
Abstract
Three new metal−organic frameworks based on scandium(III) cations and 2,5-thiophenedicarboxylic acid (H2Tdc) are synthesized: [Sc(Tdc)(OH)]·1.2DMF (I), [Sc(Tdc)(OH)]·2/3DMF (II), and (Me2NH2)[Sc3(Tdc)4(OH)2]·DMF (III) (DMF is N,N-dimethylformamide). The structures of the compounds are determined by single-crystal X-ray structure analysis (CIF file CCDC nos. 2067819 (I), 2067820 (II), and 2067821 (III)). The chemical and phase purity of compound I is proved by elemental analysis, thermogravimetry, X-ray diffraction analysis, and IR spectroscopy.
Similar content being viewed by others


INTRODUCTION
Metal−organic frameworks (MOF) represent an already known class of inorganic compounds consisting of metal ions or clusters bound to each other by bridging ligands with the formation of one- (1D), two- (2D), or three-dimensional (3D) periodic structures [1–3]. These compounds can retain porosity after the removal of guest solvent molecules and can be used as sorbents for the very broad range of applications [4–6]. A significant part of applied problems, such as heterogeneous catalysis [7], decontamination of anthropogenic wastes from harmful substances and heavy and radioactive metals [8], selective recovery of metal ions from natural sources [9], etc., requires a relatively high hydrolysis stability of a porous material. Several thousands of diverse MOF are known nowadays, but the high hydrolytic stability is shown only for a restricted scope of systems among which the frameworks based on bridging carboxylate ligands and highly charged cations of oxophilic metals, namely, Sc(III), Cr(III), Al(III), Ti(IV), and Zr(IV), can be distinguished [10–14]. Evidently, when the choice of the metal is restricted, the use of organic bridging ligands should become the main method for the variation of functional properties of porous MOF. It has recently been shown that the inclusion of the thiophene-based heterocyclic fragments in the MOF structure substantially enhances their sorption and luminescence characteristics [15–18]. However, it should be mentioned that the number of the known MOF based on highly charged ions and containing heterocyclic thiophene cycles is low, for example, only two such MOF with the 2,5-thiophenedicarboxylate anions are known for scandium(III) [19, 20]. Therefore, the development of synthetic approaches to the crystallization of new MOF based on the above listed metal cations and carboxylate ligands with heterocyclic moieties is of doubtless scientific interest and perspective practical significance.
The syntheses and crystal structures of three new MOF based on scandium(III) and 2,5-thiophenedicarboxylic acid (H2Tdc) are described in this work.
EXPERIMENTAL
The following initial substances were used as received: scandium(III) trifluoromethanesulfonate (DAlKhIM), 2,5-thiophenedicarboxylic acid (Sigma-Aldrich), sodium hydroxide (Vekton), formic acid (Reaktiv), N,N-dimethylformamide (Vekton), acetone (Vekton), and acetonitrile (Kriokhrom, not lower than reagent grade). IR spectra were recorded in a range of 4000–400 cm–1 in KBr pellets on a VERTEX 80 FT-IR spectrometer. Elemental analysis was carried out on a Vario MICRO cube CHNS analyzer at the analytical laboratory of the Nikolaev Institute of Inorganic Chemistry (Siberian Branch, Russian Academy of Sciences). The powder X-ray diffraction data (PXRD) were obtained on a Shimadzu XRD 7000S powder diffractometer (CuKα radiation). Thermogravimetric analysis (TG) was carried out in an Ar atmosphere on a NETZSCH TG 209 F1 thermal analyzer with linear heating at a rate of 10°C/min.
Synthesis of [Sc(Тdc)(OH)]·1.2DMF (I). A 0.1 M solution of scandium(III) trifluoromethanesulfonate in DMF (0.25 mL), a 0.1 M solution of 2,5-thiophenedicarboxylic acid in DMF (0.25 mL), a 10–4 M solution of NaOH in water (0.25 mL), and acetone (0.25 mL) were sealed in a glass ampule and kept at 60°С for 5 days. The composition and structure of transparent rhombic crystals of compound I were determined by single-crystal X-ray diffraction (SCXRD). The crystals were filtered off, washed with DMF (3 × 1 mL), and dried in air. The yield was 6.1 mg (69%).
For C10.5H13.5N1.5O7SSc | |||
Anal. calcd., % | C, 36.0 | H, 4.2 | N, 6.0 |
Found, % | C, 35.9 | H, 3.9 | N, 5.7 |
IR (ν, cm–1): 3384 br.m νs(O–H), 3080 w ν(C–H), 2940 w ν(C–H), 2854 w ν(C–H), 1666 s νas(C=O), 1561 s γ(C=C), 1529 s γ(C=C), 1380 s νs(C=O), 1244 w ν(C–N), 1089 m ν(C–N), 1035 w ν(C–N), 772 s δoop(C–H), 678 m δoop(C–H), 504 w δoop(C–H) (oop means out-of-plane vibrations).
Synthesis of [Sc(Tdc)(OH)]·2/3DMF (II). A 0.1 M solution of scandium(III) trifluoromethanesulfonate in DMF (0.25 mL), 0.1 M solution of 2,5-thiophenedicarboxylic acid in DMF (0.25 mL), a 10–2 M solution of NaOH in water (0.25 mL), and acetonitrile (0.25 mL) were sealed in a glass ampule and kept at 130°С for 2 days. The composition and structure of transparent rhombic crystals of compound II were determined by SCXRD.
Synthesis of (Me2NH2)[Sc3(Tdc)4(OH)2]·DMF (III). A 0.1 M solution of scandium(III) trifluoromethanesulfonate in DMF (0.25 mL), a 0.1 M solution of 2,5-thiopenedicarboxylic acid in DMF (0.25 mL), water (0.25 mL), acetonitrile (0.25 mL), and formic acid (9.5 μL) were sealed in a glass ampule and kept at 120°С for 2 days. The composition and structure of transparent rhombic crystals of compound III were determined by SCXRD.
SCXRD. The diffraction data for single crystals of compounds I–III were obtained on an Agilent Xcalibur automated diffractometer equipped with an area AtlasS2 detector (graphite monochromator, λ(MoKα) = 0.71073 Å, ω scan mode with an increment of 0.5°) at 130 K. Integration was performed, an absorption correction was applied, and unit cell parameters were determined using the CrysAlisPro program package [21]. The crystal structures were solved using the SHELXT program [22] and refined by full-matrix least squares in the anisotropic (except for hydrogen atoms) approximation using the SHELXL program [23]. The positions of the hydrogen atoms of the organic ligands were calculated geometrically and refined by the riding model. The guest composition of compound I was determined using the PLATON/SQUEEZE procedure [24] (1502 e− in 7330 Å3) and elemental analysis data (C, H, N). The crystallographic data and details of diffraction experiments are given in Table 1.
The full tables of interatomic distances and bond angles, atomic coordinates, and atomic displacement parameters were deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 2067819 (I), 2067820 (II), and 2067821 (III); deposit@ccdc. cam.ac.uk or https://www.ccdc. cam.ac.uk/structures) and also can be requested from the authors.
RESULTS AND DISCUSSION
All compounds were synthesized under close conditions (equimolar ratio Sc : H2Tdc, the same volume and solvent composition with an accuracy to the nature of the weakly coordinated additive: acetone (synthesis of compound I) or acetonitrile (syntheses of compounds II and III). The key distinctions are the synthesis temperature and the presence of acidity modulators. Compound [Sc(Tdc)(OH)]·1.2DMF (I) crystallizes at low temperatures (40–70°С). At a higher temperature (130°С), compound [Sc(Tdc)-(OH)]·2/3DMF (II) is formed with the same stoichiometry of the coordination framework as compound I but with a different crystal structure. It was found during the optimization of the synthesis conditions for compound I that the addition of minor amounts of NaOH (from 0.1 to 10% compared to the amount of Sc(III) or H2Tdc) to the reaction mixture enhanced the yield and crystallinity of the product. However, pure compound I can also be obtained in the form of orthorhombic crystals without these additives. When higher concentrations of NaOH were used, an amorphous precipitate and a new phase with needle-like crystals were formed, but attempts to determine their composition and structure were unsuccessful. The addition of formic acid (20-fold excess over Sc(III) and H2Tdc) to the system results in the formation of compound (Me2NH2)-[Sc3(Tdc)4(OH)2]·DMF (III) at 120°С. Obviously, the acidic medium and high temperature leads to the partial hydrolysis of N,N-dimethylformamide, and the formed \({\text{M}}{{{\text{e}}}_{{\text{2}}}}{\text{NH}}_{2}^{ + }\) cations are incorporated into the structure of compound III during its crystallization. The compositions and structures of all compounds were determined by single-crystal XRD. These data for compound I were additionally confirmed by chemical, TG, and PXRD analyses and IR spectroscopy. The low yield and impossibility of scaling syntheses of compounds II and III did not allow us to characterize them additionally.
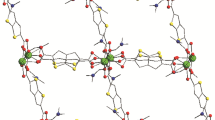
According to SCXRD data, the structure of compound I contains five crystallographically independent scandium(III) cations, each of which exists in the octahedral coordination environment of six oxygen atoms of two hydroxyl OH– groups and four carboxylate groups of the Tdc2– ligands occupying the bridging position between two adjacent scandium(III) cations (Fig. 1a). The bridging OH– and carboxylate groups bind the scandium(III) cations into chains arranged along the crystallographic axis b of the orthorhombic Pbam group (sequence –Sc(1)–Sc(2)–Sc(3)–Sc(4)–Sc(5)–Sc(4)–Sc(3)–Sc(2)–Sc(1)–). The Sc–O bond lengths are as follows: Sc(1)–O 2.0321(13)–2.1471(13), Sc(2)–O 2.0462(13)–2.1322(13), Sc(3)–O 2.0277(13)–2.1228(13), Sc(4)–O 2.0413–2.1404(13), and Sc(5)–O 2.0354(13)–2.1222(13) Å. The bridging Tdc2– ligands link such chain motifs along the crystallographic axes а and с to form the 3D MOF (Figs. 1b, 1c). The channels 5 × 6 Å in size occupied by the guest DMF molecules are observed in the porous structure in the direction of the b axis (Fig. 1c). After the guest solvent molecules were removed, the volume of the free space was 56.7% (according the PLATON program estimation [24]).

(a) Coordination environment of scandium(III) cations and (b, c) fragment of the crystal structure of compound I: in projection to planes (b) bc and (c) ac.
Rhombic crystals of compound II contain two crystallographically independent scandium(III) cations each, and each scandium(III) cation exists in the octahedral coordination environment of six oxygen atoms of two OH– groups and four carboxylate groups of the Tdc2– ligands that occupy the bridging position between two adjacent scandium(III) cations (Fig. 2a). Two bridging OH– and four carboxylate Tdc2– groups bind two Sc(2) cations and one Sc(1) cation into complicated double chains arranged along the crystallographic a axis (Fig. 2b). The linear sequence of cations –Sc(2)–Sc(1)–Sc(2)–Sc(2)–Sc(1)–Sc(2)– linked by the bridging carboxylate groups and μ2-ОН– ligands can be distinguished in each chain. The Sc(1) and Sc(2) cations are bound through one ОН– ligand and two RCOO– groups, whereas the Sc(2) and Sc(2) cations are linked through two RCOO– groups. These sequences are connected by additional lateral bridges μ2-ОН– that bind the adjacent Sc(2) cations along the b direction to form the aforementioned double chains. The Sc–O bond lengths are as follows: Sc(1)–O 2.068(2)–2.117(2) and Sc(2)–O 2.0700(19)–2.166(2) Å. When the crystal packing is formed, the double bonds are linked through the bridging Tdc2– ligands in six directions perpendicular to the а axis to form trigonal channels 4 × 3 Å in size occupied by disordered DMF molecules. According to the PLATON program data [24], the estimated volume of the free space of the channels is 31.8%.

Fragments of the (a) double chain of scandium(III) cations and (b) crystal structure of compound II: in projection to plane bc. Guest solvent molecules are omitted.
Compounds I and II have an identical formula of the coordination framework and are polymorphous modifications. A comparison of their crystallization conditions and structures shows a regularity often observed for other MOF: a lower synthesis temperature results in looser structures compared to those formed at higher temperatures, which lead, as a rule, to the formation of more compact phases with lower porosity [25–27]. Indeed, the low-temperature phase I has the crystallographic density of the framework ρ = 0.97 g/cm3 and the pore volume equal to 56.7%, whereas the high-temperature phase II has the density ρ = 1.42 g/cm3 and the pore volume equal to 31.8%.
The acidification of the reaction solution with formic acid excess at the high temperature (120°С) favors the hydrolysis of DMF to form the dimethylammonium cation \({\text{M}}{{{\text{e}}}_{{\text{2}}}}{\text{NH}}_{2}^{ + },\) which acts as a counterion for the anionic framework of compound III. According to the SCXRD data, the structure of compound III contains three crystallographically independent scandium(III) cations. The Sc(1) and Sc(3) cations exist in the octahedral coordination environment of six oxygen atoms of one bridging OH– group and five carboxylate groups of the Tdc2– ligands, two of which occupy the bridging positions between the Sc(1) and Sc(2) or Sc(3) and Sc(2) cations (Fig. 3a). The Sc(2) cation exists in the octahedral coordination environment of six oxygen atoms of two bridging OH– groups and four bridging carboxylate groups of the Tdc2– ligands linking the Sc(1) and Sc(2) cations as well as Sc(3) and Sc(2) cations. The Sc(1), Sc(2), and Sc(3) cations form a trinuclear secondary building unit {Sc3(RCOO)8(OH)2}. Using the carboxylate bridging groups of the Tdc2– ligands, these units are connected to each other according to the head-to-head principle to form the carboxylate chains (Fig. 3a). The chains are linked to each other by the bridging Tdc2– ligands to form the MOF containing rhombus-like channels (Fig. 3b) 8 × 3 Å in size occupied by the guest DMF molecules and dimethylammonium cations. According to the PLATON program estimation [24], the volume of the free space is 25.5% after the removal of the guest solvent molecules and dimethylammonium cations. The Sc–O bond lengths are as follows: Sc(1)–O 2.069(2)–2.137(2) Å, Sc(2)–O 2.0401(19)–2.1040(19) Å, and Sc(3)–O 2.0596(18)–2.158(2) Å.

Fragments of the (a) chain of scandium(III) cations and (b) crystal structure of compound III. Guest solvent molecules and dimethylammonium cations are omitted.
The synthesis procedure for compound I was optimized for the preparation of representative amounts of the product sufficient for characterization by PXRD, elemental, and TG analyses and IR spectroscopy. As can be seen from Fig. 4, the experimental PXRD pattern is well consistent with that calculated from the single-crystal SCXRD data, which proves the phase purity of the sample. According to the TG curve, compound I loses 30% of the weight at temperatures below 310°С (Fig. 5), which corresponds to the loss of 1.4 guest DMF molecules. This result is well consistent with the XSA and chemical analyses data. The MOF irreversibly decomposes at the temperature higher than 310°С.

Comparison of the (black) experimental and (red) simulated PXRD patterns for compound I.

TG curve for compound I.
The IR spectrum of compound I contains a broad band at 772 cm–1 attributed to bending out-of-plane vibrations of the C–H bond in the thiophene moiety of the Tdc2– ligand. The group of bands in the range from 1035 to 1244 cm–1 belongs to stretching vibrations of the C–N bond in the guest DMF molecule. The characteristic intense absorption bands of symmetric and asymmetric vibrations of the С=О bond are observed at 1380 and 1666 cm–1, respectively. The intense bands in the range from 1529 to 1561 cm–1 are assigned to skeletal vibrations of the С=С bond in the heterocycle of the Tdc2– ligand. Several bands of various intensity from 2854 to 3080 cm–1 belong to stretching vibrations of the С–Н bond, and the broad band at 3384 cm–1 corresponds to stretching vibrations of the О–Н bond in the coordinated hydroxyl group and in the guest water molecule.
It should be mentioned that only two MOF based on scandium(III) and thiophene ligands, namely, 2,5-thiophenedicarboxylate, are known [19, 20]. In 2011 Ibarra and coauthors synthesized the 3D MOF [Sc(Tdc)(OH)]·2.6H2O named NOTT-401 [19], which resembles compounds I and II in chemical composition but differs from them in structure: NOTT-401 crystallizes in the tetragonal space group I41/amd and consists of the binuclear secondary building units {Sc2(μ2-OH)(RCOO)4}. In 2020 Wang and coauthors synthesized the 3D MOF [Sc2(Tdc)2(OH)2]·4H2O, whose structure resembles that of MOF II. However, in this case, the OH– groups in the {Sc(OH)2O4} octahedra are located in the trans position, whereas in MOF II the OH– groups occupy the cis position, due to which the packing of the MOF differs from that described earlier [20].
Thus, the results of this work substantially extend a very restricted class of the MOF based on Sc(III) and heterocyclic thiophene ligands, which can be used as stable adsorbents and catalysts.
REFERENCES
Maurin, G., Serre, C., Cooper, A., and Férey, G., Chem. Soc. Rev., 2017, vol. 46, p. 3104.
Morris, R.E. and Brammer, L., Chem. Soc. Rev., 2017, vol. 46, p. 5444.
Kirchon, A., Feng, L., Drake, H.F., et al., Chem. Soc. Rev., 2018, vol. 47, p. 8611.
Wu, D., Zhang, P.-F., Yang, G.-P., et al., Coord. Chem. Rev., 2021, vol. 434, p. 213709.
Mukherjee, S., Sensharma, D., Qazvini, O.T., et al., Coord. Chem. Rev., 2021, vol. 437, p. 213852.
Zhang, G., Hua, B., Dey, A., et al., Acc. Chem. Res., 2021, vol. 54, no. 1, p. 155.
Gao, C., Lyu, F., and Yin, Y., Chem. Rev., 2021, vol. 121, p. 834.
Hasanpour, M. and Hatami, M., Adv. Colloid Interface Sci., 2020, vol. 284, p. 102247.
Ghomi, G.A., Asasian-Kolur, N., Sharifian, S., et al., J. Environ. Chem. Eng., 2020, vol. 8, p. 103996.
Barsukova, M.O., Sapchenko, S.A., Dybtsev, D.N., et al., Usp. Khim., 2018, vol. 87, p. 1139.
Serre, C., Millange, F., Thouvenot, C., et al., J. Am. Chem. Soc., 2002, vol. 124, p. 13519.
Loiseau, T., Serre, C., Huguenard, C., et al., Chem.-Eur. J., 2004, vol. 10, p. 1373.
Assi, H., Mouchaham, G., Steunou, N., et al., Chem. Soc. Rev., 2017, vol. 46, p. 3431.
Cavka, J.H., Jakobsen, S., Olsbye, U., et al., J. Am. Chem. Soc., 2008, vol. 130, p. 13850.
Bolotov, V.A., Kovalenko, K.A., Samsonenko, D.G., et al., Inorg. Chem., 2018, vol. 57, p. 5074.
Lysova, A.A., Samsonenko, D.G., Dorovatovskii, P.V., et al., J. Am. Chem. Soc., 2019, vol. 141, p. 17260.
Hua, C. and D’Alessandro, D.M., Cryst. Growth Des., 2017, vol. 17, p. 6262.
Zhao, J., Shi, X., Li, G., et al., Inorg. Chim. Acta, 2012, vol. 383, p. 185.
Ibarra, I.A., Yang, S., Lin, X., et al., Chem. Commun., 2011, vol. 47, p. 8304.
Wang, P., Zhang, L., Zhu, Z., et al., Inorg. Chim. Acta, 2020, p. 119304.
CrysAlisPro Software System. Version 1.171.38.46, Rigaku Oxford Diffraction, Wrocław: Rigaku Corporation, 2015.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. 71, p. 3.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Spek, A.L., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 9.
Dubskikh, V.A., Lysova, A.A., Samsonenko, D.G., et al., CrystEngComm, 2020, vol. 22, p. 6295.
Lysova, A., Samsonenko, D., Dybtsev, D., et al., Crystals, 2018, vol. 8, p. 7.
Hou, S.-S., Huang, X., Guo, J.-G., et al., CrystEngComm, 2015, vol. 17, p. 947.
Funding
This work was supported by the Russian Foundation for Basic Research and Government of the Novosibirsk oblast, project no. 20-43-540010.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Dedicated to Academician N.T. Kuznetsov on the occasion of his 90th birthday
Translated by E. Yablonskaya
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lysova, A.A., Dubskikh, V.A., Abasheeva, K.D. et al. Coordination Polymers of Scandium(III) and Thiophenedicarboxylic Acid. Russ J Coord Chem 47, 593–600 (2021). https://doi.org/10.1134/S1070328421090062
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328421090062