43.6 ± 8.6 | 54.2 ± 8.9 | 59.3 ± 7.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huis in ‘t Veld, R.V.; Da Silva, C.G.; Jager, M.J.; Cruz, L.J.; Ossendorp, F. Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models. Pharmaceutics 2021, 13, 1470. https://doi.org/10.3390/pharmaceutics13091470
Huis in ‘t Veld RV, Da Silva CG, Jager MJ, Cruz LJ, Ossendorp F. Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models. Pharmaceutics. 2021; 13(9):1470. https://doi.org/10.3390/pharmaceutics13091470
Chicago/Turabian StyleHuis in ‘t Veld, Ruben Victor, Candido G. Da Silva, Martine J. Jager, Luis J. Cruz, and Ferry Ossendorp. 2021. "Combining Photodynamic Therapy with Immunostimulatory Nanoparticles Elicits Effective Anti-Tumor Immune Responses in Preclinical Murine Models" Pharmaceutics 13, no. 9: 1470. https://doi.org/10.3390/pharmaceutics13091470