
Familial Hypercholesterolemia Genetic Variations and Long-Term Cardiovascular Outcomes in Patients with Hypercholesterolemia Who Underwent Coronary Angiography

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Data Collection and Follow-up
2.3. Targeted Sequencing
2.4. Statistical Analyses
3. Results
3.1. High Prevalence of FH in Patients with High Blood LDL-C Levels Who Underwent Coronary Angiography
3.2. FH Pathogenica Vatiants on LDLR, APOB, and PCSK9
3.3. Association of FH Pathogenic Variants with the Incidence of CVD or Mortality
3.4. Association of the LDLR c.986G>A and LDLR c.268G>A Variants with High Blood LDL-C Levels and Early-Onset CAD
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [Green Version]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the prevalence of heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef]
- Chiou, K.R.; Charng, M.J. Genetic diagnosis of familial hypercholesterolemia in Han Chinese. J. Clin. Lipidol. 2016, 10, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Kawashiri, M.A.; Nohara, A.; Inazu, A.; Mabuchi, H.; Yamagishi, M. Impact of clinical signs and genetic diagnosis of familial hypercholesterolaemia on the prevalence of coronary artery disease in patients with severe hypercholesterolaemia. Eur. Heart J. 2017, 38, 1573–1579. [Google Scholar] [CrossRef]
- Akioyamen, L.E.; Genest, J.; Chu, A.; Inibhunu, H.; Ko, D.T.; Tu, J.V. Risk factors for cardiovascular disease in heterozygous familial hypercholesterolemia: A systematic review and meta-analysis. J. Clin. Lipidol. 2019, 13, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beheshti, S.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. 2.6-fold risk of ischemic stroke in individuals with clinical familial hypercholesterolemia: The copenhagen general population study with 102,961 individuals. Atherosclerosis 2017, 263, e235. [Google Scholar] [CrossRef]
- Akioyamen, L.E.; Tu, J.V.; Genest, J.; Ko, D.T.; Coutin, A.J.S.; Shan, S.D.; Chu, A. Risk of Ischemic Stroke and Peripheral Arterial Disease in Heterozygous Familial Hypercholesterolemia: A Meta-Analysis. Angiology 2019, 70, 726–736. [Google Scholar] [CrossRef]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrie, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Nanchen, D.; Gencer, B.; Auer, R.; Raber, L.; Stefanini, G.G.; Klingenberg, R.; Schmied, C.M.; Cornuz, J.; Muller, O.; Vogt, P.; et al. Prevalence and management of familial hypercholesterolaemia in patients with acute coronary syndromes. Eur. Heart J. 2015, 36, 2438–2445. [Google Scholar] [CrossRef]
- Rerup, S.A.; Bang, L.E.; Mogensen, U.M.; Engstrom, T.; Jorgensen, E.; Pedersen, F.; Torp-Pedersen, C.; Gislason, G.; James, S.; Hagstrom, E.; et al. The prevalence and prognostic importance of possible familial hypercholesterolemia in patients with myocardial infarction. Am. Heart J. 2016, 181, 35–42. [Google Scholar] [CrossRef]
- Wald, D.S.; Bangash, F.A.; Bestwick, J.P. Prevalence of DNA-confirmed familial hypercholesterolaemia in young patients with myocardial infarction. Eur. J. Intern. Med. 2015, 26, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Harada-Shiba, M.; Ako, J.; Arai, H.; Hirayama, A.; Murakami, Y.; Nohara, A.; Ozaki, A.; Uno, K.; Nakamura, M. Prevalence of familial hypercholesterolemia in patients with acute coronary syndrome in Japan: Results of the EXPLORE-J study. Atherosclerosis 2018, 277, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Pirazzi, C.; Hakansson, L.; Gustafsson, C.; Omerovic, E.; Wiklund, O.; Mancina, R.M. High prevalence of genetic determined familial hypercholesterolemia in premature coronary artery disease. Appl. Clin. Genet. 2019, 12, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarenco, P.; Labreuche, J. Lipid management in the prevention of stroke: Review and updated meta-analysis of statins for stroke prevention. Lancet Neurol. 2009, 8, 453–463. [Google Scholar] [CrossRef]
- Beheshti, S.; Madsen, C.M.; Varbo, A.; Benn, M.; Nordestgaard, B.G. Relationship of Familial Hypercholesterolemia and High Low-Density Lipoprotein Cholesterol to Ischemic Stroke: Copenhagen General Population Study. Circulation 2018, 138, 578–589. [Google Scholar] [CrossRef]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [Green Version]
- van Loon, J.E.; de Maat, M.P.; Deckers, J.W.; van Domburg, R.T.; Leebeek, F.W. Prognostic markers in young patients with premature coronary heart disease. Atherosclerosis 2012, 224, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Guardamagna, O.; Restagno, G.; Rolfo, E.; Pederiva, C.; Martini, S.; Abello, F.; Baracco, V.; Pisciotta, L.; Pino, E.; Calandra, S.; et al. The type of LDLR gene mutation predicts cardiovascular risk in children with familial hypercholesterolemia. J. Pediatrics 2009, 155, 199–204.e2. [Google Scholar] [CrossRef]
- Galaska, R.; Kulawiak-Galaska, D.; Chmara, M.; Chlebus, K.; Mickiewicz, A.; Rynkiewicz, A.; Wasag, B.; Studniarek, M.; Fijalkowski, M.; Gruchala, M. Carotid intima-media thickness (IMT) in patients with severe familial and non-familial hypercholesterolemia: The effect of measurement site on the IMT correlation with traditional cardiovascular risk factors and calcium scores. Cardiol. J. 2021, 28, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Liu, B.; Xiong, T.; Yu, W.; She, Q. Investigation of the underlying genes and mechanism of familial hypercholesterolemia through bioinformatics analysis. BMC Cardiovasc. Disord. 2020, 20, 419. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Humphries, S.E.; Neely, R.D.; Whittall, R.A.; Troutt, J.S.; Konrad, R.J.; Scartezini, M.; Li, K.W.; Cooper, J.A.; Acharya, J.; Neil, A. Healthy individuals carrying the PCSK9 p.R46L variant and familial hypercholesterolemia patients carrying PCSK9 p.D374Y exhibit lower plasma concentrations of PCSK9. Clin. Chem. 2009, 55, 2153–2161. [Google Scholar] [CrossRef]
- Palacios, L.; Grandoso, L.; Cuevas, N.; Olano-Martin, E.; Martinez, A.; Tejedor, D.; Stef, M. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis 2012, 221, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Chora, J.R.; Medeiros, A.M.; Alves, A.C.; Bourbon, M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: Application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet. Med. 2018, 20, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, K.R.; Charng, M.J. Common mutations of familial hypercholesterolemia patients in Taiwan: Characteristics and implications of migrations from southeast China. Gene 2012, 498, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Won, H.H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef]
- Sharifi, M.; Higginson, E.; Bos, S.; Gallivan, A.; Harvey, D.; Li, K.W.; Abeysekera, A.; Haddon, A.; Ashby, H.; Shipman, K.E.; et al. Greater preclinical atherosclerosis in treated monogenic familial hypercholesterolemia vs. polygenic hypercholesterolemia. Atherosclerosis 2017, 263, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esser, V.; Limbird, L.; Brown, M.S.; Goldstein, J.L.; Russell, D.W. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J. Biol. Chem. 1988, 263, 13282–13290. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ryan, R.O. Domain swapping reveals that low density lipoprotein (LDL) type A repeat order affects ligand binding to the LDL receptor. J. Biol. Chem. 2009, 284, 13396–13400. [Google Scholar] [CrossRef] [Green Version]
- Mosca, L.; Barrett-Connor, E.; Kass Wenger, N. Sex/gender differences in cardiovascular disease prevention: What a difference a decade makes. Circulation 2011, 124, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Chen, Z.; Sun, A.; Deng, X. Gender differences in cardiovascular disease. Med. Nov. Technol. Devices 2019, 4, 100025. [Google Scholar] [CrossRef]
- Rios, J.; Stein, E.; Shendure, J.; Hobbs, H.H.; Cohen, J.C. Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum. Mol. Genet. 2010, 19, 4313–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.; Zelcer, N.; Kastelein, J.J.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenarro, A.; Etxebarria, A.; de Castro-Orós, I.; Stef, M.; Bea, A.M.; Palacios, L.; Mateo-Gallego, R.; Benito-Vicente, A.; Ostolaza, H.; Tejedor, T. The p. Leu167del mutation in APOE gene causes autosomal dominant hypercholesterolemia by down-regulation of LDL receptor expression in hepatocytes. J. Clin. Endocrinol. Metab. 2016, 101, 2113–2121. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Variables | FH Pathogenic Variant Carriers | LDLR c.986G>A/c.268G>A Variant Carriers | FH Pathogenic Variant Non-Carriers | p Value 1 | p Value 2 |
|---|---|---|---|---|---|
| Number | 41 | 9 | 244 | ||
| Age, years | 59.2 ± 13.9 | 52.9 ± 13.2 | 59.7 ± 11.7 | 0.819 | 0.089 |
| Men, n (%) | 34(82.9%) | 6(66.7%) | 174(71.3%) | 0.174 | 0.720 |
| Body mass index, kg/m2 | 27.6 ± 4.3 | 27.4 ± 5.1 | 26.7 ± 3.8 | 0.178 | 0.593 |
| Waist/Hip Ratio | 0.94 ± 0.05 | 0.91 ± 0.07 | 0.94 ± 0.06 | 0.888 | 0.183 |
| sBP, mmHg | 131 ± 23 | 127 ± 18 | 132 ± 20 | 0.859 | 0.494 |
| dBP, mmHg | 77 ± 13 | 77 ± 14 | 77 ± 12 | 0.934 | 0.876 |
| Triglycerides, mg/dL | 180 ± 156 | 190 ± 80 | 178 ± 106 | 0.937 | 0.743 |
| Cholesterol, mg/dL | 239 ± 48 | 259 ± 34 | 239 ± 53 | 0.944 | 0.260 |
| LDL-C, mg/dL | 193 ± 27 | 213 ± 25 | 190 ± 30 | 0.633 | 0.026 * |
| HDL-C, mg/dL | 44 ± 13 | 40 ± 13 | 46 ± 12 | 0.280 | 0.151 |
| HbA1c, % | 6.4 ± 1.5 | 6.2 ± 0.6 | 6.3 ± 1.3 | 0.727 | 0.849 |
| Creatinine, mg/dL | 1.2 ± 1.1 | 1.0 ± 0.2 | 1.4 ± 1.6 | 0.609 | 0.437 |
| eGFR, ml/min/1.73 m2 | 76 ± 23 | 85 ± 22 | 75 ± 27 | 0.945 | 0.310 |
| Smoking, n (%) | 21(51.2%) | 4(44.4%) | 120(49.6%) | 0.980 | 1.000 |
| DM, n (%) | 12(29.3%) | 2(22.2%) | 57(23.4%) | 0.535 | 1.000 |
| Hypertension, n (%) | 27(65.9%) | 6(66.7%) | 146(60.1%) | 0.598 | 1.000 |
| CAD, n (%) | 36(87.8%) | 8(88.9%) | 175(71.7%) | 0.048 * | 0.451 |
| Early CAD, n (%) | 11(26.8%) | 5(55.6%) | 24(9.8%) | 0.005 ** | 0.001 ** |
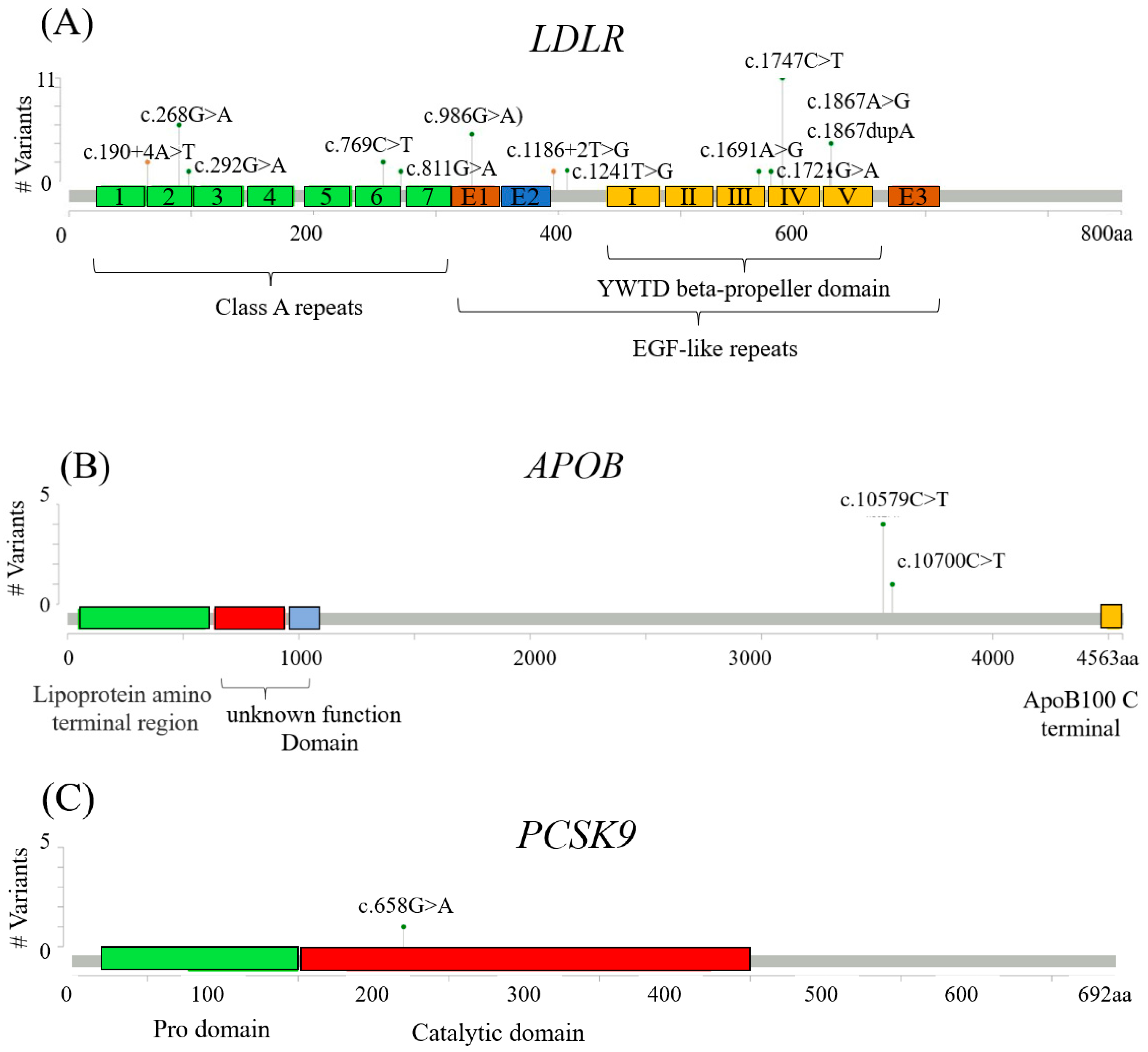
| Gene | Chromosome: Position | HGVSc | HGVSp | Variant Type | Clinical Significance | SNV ID | Number |
|---|---|---|---|---|---|---|---|
| LDLR | chr19:11227576 | c.1747C>T | p.(His583Tyr)) | missense | pathogenic | rs730882109 | 11 |
| LDLR | chr19:11221373 | c.986G>A | p.(Cys329Tyr) | missense | pathogenic | rs761954844 | 5 |
| LDLR | chr19:11213417 | c.268G>A | p.(Asp90Asn) | missense | pathogenic | rs749038326 | 4 |
| LDLR | chr19:11230789 | c.1867A>G | p.(Ile623Val) | missense | pathogenic | rs555292896 | 4 |
| LDLR | chr19:11217315 | c.769C>T | p.(Arg257Trp) | missense | pathogenic | rs200990725 | 2 |
| LDLR | chr19:11224008 | c.1241T>G | p.(Leu414Arg) | missense | likely pathogenic | rs748554592 | 1 |
| LDLR | chr19:11213441 | c.292G>A | p.(Gly98Ser) | missense | likely pathogenic | rs750474121 | 1 |
| LDLR | chr19:11227550 | c.1721G>A | p.(Arg574His) | missense | likely pathogenic | rs777188764 | 1 |
| LDLR | chr19:11217357 | c.811G>A | p.(Val271Ile) | missense | likely pathogenic | rs749220643 | 1 |
| LDLR | chr19:11226874 | c.1691A>G | p.(Asn564Ser) | missense | likely pathogenic | rs758194385 | 1 |
| LDLR | chr19:11211025 | c.190+4A>T | splice_region, intron | pathogenic | rs769446356 | 2 | |
| LDLR | chr19:11222317 | c.1186+2T>G | splice donor | likely pathogenic | rs779921498 | 2 | |
| LDLR | chr19:11230789: 11230788 | c.1867dupA | p.(Ile623AsnfsTer22) | frameshift | likely pathogenic | rs1555807206 | 1 |
| APOB | chr2:21229161 | c.10579C>T | p.(Arg3527Trp) | missense | pathogenic | rs144467873 | 4 |
| APOB | chr2:21229040 | c.10700C>T | p.(Thr3567Met) | missense | pathogenic | rs368278927 | 1 |
| PCSK9 | chr1:55518323 | c.658G>A | p.(Ala220Thr) | missense splice_region | pathogenic | rs768795323 | 1 |
| Variables | FH Pathogenic Variant Carriers | LDLR c.986G>A/c.268G>A Variant Carriers * | FH Pathogenic Variant Non-Carriers | p Value 1 | p Value 2 |
|---|---|---|---|---|---|
| Number | 41 | 9 | 244 | ||
| Revascularization on follow-up, n (%) | 21(51.2%) | 6(66.7%) | 54(22.1%) | <0.001 ** | 0.007 ** |
| PCI, n (%) | 12(29.3%) | 4(44.4%) | 48(19.7%) | 0.235 | 0.089 |
| CABG, n (%) | 8(19.5%) | 2(22.2%) | 5(2.0%) | <0.001 ** | 0.022 * |
| Variables | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| Odds Ratio | (95% CI) | p Value | Odds Ratio | (95% CI) | p Value | |
| Age, years | 1.02 | (1.00–1.04) | 0.090 | |||
| Sex, men | 2.40 | (1.35–4.28) | 0.003 ** | 2.23 | (1.06–4.68) | 0.034 * |
| Body mass index, kg/m2 | 1.04 | (0.97–1.12) | 0.253 | |||
| sBP, mmHg | 1.01 | (1.00–1.03) | 0.097 | 1.02 | (1.00–1.04) | 0.081 |
| dBP, mmHg | 1.00 | (0.98–1.02) | 0.875 | 0.98 | (0.95–1.01) | 0.219 |
| Triglycerides, mg/dL | 1.00 | (1.00–1.01) | 0.123 | |||
| Cholesterol, mg/dL | 1.00 | (1.00–1.01) | 0.262 | |||
| LDL-C, mg/dL | 1.02 | (1.00–1.03) | 0.024 * | 1.02 | (1.00–1.03) | 0.019 * |
| HDL-C, mg/dL | 0.97 | (0.95–1.00) | 0.037 * | |||
| HbA1c, % | 1.67 | (1.05–2.65) | 0.032 * | |||
| Creatinine, mg/dL | 1.33 | (0.91–1.94) | 0.143 | |||
| eGFR, mL/min/1.73 m2 | 0.99 | (0.98–1.00) | 0.011 * | 0.99 | (0.98–1.00) | 0.079 |
| Smoking | 2.51 | (1.41–4.46) | 0.002 ** | 2.09 | (1.04–4.19) | 0.039 * |
| DM | 2.93 | (1.32–6.49) | 0.008 ** | 2.42 | (1.02–5.73) | 0.045 * |
| Hypertension | 2.38 | (1.36–4.16) | 0.002 ** | |||
| FH genetic variation | ||||||
| Non-carriers | Reference | Reference | ||||
| Carriers | 3.29 | (1.13–9.59) | 0.029 * | 3.17 | (1.01–9.92) | 0.047 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, W.-J.; Chuang, H.-N.; Chen, Y.-M.; Liang, K.-W.; Tung, H.; Chen, J.-P.; Lee, I.-T.; Wang, J.-S.; Lin, C.-H.; Lin, H.-J.; et al. Familial Hypercholesterolemia Genetic Variations and Long-Term Cardiovascular Outcomes in Patients with Hypercholesterolemia Who Underwent Coronary Angiography. Genes 2021, 12, 1413. https://doi.org/10.3390/genes12091413
Lee W-J, Chuang H-N, Chen Y-M, Liang K-W, Tung H, Chen J-P, Lee I-T, Wang J-S, Lin C-H, Lin H-J, et al. Familial Hypercholesterolemia Genetic Variations and Long-Term Cardiovascular Outcomes in Patients with Hypercholesterolemia Who Underwent Coronary Angiography. Genes. 2021; 12(9):1413. https://doi.org/10.3390/genes12091413
Chicago/Turabian StyleLee, Wen-Jane, Han-Ni Chuang, Yi-Ming Chen, Kae-Woei Liang, Hsin Tung, Jun-Peng Chen, I-Te Lee, Jun-Sing Wang, Ching-Heng Lin, Hsueh-Ju Lin, and et al. 2021. "Familial Hypercholesterolemia Genetic Variations and Long-Term Cardiovascular Outcomes in Patients with Hypercholesterolemia Who Underwent Coronary Angiography" Genes 12, no. 9: 1413. https://doi.org/10.3390/genes12091413