
Abstract
Throughout the viral life cycle, interplays between cellular host factors and virus determine the infectious capacity of the virus. The pandemic of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) poses a great threat to human life and health. Extensive studies identified a number of host proviral and antiviral factors for SARS-CoV-2. In this review, we summarize the current understanding of the interplay between SARS-CoV-2 and cellular factors during virus entry and replication. Our review will highlight the future direction of study on the infection and pathogenesis of SARS-CoV-2, as well as novel therapeutic strategies and effective antiviral targets for COVID-19.
Similar content being viewed by others


Introduction
Coronavirus disease 2019 (COVID-19) caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a multi-organ disease leading to multiple clinical symptoms such as high fever, cough, dyspnea, muscle pain headache, etc. [1]. COVID-19 has resulted in over 100 million confirmed cases and only a few islands worldwide showed no SARS-CoV-2 infection as April 11 of 2021. In the past 2 decades, it has been the third emerging zoonotic coronavirus with lower mortality and increased SARS-CoV-2 infectivity compared with severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 and Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012, which facilitates its widespread transmission and pandemics [2].
As a β-coronavirus of the coronavirus family, SARS-CoV-2 contains an enveloped single-stranded positive-sense RNA genome that encodes four structural proteins consisting of the envelope protein (E), membrane protein (M), nucleocapsid protein (N), and spike protein (S) [3,4,5]. The E protein is a small integrated membrane protein determining the shape of the virus envelope, which is fundamental for virus pathogenicity [6]. The M protein functions in virus assembly and particle formation which exists in the form of a dimmer in the virion leading to curve membrane and bind to the nucleocapsid protein [6]. The N protein can bind to the RNA genome and participate in replication, assembly, and host cell response in the process of virus infection [6]. The most prominent surface protein is the S composed of two subunits, S1 and S2. Structurally, the N- and C- terminal portions of S1 are partially folded into two independent domains containing the N-terminal domain (NTD) and the C-terminal domain (CTD). The C-terminal domain acts as receptor binding domains (RBDs). The S2 subunits are key determinants for regulating membrane fusion and mediating delivery of virus RNA genome into host cell [7].
The binding of the spike glycoprotein to receptors is acknowledged as the first stage of SARS-CoV-2 life cycle, which is responsible for host range and cellular tropism [8, 9]. The hydrolysis and proteolytic cleavage of the spike protein catalyzed by cellular proteases largely determines whether SARS-CoV-2 invades host cell through plasma membrane or endocytosis [10]. Virus RNA is released into the host cytoplasm after membrane fusion, and then the host cell and its own machinery are utilized to establish viral replication and transcription complexes. The new viral particles are assembled before budding and releasing. However, the host proviral factors and antiviral factors engaging in these processes remain largely elusive.
One of the earliest studies mapped the SARS-CoV-2 host protein interplay underlying genome-wide CRISPR-Cas9 screens [11]. Then, researchers have taken into account that most proviral and antiviral genes regulated SARS-CoV-2 infection at the level of viral entry [12,13,14]. This review focuses on characterizing the role of host proviral and antiviral factors in SARS-CoV-2 infection, which reveals the infection mechanism of SARS-CoV-2 and provides a broader perspective for forthcoming research.
Host factors
Adsorption
ACE2
SARS-CoV and hCoV-NL63 employed angiotensin-converting enzyme 2 (ACE2) for host cell entry [15, 16]. The structure of the spike protein of SARS-CoV and SARS-CoV-2 displayed high similarity [7]. SARS-CoV-2 S1 CTD domain, also known as the RBD, was a critical region for interacting with ACE2 receptor and displayed stronger affinity than SARS-CoV, thus SARS-CoV-2 was much more contagious [7, 17, 18]. Anti-human ACE2 antiserum blocked SARS-S- and SARS-2-S- driven entry [19]. SARS-CoV-2 did not infect cells that unexpressed ACE2 [5]. Co-incubation of human recombinant soluble ACE2 (hrsACE2) with SARS-CoV-2 reduced the proliferation rate of the virus by 1000–5000 fold [20]. Therefore, the evidence suggests that ACE2 is a fundamental factor for SARS-CoV-2 infection. A large amount of researchers has done detailed studied on ACE2 in various aspects. Expression of ACE2 was relatively limited in gastrointestinal tract, kidney, and heart organs, accordingly it was difficult to explain the multi-organ tropism of SARS-CoV-2 [21]. Consequently, existing host factors of SARS-CoV-2 still need extra investigation.
CD147
CD147, known as Basigin or extracellular matrix metalloproteinase inducer (EMMPRIN), is a transmembrane glycoprotein that belongs to the immunoglobulin superfamily [22]. In the past, CD147 interacted with virus-associated cyclophilin A (CyPA) in the entry of host cells in SARS-CoV and human immunodeficiency virus (HIV) [22, 23]. Recently, this result was confirmed in SARS-CoV-2. Co-immunoprecipitation, ELISA, and immuno-electron microscope revealed that CD147 interacted with the spike protein RBD of SARS-CoV-2 to enhance virus invasion into host cell [24]. Knocking down CD147 decreased virus copy number markedly and overexpression of CD147 increased virus infection. Blocking CD147 by Meplazumab, an anti-CD147 humanized antibody, also had an inhibitory effect on SARS-CoV-2 [24]. Interestingly, CD147 co-localized with the spike protein, while it did not interact with ACE2. Expression levels of CD147 displayed higher than that of ACE2, suggesting that CD147 was potentially a novel receptor for SARS-CoV-2 entry through endocytosis, especially in ACE2-deficient cell types [24].
AXL
AXL, a tyrosine kinase, is one of TAM-family phosphatidylserine members that transmit signals from the extracellular into the cytoplasm [25]. Biolayer interferometry (BLI) quantification assays and binding assays have shown that AXL co-localized to the spike protein on the membrane surface and interacted with the NTD of SARS-CoV-2 S rather than RBD directly. Overexpression of AXL greatly elevated the number of SARS-CoV-2 virus pseudotype particles, indicating that AXL was a host factor of SARS-CoV-2 [26]. Other tyrosine-protein kinases, such as MER and FGFR, belong to TAM-family receptor families that cannot boost viral entry into cells, suggesting that AXL-mediated viral infection was highly specific [26]. AXL was expressed in most human tissues compared with ACE2 expression. Knocking out AXL but not ACE2 blocked viral infection in the pulmonary epithelial cells. Soluble human ACE2 failed to block viral infection in AXL-overexpression cells, revealing that AXL promoted SARS-CoV-2 attachment and entry independent of ACE2 [26]. Likewise, AXL has been investigated to promote authentic SARS-CoV-2 infection in HEK293T and H1299 cells [26]. There is abundant room for further work in determining whether co-factors exist between AXL and ACE2 involved in similar infection proceedings.
Heparin sulfate
Heparin sulfate (HS) is a linear sulfated anionic polysaccharide linking to cell membrane and extracellular matrix proteoglycans covalently [27]. HS acted as an attachment factor involved in a wide range of viruses, for instance, herpes simplex virus (HSV), HIV, and HCoV-NL63 [28,29,30]. HS also emerged as an indispensable co-factor for SARS-CoV-2 infection. Molecular modeling revealed a single RBD bound to both HS and ACE2 protein receptors on the cell surface [31]. Recombination experiments displayed HS bound to SARS-CoV-2 spike protein through the RBD domain in line with expectation that ACE2 bound to an open RBD conformation, while this binding was independent of ACE2 expression [31]. Transmission electron microscopy analyzed the total amount of ACE2 protein connects with S protein increased in the presence of dp20 oligosaccharides derived from heparin [31]. Suppression of HS synthesis reduced binding of ACE2 and S protein under treatment with protease or removal, indicating HS could improve the ability of S-ACE2 binding [31]. Similar results have been confirmed in pseudoviral systems and bona fide SARS-CoV-2 virions [31]. Hence, both heparin sulfate and ACE2 are able to bind the spike protein and heparin sulfate facilitates the interaction of SARS-CoV-2 S protein and ACE2 [31, 32].
Neuropilin-1/2
One of striking differences between SARS-CoV-2 and SARS-CoV is that S protein of SARS-CoV-2 exhibits a polybasic furin-type cleavage site, RRAR, at the S1/S2 boundary [17, 33, 34]. Furin-mediated cleavage created an amino acid sequence (682RRAR685) at the C terminus of the S1 protein conforming to the “C-end rule” (CendR) [35]. CendR peptides have been shown to bind to neuropilin transmembrane receptors (NRP1 and NRP2) [35]. The S1 protein associated with endogenous NRP1 and ACE2, whereas deletion of the CendR motif reduced this association by about 75%. Silencing of NRP1 by shRNA or knockout diminished SARS-CoV-2 infection [36]. EG00229, a selective NRP1 antagonist, blocked binding and internalization of CendR and NRP ligands efficiently thereby decreasing SARS-CoV-2 infection [37, 38]. Surprisingly, this inhibition did not affect the binding of virus and cell surface indicating NRP1 boost SARS-CoV-2 entry and infection, nevertheless, a CendR-independent mechanism involved in affecting infection via NRP1 may present additionally [36]. The crystal structure of the complex of the NRP1 b1 domain and the S1 CendR peptides also implemented the binding of them [36]. NRP1 could play a considerable role in addressing the issue that SARS-CoV-2 infected the cell with low ACE2 expression.
SR-BI
As a cell surface high-density lipoprotein (HDL) receptor, the scavenger receptor B type 1 (SR-BI) regulated uptake of cholesterol selectively by a mechanism different from the classical low-density lipoprotein (LDL) receptor pathway [39]. SR-BI mediated the increase of cell membrane cholesterol content while cholesterol was regarded as a crucial determinant for viral uptake and fusion, including influenza A virus (IAV) and respiratory syncytial virus (RSV), flavivirus, porcine rotavirus (PRV), SARS-CoV, and hepatitis C virus (HCV) [40,41,42,43,44,45]. SR-BI likewise plays a valuable role upon SARS-CoV-2 infection [46]. Researchers investigated the specific affinity of SARS-CoV-2 RBD to cholesterol and possible HDL components and observed HDL could increase SARS-CoV-2 attachment on the cell surface [46]. Depletion or inhibition of SR-BI decreased HDL-mediated enhancement of SARS-CoV-2 attachment, while SR-BI overexpression elevated SARS-CoV-2 entry [46]. Given that SARS-CoV-2 S could not bind to SR-BI directly, there may be a linker between SARS-CoV-2 and SR-BI. Combination of SR-BI and ACE2 expression displayed higher susceptibility than each individual. Especially, SR-BI affected SARS-CoV-2 infection only when ACE2 was overexpressed, suggesting that SR-BI was a cofactor with ACE2 [46]. The molecular mechanisms of the interaction between HDL and SARS-2-S1 protein require to be further measured.
ASGR1 and KREMEN1
ASGR1 and KREMEN1 as the alternative entry receptors associating with ACE2 displayed multi-organ tropism to interact with SARS-CoV-2 [47]. Cells expressing KREMEN1 or ASGR1 exhibited SARS-CoV-2 infection when knocking out ACE2, indicating these receptors modulated viral entry independent of ACE2. Researchers demonstrated ACE2 only bound to the RBD, while ASGR1 and KREMEN1 could bind to two domains at least. They were termed as ASK (ACE2/ASGR1/KREMEN1) entry receptors [47]. Subsequent experiments suggested that ASK combination was generally more relevant to viral infection than individual receptors underlying the multi-organ tropism of SARS-CoV-2 [21, 48]. After analyzing a recently published single-cell sequencing map of the upper respiratory tract, ACE2 was primarily expressed in epithelial populations, while ASGR1 and KREMEN1 were enriched in both epithelial and immune populations [47, 49]. Given that SARS-CoV-2 was mainly found in epithelial ciliated and secretory cells as well as immune non-resident macrophages, ASGR1 and KREMEN1 bound to the extracellular domain of S protein together with ACE2, thereby promoting SARS-CoV-2 and host interactions [49, 50].
HMGB1
High mobility group box 1 protein (HMGB1) is an evolutionarily conserved chromatin-binding protein, which exists in nucleated cells ubiquitously and serves as a typical alarm for innate immunity in viral infection [51,52,53]. The functional screening revealed a proviral role for HMGB1 in SARS-CoV-2 infection. HMGB1 depletion improved cell survival against SARS-CoV-2 infection, the degree of which was correlated to the abundance of HMGB1 protein [13]. The production of SARS-CoV-2 has been reduced by ~ 2-log and ACE2 transcripts were reduced in HMGB1 knockout cells, indicating that HMGB1 regulated ACE2 expression to affect SARS-CoV-2 infectivity [13]. The H3K27ac level displayed a significant reduction at the ACE2 transcription start site downstream by ChIP-seq analysis and the chromatin accessibility decreasing trend has been observed by ATAC-seq analysis, also indicating that HMGB1 was important for ACE2 expression [13]. HMGB1 levels have elevated in the nucleus and cytoplasm during SARS-CoV-2 infection, however, the addition of recombinant HMGB1 protein had no effect on SARS-CoV-2 infection, suggesting that HMGB1 regulated SARS-CoV-2 infection through acting cells intrinsically, not as an alarm [13]. Hence, HMGB1 exerts a novel role in regulating ACE2 expression thereby affecting SARS-CoV-2 susceptibility in a cell-intrinsic manner.
RAB7A
Recent observations have shown that SARS-CoV-2 with intact spike protein was endocytosed into the TMPRSS-negative Vero cells, rather than through the plasma membrane fusion pathway [54]. Likewise, when multi-basic residues (RRAR) of the SARS-CoV-2 spike protein were deleted, virus entered the host cells via the endosomal pathway regulated by the endosomal cargo sorting complexes [54]. Genome-wide CRISPR screening of RRAR deletion mutants investigated that a wide range of host factors was required for the entry of SARS-CoV-2 through the endosomal pathway, including the retromer complex, the COMMD/CCDC22/CCDC93 (CCC) complex, Wiskott–Aldrich syndrome protein, and SCAR homologue (WASH) complex, and actin-related protein 2/3 (Arp2/3) complex, etc. In parallel, silencing CCC, retromer, and WASH complexes perturbed the surface expression of ACE2 [54]. Co-immunofluorescence images provided results of co-localization of ACE2-staining RAB7A knockout cells with early endosome marker rather than with lysosomal markers, confirming that RAB7A regulated cell surface expression of ACE2 involved in viral entry, probably via isolating ACE2 receptor in endosomal vesicles [14]. Surprisingly, a recent study reported RAB7A might be partially dispensable for SARS-CoV-2 infection, whereas RAB10 and RAB14 GTPases were critical for infection [55]. The role of RAB7A involved in SARS-CoV-2 infection needs to be further investigated.
Penetration
TMPRSS2/4/11
TMPRSS2 primed spike protein resulting in association with ACE2 to invade host cell. In the prevailing view, priming of SARS-CoV-2 S protein by cellular serine protease TMPRSS2 was pivotal for virus infection in the host cell [56, 57]. TMPRSS2 inhibitors, such as camostat mesylate, blocked SARS-CoV-2 S protein driven entry while overexpression of TMPRSS2 rescued entry from inhibition, suggesting that SARS-CoV-2 S protein bound to host cells primed by TMPRSS2 [19, 58]. Expression of both ACE2 and TMPRSS2 was observed in less than 10% of target cells which were infected SARS-CoV-2 more sensitive through datasets [59, 60]. Ou et al. found that TMPRSS2/4/11 was believed to be a host cell factor that had an effect on S protein activation mediating SARS-CoV-2 entry into cell lines [10].
Furin
The processing mediated by furin expanded the tropism of SARS-CoV-2 cell and tissue, as well as increased its transmissibility and infectivity [33, 34]. Previous analyses have shown that a group of viruses were cleaved by furin in infected cells contributing to fusion activity and virus entry [61, 62]. Furin cleavage at the S1/S2 site activated SARS-CoV-2 S protein leading to undergo a conformational change that exposed the receptor binding site and boost membrane fusion [34]. Inhibition of furin blocked SARS-CoV-2 S protein cleavage resulting in suppression of viral entry and syncytium formation, suggesting that furin was a crucial host cell protease to proactive viral infection through cleaving SARS-CoV-2 S protein [34, 63]. Furin could accumulate with TMPRSS2 and cathepsin affecting cell invasion of SARS-CoV-2 [9]. Interestingly, a high concentration of trypsin promoted membrane fusion mediated by S mutant without furin cleavage site [64]. The next challenge is to evaluate whether SARS-CoV-2 infects host cells with high efficiency in the absence of the furin cleavage site [64]. Moreover, a recent study on SARS-CoV-2 replication showed furin cleavage was required for efficient SARS-CoV-2 replication in Calu3 cells which expressed a low level of TMPRSS2 and cathepsin L [32].
Cathepsin L
The endosomal cathepsin L in the lysosome has been identified to be required for SARS-CoV infection [65, 66]. Accordingly, researchers have analyzed treatment by ammonium chloride, which blocked cathepsin B and L activity, strongly inhibited SARS-2-S-driven entry into host cell [19]. However, Ou et al. showed that the inhibitor of cathepsin B (CA-074) had no significant effect on virus entry, whereas cathepsin L inhibition treatment (SID 26681509) decreased entry by more than 76%, suggesting cathepsin L rather than cathepsin B should be a key determinant of S protein priming in lysosome for SARS-CoV-2 entry through endocytosis [10]. Further research should be undertaken to investigate the role of Cathepsin B/L involved in SARS-CoV-2 infection.
PIKfyve
3-Phosphoinositides are particularly essential for endocytosis through recruiting multiple effector proteins. One of them, phosphatidylinositol-3,5-bisphosphate [PI(3,5)P2], has core cellular functions in endomembrane homeostasis regulating early endosome to late endosome dynamics [67]. Phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) is a critical enzyme synthesizing PI(3,5)P2 for the association with the early endosome [67, 68]. Suppression of PIKfyve by potent inhibitors (apilimod or YM201636) reduced SARS-CoV-2 S pseudovirions entry through endocytosis, indicating that PIKfyve might be a potential drug target for SARS-CoV-2 [10, 69].
TPC2
TPC2, a calcium channel expressed on the lysosomal membrane, is activated by the phosphoinositide PI(3,5)P2 in lysosome, and TPC proteins play a key role in Ebolavirus infection [70,71,72]. The entry of SARS-CoV-2 S pseudovirions was restrained by tetrandrine, an inhibitor for TPC2, supporting the notion that TPC2 was effective for SARS-CoV-2 entry [10].
Uncoating
TMEM106B
Transmembrane Protein 106B (TMEM106B), a glycosylated transmembrane protein, is localized in the late endosome and lysosome compartments regulating lysosomal functions [73,74,75]. There is evidence that TMEM106B plays a crucial role in SARS-CoV-2 infection. sgRNAs targeting TMEM106B protected multiple human cells from the SARS-CoV-2-induced cytopathic effect and the release of virus progeny in the supernatant was also reduced, indicating a crucial determinant for TMEM106B in SARS-CoV-2 infection [76, 77]. Knocking out TMEM106B did not decrease the number of entry virions suggesting that TMEM106B has no effect on S-mediated entry [76]. Additionally, single-cell sequencing analysis has shown TMEM106B was highly expressed in SARS-CoV-2 susceptible airway cells, especially ciliated and secretory cells [77]. Combining with previous studies that NPC1, the endo/lysosomal cholesterol transporter protein has been considered as a lysosomal receptor for Ebolavirus [78]. TMEM106B has functioned in lysosomal acidification via binding to vacuolar-ATPase accessory protein 1 (AP1) [79]. Consequently, TMEM106B may boost cytoplasmic vesicle acidification on the endosomal–lysosomal pathway thereby promoting the delivery of SARS-CoV-2 genome to the cytoplasm [77].
Biosynthesis
SRPK1/2
SRSF Protein Kinase 1 and 2(SRPK1/2) are serine/arginine-rich protein kinases that express in most human tissues and were involved in regulating alternative transcript splicing and cellular signaling [80, 81]. SRPK2 is highly correlated with SRPK1 in sequence, kinase activity, and substrate specificity [82]. Previously, SRPK1/2 kinase activity has been identified to be a key determinant in the replication of viruses, including Eola virus (EBOV), HCV, human cytomegalovirus (HCMV), human papillomavirus (HPV), HIV-1, hepatitis B virus (HBV), and SARS-CoV [83,84,85,86,87,88,89]. As for SARS-CoV-2, SRPK1/2 is likewise crucial to regulate viral infection. The SRPK1/2 inhibitor, SPRIN340, decreased expression of the viral S protein and RNA and infectious viral titer was reduced by more than three orders of magnitude [90]. Using another small molecule (SPHINX31) that only inhibited SRPK1 also suppressed SARS-CoV-2 replication, indicating SRPK1 was the major kinase for viral infection [90]. The global phosphorylation of SARS-CoV-2 proteins has been reported, and researchers predicted SRPK1/2 phosphorylation sites around SARS-CoV-2 genome in a bioinformatic way [91, 92]. Phosphoproteomic studies have shown that the viral nucleoprotein was phosphorylated at predicted SRPK1 phosphorylation sites [90]. Subsequent phosphorylation assays have demonstrated that N protein phosphorylation was enhanced with the increase of kinase concentrations, suggesting that SRPK1/2 is essential for SARS-CoV-2 replication through altering phosphorylation of the viral nucleocapsid proteins.
VPS34
Vacuolar protein sorting 34 (VPS34) is a member of the phosphoinositide 3-kinase (PI3K) family of lipid kinases that modulates endocytic trafficking, macroautophagy, phagocytosis, and other cellular functions [93]. The effects of VPS34 in the replication of HCV and tomato bushy stunt virus (TBSV) have been reported [94, 95]. Researchers demonstrated that inhibition of VPS34 caused a significant decrease in viral RNA production in agreement with the study of human lung tissue culture in vitro, indicating that VPS34 was critical for SARS-CoV-2 replication [77, 96, 97].
One of the characteristics of CoV infection was the establishment of numerous replication organelles composed of double-membrane vesicles (DMV), double-membrane spherules (DMSs), and convoluted membranes (CM) [98]. DMVs were considered as the main site for viral RNA synthesis where the autophagy membrane marker LC3 localized [99]. SARS-CoV-2 nucleoprotein and nascent viral RNA co-localized with LC3, indicating that the formation of SARS-CoV-2 replication centers was related to LC3 [96]. Subsequent experiments have shown that dsRNA associated with N protein was distributed in the cytoplasm and did not form large inclusion like formations in the presence of VPS34 inhibitors (VPS34-IN1 or PIK-III), probably suggesting that VPS34 has functions for promoting SARS-CoV-2 replication organelles formation [96]. In addition, autophagy activation could suppress viral replication, while inhibition of VPS34 has been identified to inhibit autophagy [100, 101]. The destruction of SARS-CoV-2 replication caused by VPS34 inhibition might reflect the inhibition of VPS34 non-autophagy-related functions [77, 96].
SCAP
Sterol regulatory element-binding protein (SREBP) transports from ER to Golgi apparatus in the presence of sterols for processing and activation via COPII-coated vesicles [102]. Its escort protein SCAP sequesters SREBPs in the ER to prevent transport thereby regulating lipid and cholesterol homeostasis [103]. Researchers identified that SCAP was required for all four of the coronaviruses [55]. Knocking out SCAP led to a decrease of SARS-CoV-2 RNA levels, indicating that SCAP was critical for SARS-CoV-2 replication [55]. Additionally, there were a series of genes found in the genome-scale CRISPR screens up-regulating the cholesterol synthesis pathway such as NPC2, EMC1, SREBF2, etc. [14, 55]. The relationship between the cholesterol synthesis pathway and SARS-CoV-2 will be a significant question for future investigation.
Antiviral factors
Adsorption
Human defensin 5
Human defensin 5 (HD5) is specifically synthesized and expressed in Paneth cell of the small intestine among α defensins, which can bind lipids and glycosylated proteins [104,105,106]. Binding of HD5 to the ligand-binding domain (LBD) of ACE2 weakened the binding of SARS-CoV-2 S1 to ACE2 in a high content of HD5 [107]. There was a hypothesis that the glycosylated residue of the HD5 could attach to S1 protein leading to prevent viral invasion theoretically [17, 108]. The adherence and recruitment of SARS-CoV-2 S1 to host cell were reduced significantly in the presence of HD5, indicating HD5 prohibited entry of SARS-CoV-2 [107]. A structure-dependent interaction between HD5 and ACE2 demonstrated human intestine was required for the innate defense against SARS-CoV-2 [107].
PSGL-1
P-Selectin glycoprotein ligand-1 (PSGL-1) is an integral membrane protein binding to P-, E-, and L-selectins, which promotes immune cell initial attachment and subsequent movement resulting in cell migration into inflamed tissues [109, 110]. PSGL-1 served as a functional receptor for enterovirus A71 (EV-A71) penetration and replication; however, PSGL-1 has been illustrated to block viral infection as an HIV inhibitory factor induced by IFN-γ [111, 112]. Based on pseudovirus infection system, the infectivity of PSGL-1-imprinted SARS-CoV-2 particles was much lower than SARS-CoV in the virus-producer cells, suggesting that PSGL-1 potently inhibited SARS-CoV-2 infectivity [113]. The virion attachment assay displayed PSGL-1 impaired virus infectivity through a structural hindrance of attachment to target cells [113]. The release of SARS-CoV and SARS–CoV-2 pseudovirions has been slightly blocked in PSGL-1-expressed cells, indicating that expression of PSGL-1 had minimal effects on viral release [113, 114]. In general, PSGL-1 has been identified to impair the spike proteins incorporation and block virus attachment primarily by interfering with particle binding to host cell, thereby suppressing SARS-CoV or SARS-CoV-2 infectivity [113]. Interestingly, the viral accessory proteins downregulated PSGL-1 levels to antagonize effects during HIV-1 infection [112, 114]. Whether the viral evasion of PSGL-1 exists in coronaviruses infection needs more studies to clarify.
Sialic acids
Previously, sialic acids have been identified to act as host factors for MERS-CoV [115]. Treatment with neuraminidase (NA) that removes sialic acids on the cell surface significantly enhanced SARS-CoV attachment and replication, which was confirmed by both real-time quantitative PCR (qRT-PCR) and plaque assays. The production of SARS-CoV-2 infectious particles was increased modestly, suggesting that sialic acids restricted SARS-CoV-2 infection and yet SARS-CoV-2 could overcome this restriction partly in Calu3 and Caco2 cells [32]. Knocking out sialic acids on the cell surface has been shown to boost SARS-CoV-2 attachment marginally matched with SARS-CoV, validating that sialic acids limited SARS-CoV-2 infection to a lesser extent [32]. Molecular glycan interactions were predicted between the glycans at N90, N322, and N546 of ACE2 and the spike protein [116].
Transfection of ACE2 mutants that abolished all N- and all O-linked glycosylation sites dramatically reduced SARS-CoV-2 genome copy, whereas N90Q mutant demonstrated an increase in the cell lysate, suggesting that glycans including sialic acids at the N90 site of ACE2 had indispensable functions on ACE2-spike interaction during SARS-CoV-2 infection [32]. Accordingly, sialic acids precluded the interaction between ACE2 and the spike protein during SARS-CoV-2 infection contributed by presenting on the N90 position of ACE2 possibility [32].
Penetration
CH25H
Coronavirus infections induced interferons (IFNs) production, which initiated innate immune signaling cascades as the first line of defense [117]. Recognition of viral invasion by sensing of molecular patterns associated with innate immune activated IFN pathways, further leading to activate IFN-stimulated genes (ISGs) [118]. Cholesterol-25-hydroxylase (CH25H) was one of the antiviral ISGs, which encoded an ER-associated enzyme catalyzing formation of the oxysterol 25-hydroxycholesterol (25HC) from cholesterol [118, 119]. In previous studies, 25HC has been shown to inhibit the host cell entry of enveloped DNA and RNA viruses [118, 120]. Consistently, overexpression of CH25H restricted SARS-CoV-2 entry. 25HC activated the ER-localized acyl-CoA: cholesterol acyltransferase (ACAT) which mobilized the depletion of accessible cholesterol from the plasma membrane and did not form spike-induced syncytia, causing SARS-CoV-2 virus-cell fusion to be suppressed [121]. 25HC treatment downregulated the expression of ALOD4 which could trap accessible cholesterol at the plasma membrane, suggesting that 25HC depleted plasma membrane cholesterol to prohibit SARS-CoV-2 virus-cell fusion [121,122,123]. Similar effects have been applied for both SARS-CoV and MERS-CoV pseudoviruses [121]. Therefore, 25HC might be a pan-antiviral drug to intervene in SARS-CoV-2 and other possible emerging coronaviruses therapies in the future.
LY6E
Lymphocyte antigen 6 complex, locus E (LY6E) is another ISG modulating viral infection, particularly viral entry [124]. LY6E acted a pivotal part in restraining the infectious entry of human CoVs [125]. LY6E is a glycosylphosphatidyl-inositol (GPI)-anchored protein on the cell surface containing a conserved functional site (L36) [126]. L36A mutation had no restriction impact on the entry of human CoVs, and the mutant LY6E that disrupts the addition of GPI anchor also did the same results, suggesting the conserved residue and location were responsible for LY6E modulation of viral attachment [125, 127]. LY6E potently blocked syncytia formation mediated by the spike protein, demonstrating that LY6E suppresses virus entry through viral and cellular membrane fusion. This finding was also consistent with the results from a quantitative fusion assay [127]. Surprisingly, LY6E repressed viral infection regardless of the absence or presence of cell surface and endosomal proteases, suggesting restriction of viral fusion did not depend on spike proteins activation [125, 127]. In conclusion, LY6E is a broad-spectrum CoV antiviral factor inhibiting CoV spike protein-mediated membrane fusion to control a series of CoVs infection and pathogenesis, including SARS-CoV-2.
Replication
ZAP
The binding of zinc-finger antiviral protein (ZAP) to the viral RNA sequence containing cytosine-phosphate-guanine (CpG) dinucleotides has been recently detected to attenuate virus replication [128,129,130]. It was previously found that coronaviruses exhibit striking CpG dinucleotides suppression driven by ZAP to some extent, as did SARS-CoV-2 [131, 132]. ZAP knockdown elevated the level of SARS-CoV-2 RNA by 1.4-fold in the absence of IFN, whereas the enhancing effect was more pronounced in the presence of IFN-γ(6.8 ×) or IFN-α(3.1 ×), suggesting SARS-CoV-2 replication was restricted by endogenous ZAP especially in the presence of type II IFN [133]. Cellular nucleic acid-binding protein (CNBP) is a highly conserved zinc-finger protein [134]. The level of viral RNA was prompted in the CNBP-knockout cells during SARS-CoV-2 infection and enhanced version of the crosslinking and immunoprecipitation (eCLIP) assays displayed CNBP bound to SARS-CoV-2 RNA at several strongly enriched binding sites [135].
ZAP exists two major spliced isoforms on the C-terminal, ZAP-L and ZAP-S [136]. Overexpression of ZAP-L diminished viral RNA by up to 2 orders of magnitude or more, while ZAP-S only achieved modest inhibition by about tenfold at the highest dose, confirming that ZAP-L restricted SARS-CoV-2 more efficiently than ZAP-S [133]. ZAP plays an inhibitory role in SARS-CoV-2 replication, despite the fact that SARS-CoV-2 genome is relatively deficient in CpG dinucleotides [133].
LARP1
Inhibition of PI3K/Akt/mTOR prevented SARS-CoV-2 replication in cells [137]. La-related protein 1 (LARP1) is a novel target of mTORC1 and restricts TOP mRNA translation [138]. Thus, LARP1 might have a critical negative influence on SARS-CoV-2 replication. The eCLIP experiment on host RNAs has shown that LARP1 bound to the SARS-CoV-2 5’-leader sequence which contains a terminal oligopyrimidine like (TOP-like) sequence motif, indicating a direct association of LARP1 with subgenomic mRNAs [135]. LARP1 knockout cells increased levels of intracellular viral RNA and the production of infectious virus by around fivefold higher than wild-type cells, while overexpression of LARP1 decreased viral RNA and infectious virus subsequently [135]. In addition, other proteins involved in SARS-CoV-2 RNA binding and infectious pathways have been also observed [135]. The detailed understanding of the roles for those SARS-CoV-2 RNA binding proteins in SARS-CoV-2 biology need to be further investigated.
Conclusion
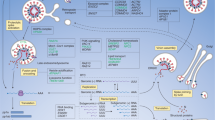
With the current identification of host proteins associated with SARS-CoV-2, the life cycle of SARS-CoV-2 is gradually being elucidated (Fig. 1). Through high-throughput functional screening including CRISPR, RNA interference, or small molecule inhibitors, plenty of host factors and antiviral factors for SARS-CoV-2 were revealed [12,13,14]. Some of these findings are summarized in Tables 1 and 2. Further biochemical research should be undertaken to dissect the detailed mechanisms of those factors. Most of the host proteins are involved in the adsorption, penetration, uncoating, and biosynthesis steps of SARS-CoV-2 life cycle. Relatively little is known on virus assembly and release. Some host proteins are assumed to contribute to the serious clinical outcome of SARS-CoV-2.

Schematic representation of SARS-CoV-2 life cycle and host proteins. The host proviral factors of various steps in the SARS-CoV-2 life cycle are shown in green. The host antiviral factors are shown in red
Understanding the relationship between host and virus provides broad insights into exploring the ambiguous mechanism by which host factors confer virus infectivity and investigating the treatment strategies of COVID-19. All the factors described in this review as well as many host proteins that have not yet been identified are potential targets for drug intervention strategies, which can be applied for developing methods to suppress the viral cycle during SARS-CoV-2 infection.
Data availability
Not applicable.
Code availability
Not applicable.
References
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet 395(10223):497–506
Liu Y, Gayle AA, Wilder-Smith A, Rocklöv J (2020) The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med 27(2):taaa021
Wu A, Peng Y, Huang B, Ding X, Wang X, Niu P et al (2020) Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27(3):325–328
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395(10224):565–574
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273
Liang Y, Wang ML, Chien CS, Yarmishyn AA, Yang YP, Lai WY et al (2020) Highlight of immune pathogenic response and hematopathologic effect in SARS-CoV, MERS-CoV, and SARS-Cov-2 infection. Front Immunol 11:1022
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z et al (2020) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181(4):894-904.e9
Letko M, Marzi A, Munster V (2020) Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 5(4):562–569
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A et al (2020) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA 117(21):11727–11734
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L et al (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11(1):1620
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM et al (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583(7816):459–468
Schneider WM, Luna JM, Hoffmann HH, Sánchez-Rivera FJ, Leal AA, Ashbrook AW et al (2021) Genome-scale identification of SARS-CoV-2 and pan-coronavirus host factor networks. Cell 184(1):120–32.e14
Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu-Culligan WJ, Cai WL et al (2020) Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell S0092–8674(20):31392–31401
Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M et al (2020) Identification of required host factors for SARS-CoV-2 infection in human cells. Cell S0092–8674(20):31394–31395
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426(6965):450–454
Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S (2005) Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci USA 102(22):7988–7993
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H et al (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807):221–224
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–80.e8
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M et al (2020) Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181(4):905–13.e7
Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C (2020) The protein expression profile of ACE2 in human tissues. Mol Syst Biol 16(7):e9610
Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M et al (2011) Basigin is a receptor essential for erythrocyte invasion by plasmodium falciparum. Nature 480(7378):534–537
Chen Z, Mi L, Xu J, Yu J, Wang X, Jiang J et al (2005) Function of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J Infect Dis 191(5):755–760
Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P et al (2020) CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther 5(1):283
O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C et al (1991) Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol 11(10):5016–5031
Wang S, Qiu Z, Hou Y, Deng X, Xu W, Zheng T et al (2021) AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res 31(2):126–140
Cagno V, Tseligka ED, Jones ST, Tapparel C (2019) Heparan sulfate proteoglycans and viral attachment: true receptors or adaptation bias? Viruses 11(7):596
WuDunn D, Spear PG (1989) Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J Virol 63(1):52–58
Roderiquez G, Oravecz T, Yanagishita M, Bou-Habib DC, Mostowski H, Norcross MA (1995) Mediation of human immunodeficiency virus type 1 binding by interaction of cell surface heparan sulfate proteoglycans with the V3 region of envelope gp120-gp41. J Virol 69(4):2233–2239
Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K (2014) Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J Virol 88(22):13221–13230
Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD et al (2020) SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 183(4):1043–57.e15
Chu H, Hu B, Huang X, Chai Y, Zhou D, Wang Y et al (2021) Host and viral determinants for efficient SARS-CoV-2 infection of the human lung. Nat Commun 12(1):134
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281–92.e6
Hoffmann M, Kleine-Weber H, Pöhlmann S (2020) A Multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell 78(4):779–84.e5
Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E (2009) C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci USA 106(38):16157–16162
Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Antón-Plágaro C et al (2020) Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 370(6518):861–865
Jarvis A, Allerston CK, Jia H, Herzog B, Garza-Garcia A, Winfield N et al (2010) Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J Med Chem 53(5):2215–2226
Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S et al (2020) Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370(6518):856–860
Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271(5248):518–520
Shen WJ, Azhar S, Kraemer FB (2018) SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol 80:95–116
Bajimaya S, Frankl T, Hayashi T, Takimoto T (2017) Cholesterol is required for stability and infectivity of influenza A and respiratory syncytial viruses. Virology 510:234–241
Osuna-Ramos JF, Reyes-Ruiz JM, Del Ángel RM (2018) The role of host cholesterol during flavivirus infection. Front Cell Infect Microbiol 8:388
Dou X, Li Y, Han J, Zarlenga DS, Zhu W, Ren X et al (2018) Cholesterol of lipid rafts is a key determinant for entry and post-entry control of porcine rotavirus infection. BMC Vet Res 14(1):45
Li GM, Li YG, Yamate M, Li SM, Ikuta K (2007) Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes Infect 9(1):96–102
Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK et al (2010) Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol 84(1):34–43
Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X et al (2020) HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat Metab 2(12):1391–1400
Gu Yunqing, Cao Jun, Zhang Xinyu, Gao Hai, Wang Yuyan, Wang Jia, et al (2020) Interaction network of SARS-CoV-2 with host receptome through spike protein. bioRxiv:2020.09.09.287508
Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L et al (2020) Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med 383(6):590–592
Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F et al (2020) COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol 38(8):970–979
Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M et al (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 26(5):681–687
Andersson U, Yang H, Harris H (2018) High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol 38:40–48
Andersson U, Ottestad W, Tracey KJ (2020) Extracellular HMGB1: a therapeutic target in severe pulmonary inflammation including COVID-19? Mol Med 26(1):42
Simpson J, Loh Z, Ullah MA, Lynch JP, Werder RB, Collinson N et al (2020) Respiratory syncytial virus infection promotes necroptosis and HMGB1 release by airway epithelial cells. Am J Respir Crit Care Med 201(11):1358–1371
Zhu Y, Feng F, Hu G, Wang Y, Yu Y, Zhu Y et al (2021) A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat Commun 12(1):961
Hoffmann HH, Sánchez-Rivera FJ, Schneider WM, Luna JM, Soto-Feliciano YM, Ashbrook AW et al (2021) Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 29(2):267–80.e5
Beeraka NM, Sadhu SP, Madhunapantula SV, Rao Pragada R, Svistunov AA, Nikolenko VN et al (2020) Strategies for targeting SARS CoV-2: small molecule inhibitors-the current status. Front Immunol 11:552925
Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T (2011) A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol 85(2):873–882
Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S (2012) Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol 86(12):6537–6545
Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN et al (2020) SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181(5):1016–35.e19
Zhou L, Xu Z, Castiglione GM, Soiberman US, Eberhart CG, Duh EJ (2020) ACE2 and TMPRSS2 are expressed on the human ocular surface, suggesting susceptibility to SARS-CoV-2 infection. Ocul Surf 18(4):537–544
Braun E, Sauter D (2019) Furin-mediated protein processing in infectious diseases and cancer. Clin Transl Immunol 8(8):e1073
Izaguirre G (2019) The proteolytic regulation of virus cell entry by furin and other proprotein convertases. Viruses 11(9):837
Cheng YW, Chao TL, Li CL, Chiu MF, Kao HC, Wang SH et al (2020) Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep 33(2):1254
Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z et al (2020) The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther 5(1):92
Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P (2005) Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA 102(33):11876–11881
Huang IC, Bosch BJ, Li F, Li W, Lee KH, Ghiran S et al (2006) SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem 281(6):3198–3203
de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbé S et al (2009) PIKfyve regulation of endosome-linked pathways. Traffic 10(7):883–893
Rutherford AC, Traer C, Wassmer T, Pattni K, Bujny MV, Carlton JG et al (2006) The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci 119:3944–3957
Kang YL, Chou YY, Rothlauf PW, Liu Z, Soh TK, Cureton D et al (2020) Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc Natl Acad Sci USA 117(34):20803–20813
She J, Zeng W, Guo J, Chen Q, Bai XC, Jiang Y (2019) Structural mechanisms of phospholipid activation of the human TPC2 channel. eLife 8:e45222
Li P, Gu M, Xu H (2019) Lysosomal ion channels as decoders of cellular signals. Trends Biochem Sci 44(2):110–124
Sakurai Y, Kolokoltsov AA, Chen CC, Tidwell MW, Bauta WE, Klugbauer N et al (2015) Ebola virus. Two-pore channels control ebola virus host cell entry and are drug targets for disease treatment. Science 347(6225):995–8
Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D et al (2012) Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 287(23):19355–19365
Brady OA, Zheng Y, Murphy K, Huang M, Hu F (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 22(4):685–695
Nicholson AM, Rademakers R (2016) What we know about TMEM106B in neurodegeneration. Acta Neuropathol 132(5):639–651
Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM et al (2021) Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell 184(1):106–19.e14
Baggen J, Persoons L, Vanstreels E, Jansen S, Van Looveren D, Boeckx B et al (2021) Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat Genet 53:435–444
Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N et al (2011) Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477(7364):340–343
Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT et al (2017) Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia-related phenotypes in progranulin-deficient mice. Neuron 95(2):281–96.e6
Aubol BE, Jamros MA, McGlone ML, Adams JA (2013) Splicing kinase SRPK1 conforms to the landscape of its SR protein substrate. Biochemistry 52(43):7595–7605
Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD (2009) Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev 23(4):482–495
Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC et al (1998) SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol 140(4):737–750
Takamatsu Y, Krähling V, Kolesnikova L, Halwe S, Lier C, Baumeister S et al (2020) Serine-arginine protein kinase 1 regulates ebola virus transcription. mBio 11(1):e02565-19
Karakama Y, Sakamoto N, Itsui Y, Nakagawa M, Tasaka-Fujita M, Nishimura-Sakurai Y et al (2010) Inhibition of hepatitis C virus replication by a specific inhibitor of serine-arginine-rich protein kinase. Antimicrob Agents Chemother 54(8):3179–3186
Gaddy CE, Wong DS, Markowitz-Shulman A, Colberg-Poley AM (2010) Regulation of the subcellular distribution of key cellular RNA-processing factors during permissive human cytomegalovirus infection. J Gen Virol 91:1547–1559
Prescott EL, Brimacombe CL, Hartley M, Bell I, Graham S, Roberts S (2014) Human papillomavirus type 1 E1^E4 protein is a potent inhibitor of the serine-arginine (SR) protein kinase SRPK1 and inhibits phosphorylation of host SR proteins and of the viral transcription and replication regulator E2. J Virol 88(21):12599–12611
Tunnicliffe RB, Hu WK, Wu MY, Levy C, Mould AP, McKenzie EA et al (2019) Molecular mechanism of SR protein kinase 1 inhibition by the herpes virus protein ICP27. mBio 10(5):e02551-19
Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J et al (2002) Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol 76(16):8124–8137
Peng TY, Lee KR, Tarn WY (2008) Phosphorylation of the arginine/serine dipeptide-rich motif of the severe acute respiratory syndrome coronavirus nucleocapsid protein modulates its multimerization, translation inhibitory activity and cellular localization. FEBS J 275(16):4152–4163
Heaton BE, Trimarco JD, Hamele CE, Harding AT, Tata A, Zhu X, et al (2020) SRSF protein kinases 1 and 2 are essential host factors for human coronaviruses including SARS-CoV-2. bioRxiv:2020.08.14.251207
Wang C, Xu H, Lin S, Deng W, Zhou J, Zhang Y et al (2020) GPS 50: an update on the prediction of kinase-specific phosphorylation sites in proteins. Genomics Proteomics Bioinform 18(1):72–80
Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M et al (2020) The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182(3):685-712.e19
Backer JM (2016) The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J 473(15):2251–2271
Feng Z, Xu K, Kovalev N, Nagy PD (2019) Recruitment of Vps34 PI3K and enrichment of PI3P phosphoinositide in the viral replication compartment is crucial for replication of a positive-strand RNA virus. PLoS Path 15(1):e1007530
Liefhebber JM, Hague CV, Zhang Q, Wakelam MJ, McLauchlan J (2014) Modulation of triglyceride and cholesterol ester synthesis impairs assembly of infectious hepatitis C virus. J Biol Chem 289(31):21276–21288
Silvas Jesus A, Jureka Alexander S, Nicolini Anthony M, Chvatal Stacie A, Basler Christopher F (2020) Inhibitors of VPS34 and lipid metabolism suppress SARS-CoV-2 replication. bioRxiv:2020.07.18.210211
Yuen CK, Wong WM, Mak LF, Wang X, Chu H, Yuen KY et al (2021) Suppression of SARS-CoV-2 infection in ex-vivo human lung tissues by targeting class III phosphoinositide 3-kinase. J Med Virol 93(4):2076–2083
Snijder EJ, Limpens RWAL, de Wilde AH, de Jong AWM, Zevenhoven-Dobbe JC, Maier HJ et al (2020) A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol 18(6):e3000715
Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S et al (2010) Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 7(6):500–508
Gassen NC, Niemeyer D, Muth D, Corman VM, Martinelli S, Gassen A et al (2019) SKP2 attenuates autophagy through beclin1-ubiquitination and its inhibition reduces MERS-coronavirus infection. Nat Commun 10(1):5770
Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E et al (2014) Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16(11):1069–1079
Su L, Zhou L, Chen FJ, Wang H, Qian H, Sheng Y et al (2019) Cideb controls sterol-regulated ER export of SREBP/SCAP by promoting cargo loading at ER exit sites. EMBO J 38(8):e100156
Luo J, Yang H, Song BL (2020) Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 21(4):225–245
Jones DE, Bevins CL (1992) Paneth cells of the human small intestine express an antimicrobial peptide gene. J Biol Chem 267(32):23216–23225
Ericksen B, Wu Z, Lu W, Lehrer RI (2005) Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob Agents Chemother 49(1):269–275
Wang C, Shen M, Gohain N, Tolbert WD, Chen F, Zhang N et al (2015) Design of a potent antibiotic peptide based on the active region of human defensin 5. J Med Chem 58(7):3083–3093
Wang C, Wang S, Li D, Wei DQ, Zhao J, Wang J (2020) Human intestinal defensin 5 inhibits SARS-CoV-2 invasion by cloaking ACE2. Gastroenterology 159(3):1145–7.e4
Niv Y (2020) Defensin 5 for prevention of SARS-CoV-2 invasion and Covid-19 disease. Med Hypotheses 143:110244
Sako D, Chang XJ, Barone KM, Vachino G, White HM, Shaw G et al (1993) Expression cloning of a functional glycoprotein ligand for P-selectin. Cell 75(6):1179–1186
Somers WS, Tang J, Shaw GD, Camphausen RT (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 103(3):467–479
Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H (2009) Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med 15(7):794–797
Liu Y, Fu Y, Wang Q, Li M, Zhou Z, Dabbagh D et al (2019) Proteomic profiling of HIV-1 infection of human CD4 T cells identifies PSGL-1 as an HIV restriction factor. Nat Microbiol 4(5):813–825
He S, Hetrick B, Dabbagh D, Akhrymuk IV, Kehn-Hall K, Freed EO, et al (2020) PSGL-1 blocks SARS-CoV-2 S protein-mediated virus attachment and infection of target cells. bioRxiv:2020.05.01.073387
Fu Y, He S, Waheed AA, Dabbagh D, Zhou Z, Trinité B et al (2020) PSGL-1 restricts HIV-1 infectivity by blocking virus particle attachment to target cells. Proc Natl Acad Sci USA 117(17):9537–9545
Li W, Hulswit RJG, Widjaja I, Raj VS, McBride R, Peng W et al (2017) Identification of sialic acid-binding function for the middle east respiratory syndrome coronavirus spike glycoprotein. Proc Natl Acad Sci USA 114(40):E8508–E8517
Zhao P, Praissman JL, Grant OC, Cai Y, Xiao T, Rosenbalm KE et al (2020) Virus-receptor interactions of glycosylated SARS-CoV-2 spike and human ACE2 receptor. Cell Host Microbe 28(4):586-601.e6
Park A, Iwasaki A (2020) Type I and type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27(6):870–878
Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK et al (2013) Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38(1):92–105
Lund EG, Kerr TA, Sakai J, Li WP, Russell DW (1998) cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem 273(51):34316–34327
Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY et al (2017) 25-Hydroxycholesterol protects host against zika virus infection and its associated microcephaly in a mouse model. Immunity 46(3):446–456
Wang S, Li W, Hui H, Tiwari SK, Zhang Q, Croker BA et al (2020) Cholesterol 25-hydroxylase inhibits SARS-CoV-2 and other coronaviruses by depleting membrane cholesterol. EMBO J 39(21):6057
Johnson KA, Endapally S, Vazquez DC, Infante RE, Radhakrishnan A (2019) Ostreolysin A and anthrolysin O use different mechanisms to control movement of cholesterol from the plasma membrane to the endoplasmic reticulum. J Biol Chem 294(46):17289–17300
Abrams ME, Johnson KA, Perelman SS, Zhang LS, Endapally S, Mar KB et al (2020) Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat Microbiol 5(7):929–942
Yu J, Liu SL (2019) Emerging role of LY6E in virus-host interactions. Viruses 11(11):1020
Zhao X, Zheng S, Chen D, Zheng M, Li X, Li G et al (2020) LY6E restricts entry of human coronaviruses, including currently pandemic SARS-CoV-2. J Virol 94(18):e00562-e620
Mar KB, Rinkenberger NR, Boys IN, Eitson JL, McDougal MB, Richardson RB et al (2018) LY6E mediates an evolutionarily conserved enhancement of virus infection by targeting a late entry step. Nat Commun 9(1):3603
Pfaender S, Mar KB, Michailidis E, Kratzel A, BoysV’kovski INP et al (2020) LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat Microbiol 5(11):1330–1339
Meagher JL, Takata M, Gonçalves-Carneiro D, Keane SC, Rebendenne A, Ong H et al (2019) Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc Natl Acad Sci USA 116(48):24303–24309
Ficarelli M, Antzin-Anduetza I, Hugh-White R, Firth AE, Sertkaya H, Wilson H et al (2020) CpG dinucleotides inhibit HIV-1 replication through zinc finger antiviral protein (ZAP)-dependent and -independent mechanisms. J Virol 94(6):e01337-e1419
Takata MA, Gonçalves-Carneiro D, Zang TM, Soll SJ, York A, Blanco-Melo D et al (2017) CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 550(7674):124–127
Woo PC, Wong BH, Huang Y, Lau SK, Yuen KY (2007) Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology 369(2):431–442
Xia X (2020) Extreme genomic CpG deficiency in SARS-CoV-2 and evasion of host antiviral defense. Mol Biol Evol 37(9):2699–2705
Nchioua R, Kmiec D, Müller JA, Conzelmann C, Groß R, Swanson CM et al (2020) SARS-CoV-2 is restricted by zinc finger antiviral protein despite preadaptation to the low-CpG environment in humans. mBio 11(5):e01930-20
Calcaterra NB, Armas P, Weiner AM, Borgognone M (2010) CNBP: a multifunctional nucleic acid chaperone involved in cell death and proliferation control. IUBMB Life 62(10):707–714
Schmidt N, Lareau CA, Keshishian H, Ganskih S, Schneider C, Hennig T et al (2020) The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat Microbiol 6(3):339–353
Kerns JA, Emerman M, Malik HS (2008) Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet 4(1):e21
Klann K, Bojkova D, Tascher G, Ciesek S, Münch C, Cinatl J (2020) Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. Mol Cell 80(1):164–74.e4
Fonseca BD, Zakaria C, Jia JJ, Graber TE, Svitkin Y, Tahmasebi S et al (2015) La-related protein 1 (LARP1) represses terminal oligopyrimidine (TOP) mRNA translation downstream of mTOR complex 1 (mTORC1). J Biol Chem 290(26):15996–16020
Funding
This work was supported by grants from National Key Plan for Research and Development of China [2016YFD0500300], National Natural Science Foundation of China [81871663 and 82072270], and Academic promotion programme of Shandong First Medical University [2019LJ001].
Author information
Authors and Affiliations
Contributions
LL wrote the manuscript. LZ conceived the work and revised the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Informed consent
Not applicable.
Research involving human participants and/or animals
Not applicable.
Additional information
Edited by Joachim Jakob Bugert.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lv, L., Zhang, L. Host proviral and antiviral factors for SARS-CoV-2. Virus Genes 57, 475–488 (2021). https://doi.org/10.1007/s11262-021-01869-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-021-01869-2