
Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory and Methodology
2.1. Origin of CG Dynamics: Separation of the Coarse-Grained and Fine-Grained Motions
2.2. Implementation of Coarse-Grained MD
2.3. Effective Potential Energy Functions
2.3.1. AWSEM
2.3.2. MARTINI
2.3.3. OPEP and HiRe-RNA
2.3.4. oxDNA and oxRNA
2.3.5. SIRAH
2.3.6. UNICORN
2.3.7. Coarse-Grained Potentials for Glycosoaminoglycans
2.4. Extensions of MD
3. Examples
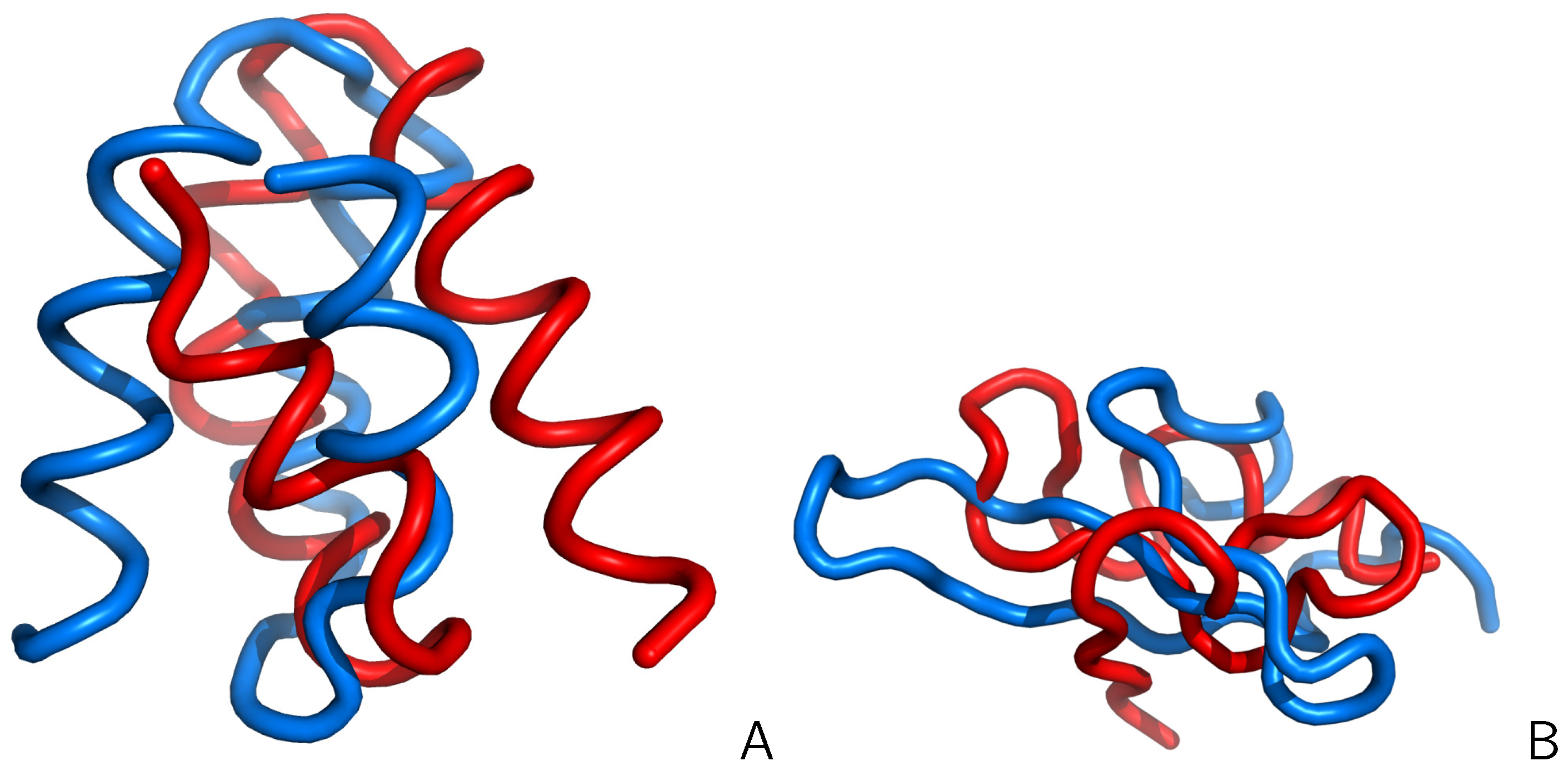
3.1. Investigation of Protein-Folding Kinetics and Pathways
3.1.1. Folding Kinetics of FBP WW Domain and Its Mutants
3.1.2. Effect of Hydrodynamic Interactions on Folding
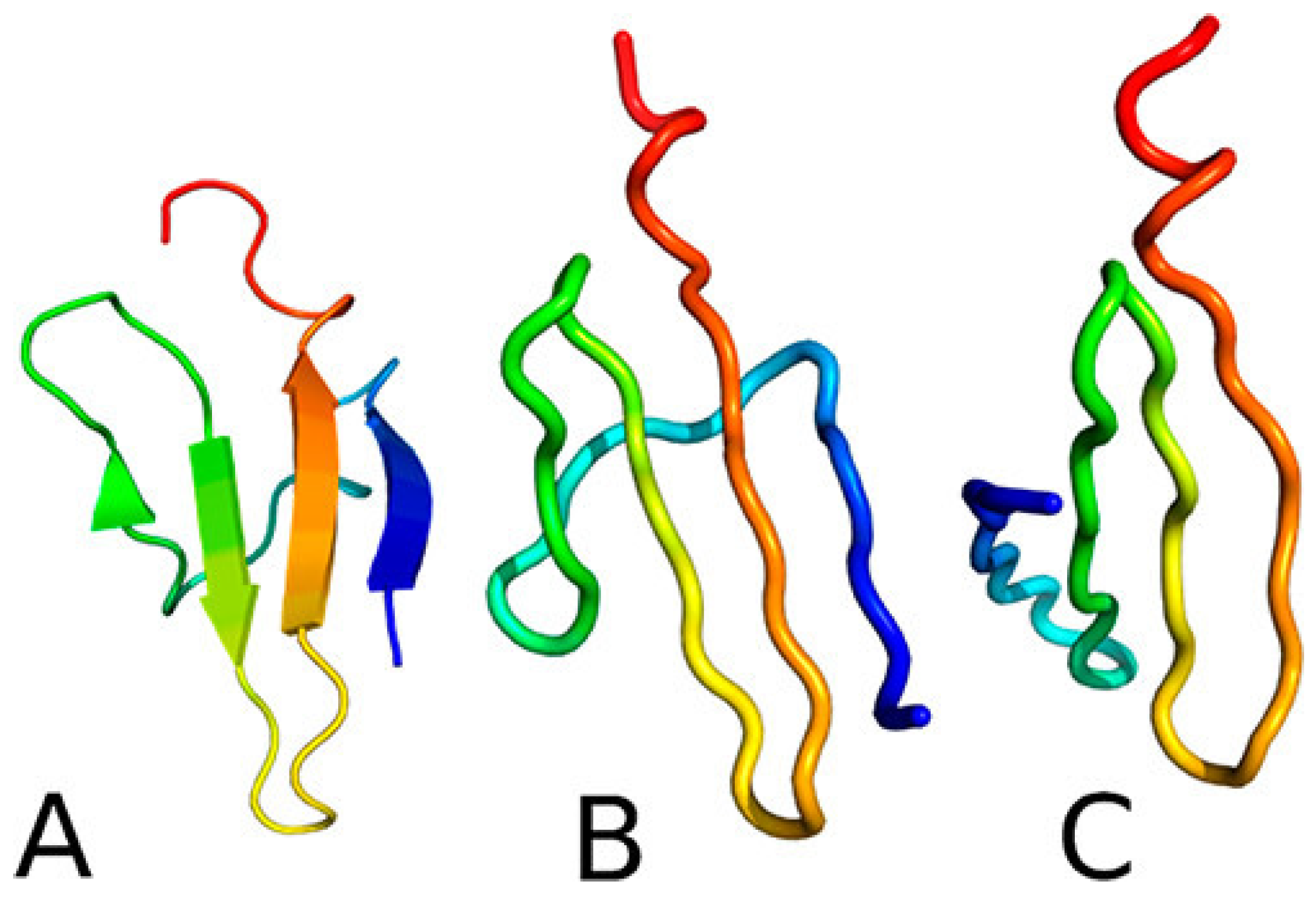
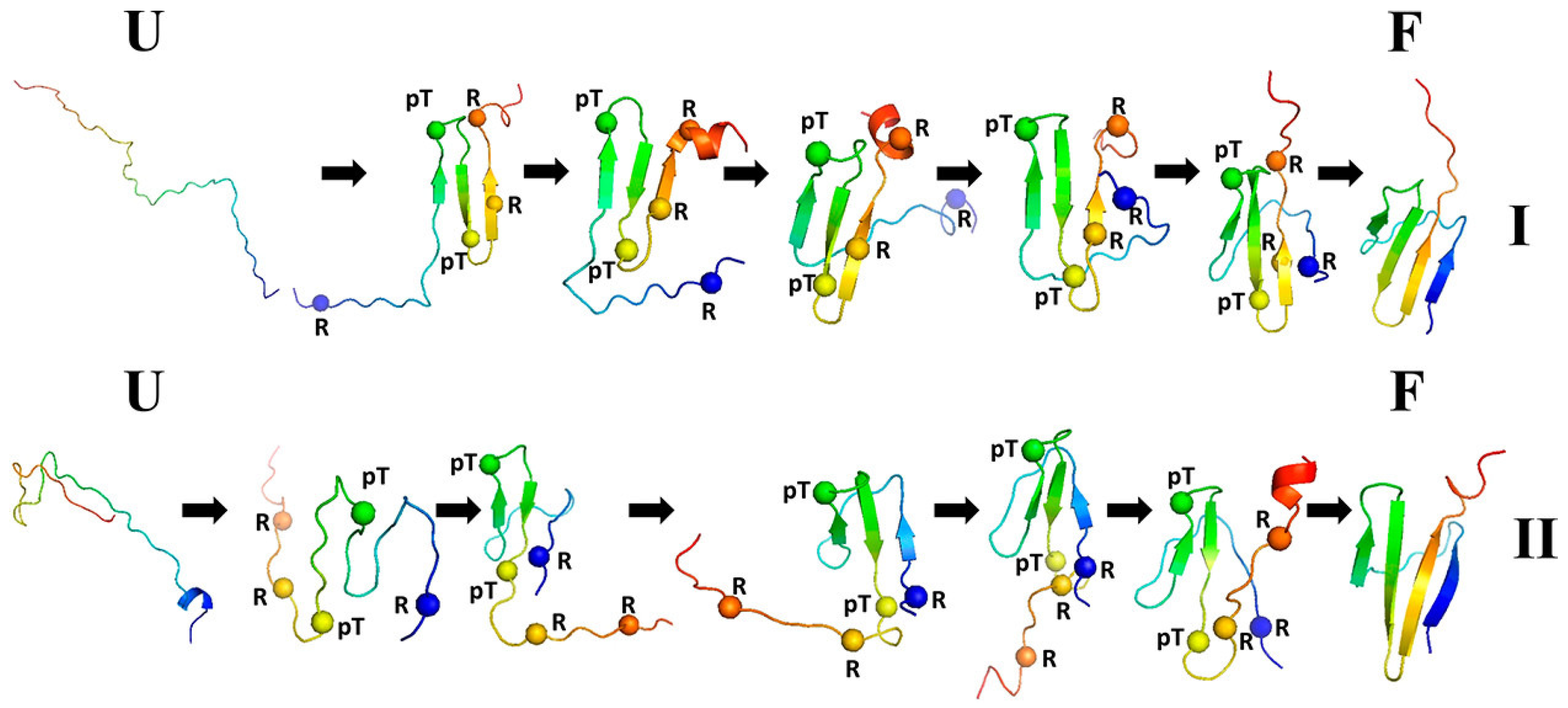
3.1.3. Phosphorylation-Induced Folding of the Intrinsically Disordered eIF4E-Binding Protein Isoform 2
3.2. Ensemble-Based Modeling of Protein Structures
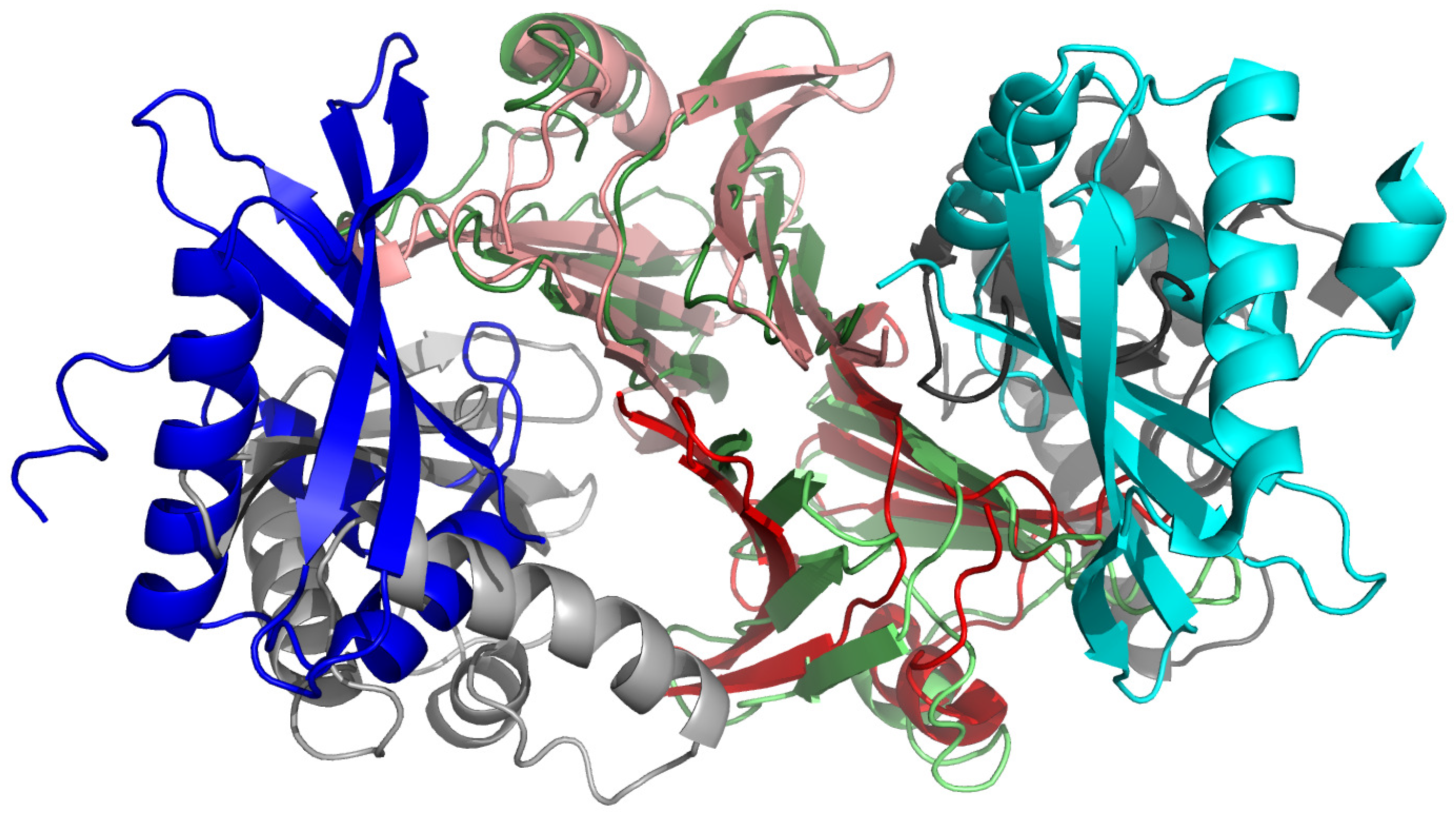
3.3. Investigation of Telomere Stability
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMBER | Assisted Model Building with Energy Refinement |
| AWSEM | Associative memory, Water mediated, Structure and Energy Model |
| CASP | Community Wide Experiment on the Critical Assessment of Techniques for Protein |
| Structure Prediction | |
| CG | Coarse-Grained |
| CHARMM | Chemistry at Harvard Molecular Mechanics |
| FEL | Free Energy Landscape |
| GAG | Glycosoaminoglycan |
| MC | Monte Carlo |
| MD | Molecular Dynamics |
| MREMD | Multiplexed Replica Exchange Molecular Dynamics |
| NARES-2P | Nucleic Acid united Residue 2-Points |
| OPEP | Optimized Potential for Efficient protein structure Prediction |
| PMF | Potential of Mean Force |
| REMD | Replica Exchange Molecular Dynamics |
| SIRAH | Southamerican Initiative for a Rapid and Accurate Hamiltonian |
| SUGRES-1P | Sugar united Residue 1-Point |
| UNRES | United Residue |
| UNICORN | Unified Coarse-Grained Model |
References
- Alder, B.J.; Wainwright, B.E. Molecular dynamics by electronic computers. In Proceedings of the International Symposium on Statistical Mechanical Theory of Transport Processes; Prigogine, I., Ed.; Wiley: New York, NY, USA, 1958; pp. 97–131. [Google Scholar]
- van Gunsteren, W.F. Molecular dynamics and stochastic dynamics: A primer. In Computer Simulation of Biomolecular Systems; van Gunsteren, W.F., Weiner, P.K., Wilkinson, A.J., Eds.; ESCOM: Leiden, The Netherlands, 1993; pp. 3–36. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: New York, NY, USA, 2000; p. 2. [Google Scholar]
- Scheraga, H.A.; Khalili, M.; Liwo, A. Protein-folding dynamics: Overview of molecular simulation techniques. Annu. Rev. Phys. Chem. 2007, 58, 57–83. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Atkins, P.; Friedman, R. Molecular Quantum Mechanics; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Leach, A.R. Molecular Modeling: Principles and Applications; Pearson Education: Harlow, UK, 2010. [Google Scholar]
- Shaw, D.E.; Deneroff, M.M.; Dror, R.O.; Kuskin, J.S.; Larson, R.H.; Salmon, J.K.; Young, C.; Batson, B.; Bowers, K.J.; Chao, J.C.; et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 2008, 51, 91–97. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Trbovic, N.; Maragakis, P.; Piana, S.; Shaw, D.E. Structure and dynamics of an unfolded protein examined by molecular dynamics simulation. J. Am. Chem. Soc. 2012, 134, 3787–3791. [Google Scholar] [CrossRef] [PubMed]
- Larsson, D.S.D.; Liljas, L.; van der Spoel, D. Virus capsid dissolution studied by microsecond molecular dynamics simulations. PLoS Comput. Biol. 2012, 8, e1002502. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M.; Warshell, A. Computer simulation of protein folding. Nature 1975, 253, 694–698. [Google Scholar] [CrossRef]
- Voth, G. Coarse-Graining of Condensed Phase and Biomolecular Systems, 1st ed.; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: Abingdon, UK, 2008. [Google Scholar]
- Kolinski, A. Multiscale Approaches to Protein Folding; Springer: New York, NY, USA, 2011. [Google Scholar]
- Papoian, G.A. Coarse-Grained Modeling of Biomolecules; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Zwanzig, R. Nonequilibrium Statistical Mechanics; Oxford University Press: New York, NY, USA, 2001; Chapter 8. [Google Scholar]
- Kinjo, T.; Hyodo, S. Equation of motion for coarse-grained simulation based on microscopic description. Phys. Rev. E 2007, 75, 051109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klippenstein, V.; Tripathy, M.; Jung, G.; Schmid, F.; van der Vegt, N.F.A. Introducing memory in coarse-grained molecular simulations. J. Phys. Chem. B 2021, 125, 4931–4954. [Google Scholar] [CrossRef]
- Han, Y.; Jin, J.; Voth, G.A. Constructing many-body dissipative particle dynamics models of fluids from bottom-up coarse-graining. J. Chem. Phys. 2021, 154, 084122. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, W.R.; Bakalarski, G.; Lesyng, B. A mezoscopic model of nucleic acids. Part 1. Lagrangian and quaternion molecular dynamics. J. Biomol. Struct. Dyn. 2000, 17, 1097–1108. [Google Scholar] [CrossRef]
- Alvarado, C.; Kazerounian, K. On the rotational operators in protein structure simulations. Prot. Eng. 2003, 16, 717–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, M.; Liwo, A.; Rakowski, F.; Grochowski, P.; Scheraga, H.A. Molecular dynamics with the united-residue (UNRES) model of polypeptide chains. I. Lagrange equations of motion and tests of numerical stability in the microcanonical mode. J. Phys. Chem. B 2005, 109, 13785–13797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozzini, V. Coarse-grained models for proteins. Curr. Opin. Struct. Biol. 2005, 15, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V. Minimalist models for proteins: A comparative analysis. Quart. Rev. Biophys. 2010, 43, 333–371. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Czaplewski, C.; Pillardy, J.; Scheraga, H.A. Cumulant-based expressions for the multibody terms for the correlation between local and electrostatic interactions in the united-residue force field. J. Chem. Phys. 2001, 115, 2323–2347. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Makowski, M.; Augustynowicz, A.; Liwo, A. A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. I. Backbone potentials of coarse-grained polypeptide chains. J. Chem. Phys. 2017, 146, 124106. [Google Scholar] [CrossRef]
- Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lubecka, E.A.; Lipska, A.G.; Golon, Ł.; Karczyńka, A.; Krupa, P.; Mozolewska, M.A.; Makowski, M.; et al. Scale-consistent approach to the derivation of coarse-grained force fields for simulating structure, dynamics, and thermodynamics of biopolymers. In Progress in Molecular Biology and Translational Science. Computational Approaches for Understanding Dynamical Systems: Protein Folding and Assembly; Strodel, B., Barz, B., Eds.; Academic Press: London, UK, 2020; Volume 170, Chapter 2; pp. 73–122. [Google Scholar]
- Ayton, G.S.; Noid, W.G.; Voth, G.A. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr. Opin. Struct. Biol. 2007, 17, 192–198. [Google Scholar] [CrossRef]
- Kolinski, A. Protein modeling and structure prediction with a reduced representation. Acta Biochim. Pol. 2004, 51, 349–371. [Google Scholar] [CrossRef] [Green Version]
- Boniecki, M.J.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysinski, T.; Rother, K.M.; Bujnicki, J.M. SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016, 44, e63. [Google Scholar] [CrossRef]
- Kmiecik, S.; Kolinski, A. Folding pathway of the B1 domain of protein G explored by multiscale modeling. Biophys. J. 2008, 94, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef]
- Zwanzig, R. Memory effects in irreversible thermodynamics. Phys. Rev. 1961, 124, 983–992. [Google Scholar] [CrossRef]
- Mori, H. Transport, collective motion, and Brownian motion. Prog. Theor. Phys. 1965, 33, 423–455. [Google Scholar] [CrossRef] [Green Version]
- Rudzinski, J.F. Recent progress towards chemically-specific coarse-grained simulation models with consistent dynamical properties. Computation 2019, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Langevin, P. Sur le théorie du mouvement brownien. C. R. Acad. Sci. 1908, 146, 530–533. [Google Scholar]
- Liwo, A.; Khalili, M.; Scheraga, H.A. Molecular dynamics with the united-residue (UNRES) model of polypeptide chains; test of the approach on model proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 2362–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, M.; Liwo, A.; Jagielska, A.; Scheraga, H.A. Molecular dynamics with the united-residue (UNRES) model of polypeptide chains. II. Langevin and Berendsen-bath dynamics and tests on model α-helical systems. J. Phys. Chem. B 2005, 109, 13798–13810. [Google Scholar] [CrossRef] [Green Version]
- Murarka, R.K.; Liwo, A.; Scheraga, H.A. Separation of time scale and coupling in the motion governed by the coarse-grained and fine degrees of freedom in a polypeptide backbone. J. Chem. Phys. 2007, 127, 155103. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Frembgen-Kesner, T.; Elcock, A.H. Striking effects of hydrodynamic interactions on the simulated diffusion and folding of proteins. J. Chem. Theory Comput. 2009, 5, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Cieplak, M.; Niewieczerzał, S. Hydrodynamic interactions in protein folding. J. Chem. Phys. 2009, 130, 124906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipska, A.G.; Seidman, S.R.; Sieradzan, A.K.; Giełdoń, A.; Liwo, A.; Scheraga, H.A. Molecular dynamics of protein A and a WW domain with a united-residue model including hydrodynamic interaction. J. Chem. Phys. 2016, 144, 184110. [Google Scholar] [CrossRef] [Green Version]
- Rotne, J.; Prager, S. Variational treatment of hydrodynamic interaction in polymers. J. Chem. Phys. 1969, 50, 4831–4837. [Google Scholar] [CrossRef]
- Levy, R.M.; Karplus, M.; McCammon, J.A. Diffusive Langevin dynamics of model alkanes. Chem. Phys. Lett. 1979, 65, 4–11. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Ouldridge, T.E.; Tretyakov, M.V. New Langevin and gradient thermostats for rigid body dynamics. J. Chem. Phys. 2015, 142, 144114. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Czaplewski, C.; Ołdziej, S.; Rojas, A.V.; Kaźmierkiewicz, R.; Makowski, M.; Murarka, R.K.; Scheraga, H.A. Simulation of protein structure and dynamics with the coarse-grained UNRES force field. In Coarse-Graining of Condensed Phase and Biomolecular Systems; Voth, G., Ed.; CRC Press: Boca Raton, MA, USA, 2008; Chapter 8; pp. 1391–1411. [Google Scholar]
- Liwo, A.; Baranowski, M.; Czaplewski, C.; Gołaś, E.; He, Y.; Jagieła, D.; Krupa, P.; Maciejczyk, M.; Makowski, M.; Mozolewska, M.A.; et al. A unified coarse-grained model of biological macromolecules based on mean-field multipole-multipole interactions. J. Mol. Model. 2014, 20, 2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieradzan, A.K.; Czaplewski, C.; Lubecka, E.A.; Lipska, A.G.; Karczyńska, A.S.; Giełdoń, A.P.; Ślusarz, R.; Makowski, M.; Krupa, P.; Kogut, M.; et al. Extension of UNRES package for physics-based coarse-grained simulations of proteins and protein complexes to very large systems. Biophys. J. 2021, 120, 83a–84a. [Google Scholar] [CrossRef]
- Kleinerman, D.S.; Czaplewski, C.; Liwo, A.; Scheraga, H.A. Implementations of Nosé – Hoover and Nosé – Poincaré thermostats in mesoscopic dynamic simulations with the united-residue model of a polypeptide chain. J. Chem. Phys. 2008, 128, 245103. [Google Scholar] [CrossRef] [PubMed]
- Paterlini, M.G.; Ferguson, D.M. Constant temperature simulations using the Langevin equation with velocity Verlet integration. Chem. Phys. 1998, 236, 243–252. [Google Scholar] [CrossRef]
- Ricci, A.; Ciccotti, G. Algorithms for Brownian dynamics. Mol. Phys. 2003, 101, 1927–1931. [Google Scholar] [CrossRef]
- Ciccotti, G.; Kalibaeva, G. Deterministic and stochastic algorithms for mechanical systems under constraints. Philos. Trans. R. Soc. Lond. A 2004, 362, 1583–1594. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Goga, N.; Rzepiela, J.; de Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C. Efficient algorithms for Langevin and DPD dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosé, S. A molecular dynamics method for simulation in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Nosé, S. An improved symplectic integrator for Nosé-Poincaré themostat. J. Phys. Soc. Jpn. 2001, 70, 75–77. [Google Scholar] [CrossRef]
- Smith, A.V.; Hall, C.K. α-Helix formation: Discontinuous molecular dynamics on an intermediate-resolution protein model. Proteins 2001, 44, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Cheon, M.; Chang, I.; Hall, C.K. Extending the PRIME model for protein aggregation to all 20 amino acids. Proteins 2010, 78, 2950–2960. [Google Scholar] [CrossRef] [Green Version]
- Sippl, M.J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990, 213, 859–883. [Google Scholar] [CrossRef]
- Kolinski, A.; Godzik, A.; Skolnick, J. A general method for the prediction of the three-dimensional structure and folding pathway of globular proteins: Application to designed helical proteins. J. Chem. Phys. 1993, 98, 7420–7433. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Gront, D.; Koliński, M.; Wieteska, L.; Dawid, A.; Koliński, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Li, W. Recent advances in coarse-grained models for biomolecules and their applications. Int. J. Mol. Sci. 2019, 20, 3774. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Minhas, V.; Korolev, N.; Mirzoev, A.; Lyubartsev, A.P.; Nordensiöld, L. Bottom-up coarse-grained modeling of DNA. Front. Biomol. Sci. 2021, 8, 645527. [Google Scholar]
- Giulini, M.; Rigoli, M.; Mattioti, G.; Menichetti, R.; Tarenzi, T.; Fiorentini, R.; Potestio, R.F. From system modeling to system analysis: The impact of resolution level and resolution distribution in the computer-aided investigation of biomolecules. Front. Mol. Biosci. 2021, 8, 676976. [Google Scholar] [CrossRef]
- Gay, J.G.; Berne, B.J. Modification of the overlap potential to mimic a linear site-site potential. J. Chem. Phys. 1981, 74, 3316–3319. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse Rgained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI coarse-grained force field. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Lopez, C.A.; Rzepiela, A.; de Vries, A.H.; Dijkhuizen, L.; Hunenberger, P.H.; Marrink, S.J. Martini coarse-grained force field: Extension to carbohydrates. J. Chem. Theory Comput. 2009, 5, 3195–3210. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [Green Version]
- Uusitalo, J.J.; Ingólfsson, H.I.; Akhshi, P.; Tieleman, D.P.; Marrink, S.J. Martini coarse-grained force field: Extension to DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.C.; Andrews, C.T.; Elcock, A.H. Toward optimized potential functions for protein-protein interactions in aqueous solutions: Osmotic second virial coefficient calculations using the MARTINI coarse-grained force field. J. Chem. Theory Comput. 2013, 9, 4176–4185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmalhorst, P.S.; Deluweit, F.; Scherrers, R.; Heisenberg, C.P.; Sikora, M.J. Overcoming the limitations of the MARTINI force field in simulations of polysaccharides. J. Chem. Theory Comput. 2017, 13, 5039–5053. [Google Scholar] [CrossRef]
- Fornasier, F.; de Souza, L.; Souza, F.; Reynaud, F.; Pimentel, A. The lipophilicity of coarse-grained cholesterol models. J. Chem. Inf. Model. 2020, 60, 569–577. [Google Scholar] [CrossRef]
- Souza, L.M.P.; Souza, F.R.; Reynaud, F.; Pimentel, A.S. Tuning the hydrophobicity of a coarse grained model of 1,2-dipalmitoyl-Sn-glycero-3-phosphatidylcholine using the experimental octanol-water partition coefficient. J. Mol. Liq. 2020, 319, 114132. [Google Scholar] [CrossRef]
- Souza, F.R.; Pereira Souza, L.M.; Pimentel, A.S. Recent open issues in coarse grained force fields. J. Chem. Inf. Model. 2020, 60, 5881–5884. [Google Scholar] [CrossRef]
- Alessandrini, R.; Souza, P.C.T.; Thallmair, S.; Melo, M.N.; de Vries, A.H.; Marrink, S.J. Pitfalls of the Martini model. J. Chem. Theory Comput. 2019, 15, 5448–5460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Kubo, R. Generalized cumulant expansion method. J. Phys. Soc. Jpn. 1962, 17, 1100–1120. [Google Scholar] [CrossRef]
- Liwo, A.; Khalili, M.; Czaplewski, C.; Kalinowski, S.; Ołdziej, S.; Wachucik, K.; Scheraga, H. Modification and optimization of the united-residue (UNRES) potential energy function for canonical simulations. I. Temperature dependence of the effective energy function and tests of the optimization method with single training proteins. J. Phys. Chem. B 2007, 111, 260–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liwo, A.; Sieradzan, A.K.; Lipska, A.G.; Czaplewski, C.; Joung, I.; Żmudzińska, W.; Hałabis, A.; Ołdziej, S. A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. III. Determination of scale-consistent backbone-local and correlation potentials in the UNRES force field and force-field calibration and validation. J. Chem. Phys. 2019, 150, 155104. [Google Scholar] [PubMed]
- Kolinski, A.; Skolnick, J. Discretized model of proteins. I. Monte Carlo study of cooperativity in homopolypeptides. J. Chem. Phys. 1992, 97, 9412–9426. [Google Scholar] [CrossRef] [Green Version]
- Maupetit, J.; Tuffery, P.; Derreumaux, P. A coarse-grained protein force field for folding and structure prediction. Proteins 2007, 69, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Chothia, C. Structural patterns in globular proteins. Nature 1976, 261, 552–558. [Google Scholar] [CrossRef]
- Liwo, A.; Ołdziej, S.; Czaplewski, C.; Kozłowska, U.; Scheraga, H.A. Parameterization of backbone-electrostatic and multibody contributions to the UNRES force field for protein-structure prediction from ab initio energy surfaces of model systems. J. Phys. Chem. B 2004, 108, 9421–9438. [Google Scholar] [CrossRef]
- Yin, Y.; Sieradzan, A.K.; Liwo, A.; He, Y.; Scheraga, H.A. Physics-based potentials for coarse-grained modeling of protein DNA interactions. J. Chem. Theory Comput. 2015, 11, 1792–1808. [Google Scholar] [CrossRef] [Green Version]
- Makowski, M.; Sobolewski, E.; Czaplewski, C.; Liwo, A.; Ołdziej, S.; No, J.H.; Scheraga, H.A. Simple physics-based analytical formulas for the potentials of mean force for the interaction of amino acid side chains in water. III. Calculation and parameterization of the potentials of mean force of pairs of identical hydrophobic side chains. J. Phys. Chem. B 2007, 111, 2925–2931. [Google Scholar] [CrossRef]
- Makowski, M.; Liwo, A.; Scheraga, H.A. Simple physics-based analytical formulas for the potentials of mean force of the interaction of amino acid side chains in water. VII. Charged-hydrophobic/polar and polar-hydrophobic/polar side chains. J. Phys. Chem. B 2017, 121, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Makowski, M. Physics-based modeling of side chain-side chain interactions in the UNRES force field. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes. From Bioinformatics to Molecular Quantum Mechanics; Liwo, A., Ed.; Springer: Cham, Switzerland, 2018; pp. 89–115. [Google Scholar]
- Soper, A.K. Empirical potential Monte Carlo simulation of fluid structure. Chem. Phys. 1996, 202, 295–306. [Google Scholar] [CrossRef]
- Reith, D.; Püt, M.; Müller-Plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003, 24, 1624–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef] [PubMed]
- Lyubartsev, A.P.; Naômé, A.; Vercauteren, D.P.; Laaksonen, A. Systematic hierarchical coarse-graining with the inverse Monte Carlo method. J. Chem. Phys. 2015, 143, 243120. [Google Scholar] [CrossRef]
- Mashayak, S.Y.; Jochum, M.N.; Koschke, K.; Aluru, N.R.; Rühle, V.; Junghans, C. Relative entropy and optimization-driven coarse-graining methods in VOTCA. PLoS ONE 2016, 10, e0131754. [Google Scholar] [CrossRef]
- Mereghetti, P.; Maccari, G.; Spampinato, G.L.B.; Tozzini, V. Optimization of analytical potentials for coarse-grained biopolymers. J. Phys. Chem. B 2016, 120, 8571–8579. [Google Scholar] [CrossRef]
- Sanyal, T.; Shell, M.S. Coarse-grained models using local-density potentials optimized with the relative entropy: Application to implicit solvation. J. Chem. Phys. 2016, 145, 034109. [Google Scholar] [CrossRef]
- Latham, A.P.; Zhang, B. Maximum entropy optimized force field for intrinsically disordered proteins. J. Chem. Theory Comput. 2020, 16, 773–781. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Zaborowski, B.; Jagieła, D.; Czaplewski, C.; Hałabis, A.; Lewandowska, A.; Żmudzińska, W.; Ołdziej, S.; Karczyńska, A.; Omieczynski, C.; Wirecki, T.; et al. A maximum-likelihood approach to force-field calibration. J. Chem. Inf. Model. 2015, 55, 2050–2070. [Google Scholar] [CrossRef]
- Hałabis, A.; Żmudzińska, W.; Liwo, A.; Ołdziej, S. Conformational dynamics of the Trp-cage miniprotein at its folding temperature. J. Phys. Chem. B 2012, 116, 6898–6907. [Google Scholar] [CrossRef]
- Krupa, P.; Hałabis, A.; Żmudzińska, W.; Ołdziej, S.; Scheraga, H.A.; Liwo, A. Maximum likelihood calibration of the UNRES force field for simulation of protein structure and dynamics. J. Chem. Inf. Model. 2017, 57, 2364–2377. [Google Scholar] [CrossRef]
- He, Y.; Liwo, A.; Scheraga, H.A. Optimization of a Nucleic Acids united-RESidue 2-Point model (NARES-2P) with a maximum-likelihood approach. J. Chem. Phys. 2015, 143, 243111. [Google Scholar] [CrossRef] [Green Version]
- Izvekov, S.; Voth, G.A. A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 2005, 109, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Židek, A.; Nelson, A.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. ‘It will change averything’: AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Behler, J. Machine learning potentials for atomistic simulations. J. Chem. Phys. 2016, 145, 170901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huan, T.D.; Batra, R.; Chapman, J.; Krishnan, S.; Chen, L.; Ramprasad, R. A universal strategy for the creation of machine learning-based atomistic force fields. NPJ Comput. Mater. 2017, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Friederich, P.; Häse, F.; Proppe, J.; Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations. Nat. Mater. 2021, 20, 750–761. [Google Scholar] [CrossRef]
- Wang, J.; Chmiela, S.; Müller, K.R.; Noé, F.; Clementi, C. Ensemble learning of coarse-grained molecular dynamics force fields with a kernel approach. J. Chem. Phys. 2020, 152, 194106. [Google Scholar] [CrossRef]
- Doerr, S.; Majewski, M.; Pérez, A.; Krämer, A.; Clementi, C.; Noe, F.; Giorgino, T.; De Fabritiis, G. TorchMD: A deep learning framework for molecular simulations. J. Chem. Theory Comput. 2021, 17, 2355–2363. [Google Scholar] [CrossRef]
- Wang, J.; Olsson, S.; Wehmeyer, C.; Pérez, A.; Charron, N.E.; de Fabritiis, G.; Noé, F.; Clementi, C. Machine learning of coarse-grained molecular dynamics force fields. ACS Cent. Sci. 2019, 5, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Taketomi, H.; Ueda, Y.; Gō, N. Studies on protein folding, unfolding and fluctuations by computer simulation. 1. Effect of specific amino-acid sequence represented by specific inter-unit interactions. Int. J. Pept. Protein Res. 1975, 7, 445–459. [Google Scholar] [CrossRef]
- Hills, R.D.; Brooks, C.L. Insights from coarse-grained Gō models for protein folding and dynamics. Int. J. Mol. Sci. 2009, 10, 889–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinitskiy, A.V.; Voth, G.A. Coarse-graining of proteins based on elastic network models. Chem. Phys. 2008, 422, 165–174. [Google Scholar] [CrossRef]
- Trylska, J. Coarse-grained models to study dynamics of nanoscale biomolecules and their applications to the ribosome. J. Phys. Cond. Matter 2010, 22, 453101. [Google Scholar] [CrossRef]
- Kmiecik, S.; Kouza, M.; Badaczewska-Dawid, A.; Kloczkowski, A.; Kolinski, A. Modeling of protein structural flexibility and large-scale dynamics: Coarse-grained simulations and elastic network models. Int. J. Mol. Sci. 2018, 19, 3496. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.C.; Onuchic, J.N.; Levine, H. Effective stochastic dynamics on a protein folding energy landscape. J. Chem. Phys. 2004, 125, 054910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schug, A.; Hyeon, C.; Onuchic, J. Coarse-grained structure-based simulations of proteins and RNA. In Coarse-Graining of Condensed Phase and Biomolecular Systems; Voth, G., Ed.; CRC Press: Boca Raton, FL, USA, 2008; Chapter 9; pp. 123–140. [Google Scholar]
- Hoang, T.X.; Cieplak, M. Molecular dynamics of folding of secondary structures in Go-like models of proteins. J. Chem. Phys. 2000, 112, 6851–6862. [Google Scholar] [CrossRef] [Green Version]
- Sułkowska, J.I.; Sułkowski, P.; Szymczak, P.; Cieplak, M. Untying knots in proteins. J. Am. Chem. Soc. 2010, 132, 13954–13956. [Google Scholar] [CrossRef]
- Cieplak, M. Mechanostability of virus capsids and their proteins in structure-based models. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes. From Bioinformatics to Molecular Quantum Mechanics; Liwo, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 295–315. [Google Scholar]
- Davtyan, A.; Schafer, N.P.; Zheng, W.; Clementi, C.; Wolynes, P.G.; Papoian, G.A. AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Lin, X.; Lu, W.; Onuchic, J.N.; Wolynes, P.G. Protein folding and structure prediction from the ground up II: AAWSEM for α/β proteins. J. Phys. Chem. B 2017, 121, 3473–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, M.; Schafer, N.P.; Wolynes, P.G. Exploring the interplay between fibrillization and amorphous aggregation channels on the energy landscapes of tau repeat isoforms. Proc. Natl. Acad. Sci. USA 2020, 117, 4125–4130. [Google Scholar] [CrossRef]
- Alessandri, R.; Grünewald, F.; Marrink, S.J. The Martini model in materials science. Adv. Mater. 2021, 33, 2008635. [Google Scholar] [CrossRef]
- Arnarez, C.; Uusitalo, J.J.; Masman, M.F.; Ingólfsson, H.I.; de Jong, D.H.; Melo, M.N.; Periole, X.; de Vries, A.H.; Marrink, S.J. Dry Martini, a coarse-grained force field for lipid membrane simulations with implicit solvent. J. Chem. Theory Comput. 2015, 11, 260–275. [Google Scholar] [CrossRef]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hou, Q.; Lensink, M.F.; Heringa, J.; Feenstra, K.A. CLUB-MARTINI: Selecting favourable interactions amongst available candidates, a coarse-grained simulation approach to scoring docking decoys. PLoS ONE 2016, 11, e0155251. [Google Scholar] [CrossRef]
- Honorato, R.V.; Roel-Touris, J.; Bonvin, A.M.J.J. MARTINI-based protein-DNA coarse-grained HADDOCKing. Front. Mol. Biosci. 2016, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Sterpone, F.; Melchionna, S.; Tuffery, P.; Pasquali, S.; Mousseau, N.; Cragnolini, T.; Chebaro, Y.; St-Pierre, J.F.; Kalimeri, M.; Barducci, A.; et al. The OPEP protein model: From single molecules, amyloid formation, crowding and hydrodynamics to DNA/RNA systems. Chem. Soc. Rev. 2014, 43, 4871–4893. [Google Scholar] [CrossRef] [Green Version]
- Barroso da Silva, F.L.; Sterpone, F.; Derreumaux, P. OPEP6: A new constant-pH molecular dynamics simulation scheme with OPEP coarse-grained force field. J. Chem. Theory Comput. 2019, 15, 3875–3888. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster denovo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [Green Version]
- Cragnolini, T.; Laurin, Y.; Derreumaux, P.; Pasquali, S. Coarse-grained HiRE-RNA model for ab initio RNA folding beyond simple molecules, including noncanonical and multiple base pairings. J. Chem. Theory Comput. 2015, 11, 3510–3522. [Google Scholar] [CrossRef] [PubMed]
- Kynast, P.; Derreumaux, P.; Strodel, B. Evaluation of the coarse-grained OPEP force field for protein-protein docking. BMC Biophys. 2016, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouldridge, T.E.; Louis, A.A.; Doye, J.P.K. DNA nanotweezers studied with a coarse-grained model of DNA. Phys. Rev. Lett. 2010, 104, 178101. [Google Scholar] [CrossRef] [PubMed]
- Ouldridge, T.E.; Louis, A.A.; Doye, J.P.K. Structural, mechanical, and thermodynamic properties of a coarse-grained DNA model. J. Chem. Phys. 2011, 134, 085101. [Google Scholar] [CrossRef] [Green Version]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Rovigatti, L.; Doye, J.P.K.; Louis, A.A. Sequence-dependent thermodynamics of a coarse-grained DNA model. J. Chem. Phys. 2012, 137, 135101. [Google Scholar] [CrossRef] [PubMed]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Doye, J.P.K.; Louis, A.A. A nucleotide-level coarse-grained model of RNA. J. Chem. Phys. 2014, 140, 235102. [Google Scholar] [CrossRef] [Green Version]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing improved structural properties and salt dependence into a coarse-grained model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [Green Version]
- Snodin, B.E.K.; Romano, F.; Rovigatti, L.; Ouldridge, T.E.; Louis, A.A.; Doye, J.P.K. Direct Simulation of the Self-Assembly of a Small DNA Origami. ACS Nano 2016, 10, 1724–1737. [Google Scholar] [CrossRef] [Green Version]
- Darré, L.; Machado, M.R.; Brandner, A.F.; González, H.C.; Ferreira, S.; Pantano, S. SIRAH: A structurally unbiased coarse-grained force field for proteins with aqueous solvation and long-range electrostatics. J. Chem. Theory Comput. 2015, 11, 723–739. [Google Scholar] [CrossRef]
- Machado, M.R.; Barrera, E.E.; Klein, F.; Sóñora, M.; Silva, S.; Pantano, S. The SIRAH 2.0 force field: Altius, Fortius, Citius. J. Chem. Theory Comput. 2019, 15, 2719–2733. [Google Scholar] [CrossRef]
- Brandner, A.; Schüller, A.; Melo, F.; Pantano, S. Exploring DNA dynamics within oligonucleosomes with coarse-grained simulations: SIRAH force field extension for protein-DNA complexes. Biochem. Biophys. Res. Commun. 2018, 498, 319–326. [Google Scholar] [CrossRef]
- Pearlman, D.; Case, D.; Caldwell, J.; Ross, W.; Cheatham, T., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Liwo, A.; Ołdziej, S.; Pincus, M.R.; Wawak, R.J.; Rackovsky, S.; Scheraga, H.A. A united-residue force field for off-lattice protein-structure simulations. I. Functional forms and parameters of long-range side-chain interaction potentials from protein crystal data. J. Comput. Chem. 1997, 18, 849–873. [Google Scholar] [CrossRef]
- He, Y.; Maciejczyk, M.; Ołdziej, S.; Scheraga, H.A.; Liwo, A. Mean-field interactions between nucleic-acid-base dipoles can drive the formation of a double helix. Phys. Rev. Lett. 2013, 110, 098101. [Google Scholar] [CrossRef] [Green Version]
- Lubecka, E.A.; Liwo, A. A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. II. Backbone-local potentials of coarse-grained O1→4-bonded polyglucose chains. J. Chem. Phys. 2017, 147, 115101. [Google Scholar] [CrossRef]
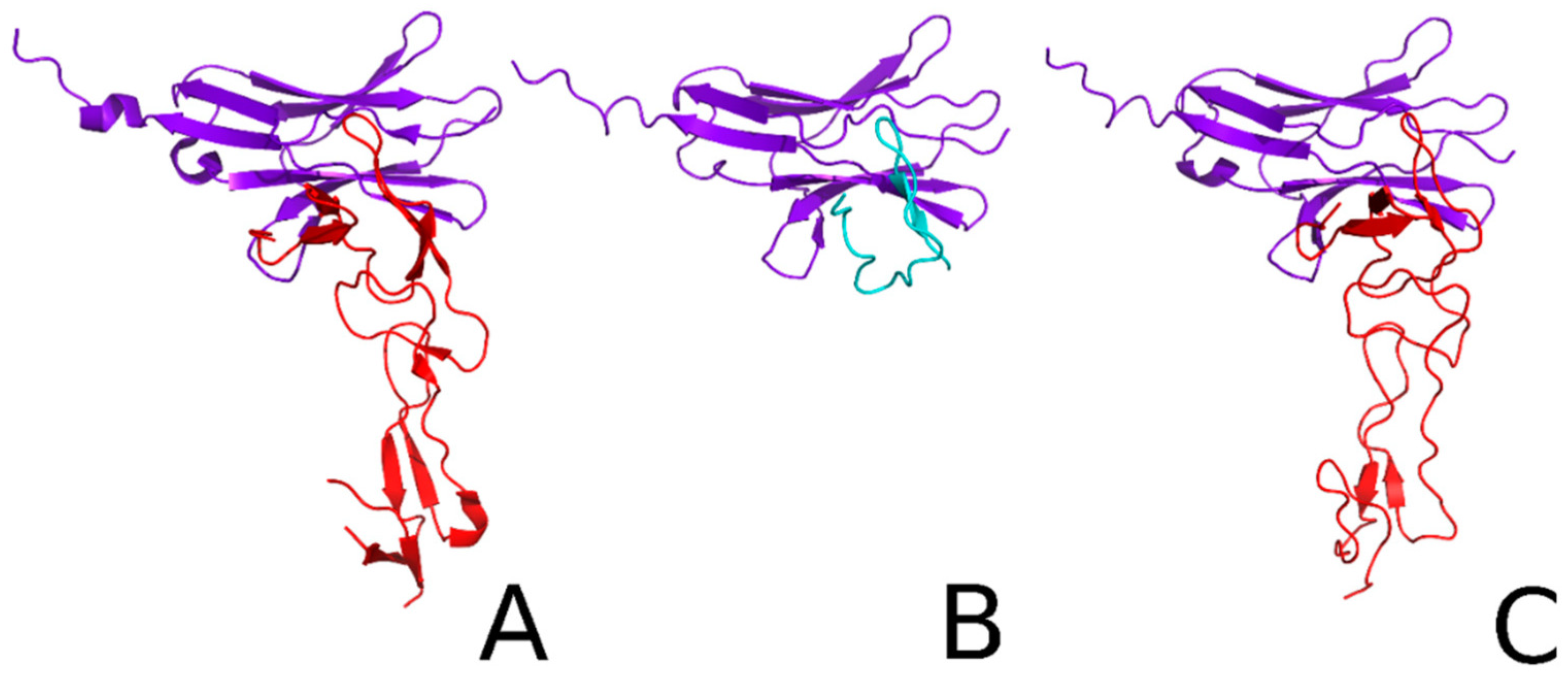
- Samsonov, S.A.; Lubecka, E.A.; Bojarski, K.K.; Ganzynkowicz, R.; Liwo, A. Local and long range potentials for heparin-protein systems for coarse-grained simulations. Biopolymers 2019, 110, e23269. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, A.K.; Giełdoń, A.; Yin, Y.; He, Y.; Scheraga, H.A.; Liwo, A. A new protein nucleic-acid coarse-grained force field based on the UNRES and NARES-2P force fields. J. Comput. Chem. 2018, 39, 2360–2370. [Google Scholar] [CrossRef]
- Liwo, A.; Kaźmierkiewicz, R.; Czaplewski, C.; Groth, M.; Ołdziej, S.; Wawak, R.J.; Rackovsky, S.; Pincus, M.R.; Scheraga, H.A. United-residue force field for off-lattice protein-structure simulations; III. Origin of backbone hydrogen-bonding cooperativity in united-residue potentials. J. Comput. Chem. 1998, 19, 259–276. [Google Scholar] [CrossRef]
- Ziȩba, K.; Ślusarz, M.; Ślusarz, R.; Liwo, A.; Czaplewski, C.; Sieradzan, A.K. Extension of the UNRES coarse-grained force field to membrane proteins in the lipid bilayer. J. Phys. Chem. B 2019, 22, 4758. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Scheraga, H.A. Conformational space annealing by parallel computations: Extensive conformational search of Met-enkephalin and of the 20-residue membrane-bound portion of melittin. Int. J. Quant. Chem. 1999, 75, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Ołdziej, S.; Czaplewski, C.; Liwo, A.; Chinchio, M.; Nanias, M.; Vila, J.A.; Khalili, M.; Arnautova, Y.A.; Jagielska, A.; Makowski, M.; et al. Physics-based protein-structure prediction using a hierarchical protocol based on the UNRES force field—Test with CASP5 and CASP6 targets. Proc. Natl. Acad. Sci. USA 2005, 102, 7547–7552. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Mozolewska, M.A.; Krupa, P.; Sieradzan, A.K.; Wirecki, T.K.; Liwo, A.; Kachlishvili, K.; Rackovsky, S.; Jagieła, D.; Ślusarz, R.; et al. Lessons from application of the UNRES force field to predictions of structures of CASP10 targets. Proc. Natl. Acad. Sci. USA 2013, 110, 14936–14941. [Google Scholar] [CrossRef] [Green Version]
- Krupa, P.; Mozolewska, M.; Wiśniewska, M.; Yin, Y.; He, Y.; Sieradzan, A.; Ganzynkowicz, R.; Lipska, A.; Karczyńska, A.; Ślusarz, M.; et al. Performance of protein-structure predictions with the physics-based UNRES force field in CASP11. Bioinformatics 2016, 32, 3270–3278. [Google Scholar] [CrossRef] [Green Version]
- Lubecka, E.A.; Karczyńska, A.S.; Lipska, A.G.; Sieradzan, A.K.; Ziȩba, K.; Sikorska, C.; Uciechowska, U.; Samsonov, S.A.; Krupa, P.; Mozolewska, M.A.; et al. Evaluation of the scale-consistent UNRES force field in template-free prediction of protein structures in the CASP13 experiment. J. Mol. Graph. Model. 2019, 92, 154–166. [Google Scholar] [CrossRef]
- Karczyńska, A.; Ziȩba, K.; Uciechowska, U.; Mozolewska, M.A.; Krupa, P.; Lubecka, E.A.; Lipska, A.G.; Sikorska, C.; Samsonov, S.A.; Sieradzan, A.K.; et al. Improved consensus-fragment selection in template-assisted prediction of protein structures with the UNRES force field in CASP13. J. Chem. Inf. Model. 2020, 60, 1844–1864. [Google Scholar] [CrossRef]
- Zhou, R.; Maisuradze, G.G.; Suñol, D.; Todorovski, T.; Macias, M.J.; Xiao, Y.; Scheraga, H.A.; Czaplewski, C.; Liwo, A. Folding kinetics of WW domains with the united residue force field for bridging microscopic motions and experimental measurements. Proc. Natl. Acad. Sci. USA 2014, 111, 18243–18248. [Google Scholar] [CrossRef] [Green Version]
- Maisuradze, G.G.; Senet, P.; Czaplewski, C.; Liwo, A.; Scheraga, H.A. Investigation of protein folding by coarse-grained molecular dynamics with the UNRES force field. J. Phys. Chem. A 2010, 114, 4471–4485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golas, E.I.; Maisuradze, G.G.; Senet, P.; Ołdziej, S.; Czaplewski, C.; Scheraga, H.A.; Liwo, A. Simulation of the opening and closing of Hsp70 chaperones by coarse-grained molecular dynamics. J. Chem. Theory Comput. 2012, 8, 1334–1343. [Google Scholar] [CrossRef]
- Mozolewska, M.; Krupa, P.; Scheraga, H.; Liwo, A. Molecular modeling of the binding modes of the iron-sulfur protein to the Jac1 co-chaperone from Saccharomyces cerevisiae by all-atom and coarse-grained approaches. Proteins Struct. Funct. Bioinf. 2015, 83, 1414–1426. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Liwo, A.; Browne, D.; Scheraga, H.A. Mechanism of fiber assembly; treatment of Aβ-peptide peptide aggregation with a coarse-grained united-residue force field. J. Mol. Biol. 2010, 404, 537–552. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Liwo, A.; Scheraga, H.A. A study of the α-helical intermediate preceding the aggregation of the amino-terminal fragment of the Aβ-amyloid peptide (1–28). J. Phys. Chem. B 2011, 115, 12978–12983. [Google Scholar] [CrossRef] [Green Version]
- Rojas, A.; Maisuradze, G.; Scheraga, H. Dependence of the formation of Tau and A beta peptide mixed aggregates on the secondary structure of the N-terminal region of A beta. J. Phys. Chem. B 2018, 122, 7049–7056. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Krupa, P.; Hai, N.M.; Linh, H.Q.; Li, M.S. Structure and physicochemical properties of the Aβ42 tetramer: Multiscale molecular dynamics simulations. J. Phys. Chem. B 2019, 123, 7253–7269. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Niadzvedtski, A.; Scheraga, H.A.; Liwo, A. Revised backbone-virtual-bond-angle potentials to Rteat the L- and D-amino acid residues in the coarse-grained united residue (UNRES) force field. J. Chem. Theory Comput. 2014, 10, 2194–2203. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Bogunia, M.; Mech, P.; Ganzynkowicz, R.; Giełdoń, A.; Liwo, A.; Makowski, M. Introduction of phosphorylated residues into the UNRES coarse-grained model: Toward modeling of signaling processes. J. Phys. Chem. B 2019, 119, 8526–8534. [Google Scholar] [CrossRef]
- Chinchio, M.; Czaplewski, C.; Liwo, A.; Ołdziej, S.; Scheraga, H.A. Dynamic formation and breaking of disulfide bonds in molecular dynamics simulations with the UNRES force field. J. Chem. Theory Comput. 2007, 3, 1236–1248. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Mozolewska, M.A. Extension of coarse-grained UNRES force field to treat carbon nanotubes. J. Mol. Model. 2018, 24, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaplewski, C.; Karczyńska, A.; Sieradzan, A.K.; Liwo, A. UNRES server for physics-based coarse-grained simulations and prediction of protein structure, dynamics and thermodynamics. Nucleic Acids Res. 2018, 46, W304–W309. [Google Scholar] [CrossRef]
- Krupa, P.; Karczyńska, A.S.; Mozolewska, M.A.; Liwo, A.; Czaplewski, C. UNRES-Dock protein-protein and peptide-protein docking by coarse-grained replica-exchange MD simulations. Bioinformatics 2021, 37, 1613–1615. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Golon, Ł.; Liwo, A. Prediction of DNA and RNA structure with the NARES-2P force field and conformational space annealing. Phys. Chem. Chem. Phys. 2018, 20, 19656–19663. [Google Scholar] [CrossRef]
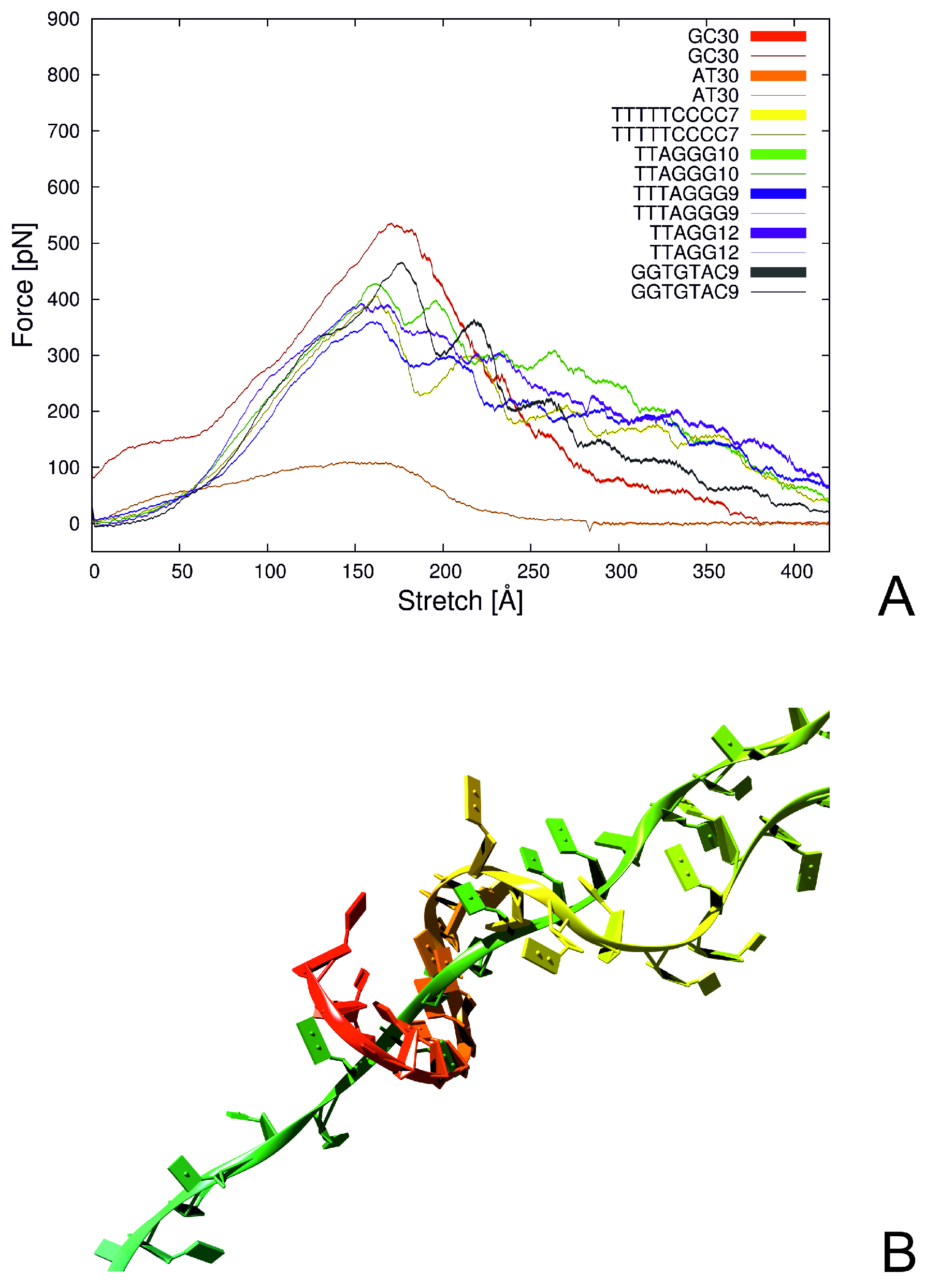
- Sieradzan, A.K.; Krupa, P.; Wales, D.J. What makes telomeres unique? J. Phys. Chem. B 2017, 121, 2207–2219. [Google Scholar] [CrossRef]
- Krupa, P.; Wales, D.J.; Sieradzan, A.K. Computational studies of the mechanical stability for single-strand break DNA. J. Phys. Chem. B 2018, 122, 8166–8173. [Google Scholar] [CrossRef]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and sulfated glycosaminoglycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring, NY, USA, 2009. [Google Scholar]
- Habuchi, H.; Habuchi, O.; Kimata, K. Sulfation pattern in glycosaminoglycan: Does it have a code? Glycoconj. J. 2004, 21, 47–52. [Google Scholar] [CrossRef]
- Peng, Y.; Yu, Y.; Lin, L.; Liu, X.; Zhang, X.; Wang, P.; Hoffman, P.; Kim, S.Y.; Zhang, F.; Linhardt, R.J. Glycosaminoglycans from bovine eye vitreous humour and interaction with collagen type II. Glycoconj. J. 2018, 35, 119–128. [Google Scholar] [CrossRef]
- Shute, J. Glycosaminoglycan and chemokine/growth factor interactions. Handb. Exp. Pharmacol. 2012, 207, 307–324. [Google Scholar]
- Li, Z.; Yasuda, Y.; Li, W.; Bogyo, M.; Katz, N.; Gordon, R.E.; Fields, G.B.; Brömme, D. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J. Biol. Chem. 2004, 279, 5470–5479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankaranarayanan, N.V.; Nagarajan, B.; Desai, U.R. So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again! Curr. Opin. Struct. Biol. 2018, 50, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Samsonov, S.A.; Pisabarro, M.T. Computational analysis of interactions in structurally available protein-glycosaminoglycan complexes. Glycobiology 2016, 26, 850–861. [Google Scholar] [CrossRef] [Green Version]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef]
- Perrimon, N.; Bernfield, M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature 2000, 404, 725–728. [Google Scholar] [CrossRef]
- Bathe, M.; Rutledge, G.C.; Grodzinsky, A.J.; Tidor, B. A coarse-grained molecular model for glycosaminoglycans: Application to chondroitin, chondroitin sulfate, and hyaluronic acid. Biophys. J. 2005, 88, 3870–3887. [Google Scholar] [CrossRef] [Green Version]
- Sattelle, B.M.; Shakeri, J.; Almond, A. Does microsecond sugar ring flexing encode 3D-shape and bioactivity in the heparanome? Biomacromolecules 2013, 14, 1149–1159. [Google Scholar] [CrossRef]
- Sattelle, B.M.; Shakeri, J.; Cliff, M.J.; Almond, A. Proteoglycans and their heterogeneous glycosaminoglycans at the atomic scale. Biomacromolecules 2015, 16, 951–961. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Yongye, A.B.; Tschampel, S.M.; González-Outeiriño, J.; Daniels, C.R.; Foley, B.L.; Woods, R.J. GLYCAM06: A generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Palmer, J.C.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Liquid-liquid transition in ST2 water. J. Chem. Phys. 2012, 137, 214505. [Google Scholar] [CrossRef]
- Samsonov, S.A.; Bichmann, L.; Pisabarro, M.T. Coarse-grained model of glycosaminoglycans. J. Chem. Inf. Model. 2015, 55, 114–124. [Google Scholar] [CrossRef]
- Christen, M.; van Gunsteren, W.F. On searching in, sampling of, and dynamically moving through conformational space of biomolecular systems: A review. J. Comput. Chem. 2008, 29, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Barducci, A.; Pfaendtner, J.; Bonomi, M. Tackling sampling challenges in biomolecular simulations. Meth. Mol. Biol. 2015, 1215, 151–171. [Google Scholar]
- Sidky, H.; Colón, Y.J.; Helfferich, J.; Sikora, B.J.; Bezik, C.; Chu, W.; Giberti, F.; Guo, A.Z.; Jiang, X.; Lequieu, J.; et al. SSAGES: Software suite for advanced general ensemble simulations. J. Chem. Phys. 2018, 148, 044104. [Google Scholar] [CrossRef]
- Allison, J.R. Computational methods for exploring protein conformations. Biochem. Soc. Trans. 2020, 48, 1707–1724. [Google Scholar] [CrossRef]
- Wang, J.; Arantes, P.R.; Bhattarai, A.; Hsu, R.V.; Pawnikar, S.; Huang, Y.M.M.; Palermo, G.; Miao, Y. Gaussian accelerated molecular dynamics: Principles and applications. WIREs Comput. Mol. Sci. 2021, 11, e1521. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kästner, J.; Thiel, W. Analysis of the statistical error in umbrella sampling simulations by umbrella integration. J. Chem. Phys. 2006, 124, 234106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, D.A. Determining the contributions of constraints in free energy calculations: Development, characterization, and recommendations. J. Chem. Phys. 1993, 98, 8946–8957. [Google Scholar] [CrossRef]
- Sieradzan, A.K.; Jakubowski, R. Introduction of steered molecular dynamics into UNRES coarse-grained simulations package. J. Comput. Chem. 2017, 38, 553–562. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Lamprakis, C.; Andreadelis, I.; Manchester, J.; Velez-Vega, C.; Duca, J.S.; Cournia, Z. Evaluating the efficiency of the Martini force field to study protein dimerization in aqueous and membrane environments. J. Chem. Theory Comput. 2021, 17, 3088–3102. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D., Jr.; Vecchi, M.P. Optimization by simulated annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef]
- Hansmann, U.H.E.; Okamoto, Y. Comparative study of multicanonical and simulated annealing algorithms in the protein folding problem. Physica A 1994, 212, 415–437. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange multicanonical algorithm and multicanonical replica-exchange method for simulating systems with rough energy landscape. Phys. Rev. Lett. 2000, 329, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 2001, 60, 96–123. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H.; Okamoto, Y. Generalized-ensemble algorithms for molecular dynamics simulations. Mol. Simul. 2007, 33, 47–56. [Google Scholar] [CrossRef]
- van Erp, T.S.; Moroni, D.; Bolhuis, P.G. A novel path sampling method for the calculation of rate constants. J. Chem. Phys. 2003, 118, 7762–7774. [Google Scholar] [CrossRef] [Green Version]
- Smit, F.X.; Luiken, J.A.; Bolhuis, P.G. Primary fibril nucleation of aggregation prone tau fragments PHF6 and PHF6*. J. Phys. Chem. B 2017, 121, 3250–3261. [Google Scholar] [CrossRef]
- Shen, H.; Liwo, A.; Scheraga, H.A. An improved functional form for the temperature scaling factors of the components of the mesoscopic UNRES force field for simulations of protein structure and dynamics. J. Phys. Chem. B 2009, 113, 8738–8744. [Google Scholar] [CrossRef] [Green Version]
- Rhee, Y.M.; Pande, V.S. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys J. 2003, 84, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Czaplewski, C.; Kalinowski, S.; Liwo, A.; Scheraga, H.A. Application of multiplexing replica exchange molecular dynamics method to the UNRES force field: Tests with α and α+β proteins. J. Chem. Theory Comput. 2009, 5, 627–640. [Google Scholar] [CrossRef] [Green Version]
- Fukunishi, H.; Watanabe, O.; Takada, S. On the Hamiltonian replica exchange method for efficient sampling, of biomolecular systems: Application to protein structure prediction. J. Chem. Phys. 2002, 116, 9058–9067. [Google Scholar] [CrossRef]
- Lee, K.H.; Chen, J. Multiscale enhanced sampling of intrinsically disordered protein conformations. J. Comput. Chem. 2015, 37, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Karczyńska, A.S.; Czaplewski, C.; Krupa, P.; Mozolewska, M.A.; Joo, K.; Lee, J.; Liwo, A. Ergodicity and model quality in template-restrained canonical and temperature/Hamiltonian replica exchange coarse-grained molecular dynamics simulations of proteins. J. Comput. Chem. 2017, 38, 2730–2746. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pezeshkian, W.; Barnoud, J.; de Vries, A.H.; Marrink, S.J. Coupling coarse-grained to fine-grained models via Hamiltonian Replica Exchange. J. Chem. Theory Comput. 2020, 16, 5313–5322. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F.A. Biomolecular force field based on the free enthalpy of hydration and solvation: The Gromos force-field parameter sets 53a5 and 53a6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Berg, B.A.; Neuhaus, T. Multicanonical ensemble: A new approach to simulate 1st order phase-transitions. Phys. Rev. Lett. 1992, 68, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, J. New Monte Carlo algorithm: Entropic sampling. Phys. Rev. Lett. 1993, 71, 211–214. [Google Scholar] [CrossRef]
- Nanias, M.; Czaplewski, C.; Scheraga, H.A. Replica exchange and multicanonical algorithms with the coarse-grained united-residue (UNRES) force field. J. Chem. Theory Comput. 2006, 2, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Macias, M.J.; Gervais, V.; Civera, C.; Oschkinat, H. Domains and design of a WW prototype. Nat. Struct. Biol. 2000, 7, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Jäger, M.; Moretto, A.; Gruebele, M.; Kelly, J.W. Tuning the free-energy landscape of a WW domain by temperature, mutation, and truncation. Proc. Natl. Acad. Sci. USA 2003, 100, 3948–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanicolas, J.; Brooks, C.L., III. The structural basis for biphasic kinetics in the folding of the WW domain from a formin-binding protein: Lessons for protein design. Proc. Natl. Acad. Sci. USA 2003, 100, 3954–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H. Roles of hydrodynamic interactions in structure formation of soft matter: Protein folding as an example. J. Phys. Condens. Matter 2005, 17, S2795–S2803. [Google Scholar] [CrossRef]
- Frembgen-Kesner, T.; Elcock, A.H. Absolute protein-protein association rate constants from flexible, coarse-grained Brownian dynamics simulations: The role of intermolecular hydrodynamic interactions in Barnase-Barstar association. Biophys. J. 2010, 99, L75–L77. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Skolnick, J. On the importance of hydrodynamic interactions in lipid membrane formation. Biophys. J. 2013, 104, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Skolnick, J. Sliding of proteins non-specifically bound to DNA: Brownian dynamics studies with coarse-grained protein and DNA models. PLoS Comput. Biol. 2014, 10, e1003990. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vernon, R.M.; Soddoqui, Z.; Krzeminski, M.; Munhandiram, R.; Zhao, C.; Senenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Mirjalili, V.; Noyes, K.; Feig, M. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins 2014, 82, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Kolinski, A.; Skolnick, J. Reduced models of proteins and their applications. Polymer 2004, 45, 511–524. [Google Scholar] [CrossRef]
- Gront, D.; Hansmann, U.H.E.; Kolinski, A. Exploring protein energy landscapes with hierarchical clustering. J. Comput. Chem. 2005, 105, 826–830. [Google Scholar] [CrossRef] [Green Version]
- Karczyńska, A.S.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Liwo, A.; Czaplewski, C. Prediction of protein structure with the coarse-grained UNRES force field assisted by small X-Ray scattering data and knowledge-based information. Proteins 2018, 86, 228–239. [Google Scholar] [CrossRef]
- Lubecka, E.; Liwo, A. ESCASA: Analytical estimation of atomic coordinates from coarse-grained geometry for NMR-assisted protein structure modeling. I. Backbone and Hβ protons. J. Comput. Chem. 2021, 42, 1579–1589. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Mozolewska, M.; Krupa, P.; Zaborowski, B.; Liwo, A.; Lee, J.; Joo, K.; Czaplewski, C. Use of restraints from consensus fragments of multiple server models to enhance protein-structure prediction capability of the UNRES force field. J. Chem. Inf. Model. 2016, 56, 2263–2279. [Google Scholar] [CrossRef]
- Lubecka, E.A.; Liwo, A. Introduction of a bounded penalty function in contact-assisted simulations of protein structures to omit false restraints. J. Comput. Chem. 2019, 40, 2164–2178. [Google Scholar] [CrossRef]
- Karczyńska, A.; Mozolewska, M.A.; Krupa, P.; Giełdoń, A.; Bojarski, K.K.; Zaborowski, B.; Liwo, A.; Ślusarz, R.; Ślusarz, M.; Lee, J.; et al. Use of the UNRES force field in template-based prediction of protein structures and the refinement of server models: Test with CASP12 targets. J. Mol. Graph. Model. 2018, 83, 92–99. [Google Scholar] [CrossRef]
- Spodzieja, M.; Kuncewicz, K.; Sieradzan, A.; Karczyńska, A.; Iwaszkiewicz, J.; Cesson, V.; Wȩgrzyn, K.; Zhukov, I.; Maszota-Zieleniak, M.; Michielin, O.; et al. Disulfide-linked peptides for blocking. Int. J. Mol. Sci. 2020, 21, 636. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Galati, A.; Micheli, E.; Cacchione, S. Chromatin structure in telomere dynamics. Front. Oncol. 2013, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- von Zglinicki, T.; Saretzki, G.; Döcke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heymann, B.; Tavan, P. Ligand binding: Molecular mechanics calculation of the streptavidin-biotin rupture force. Science 1996, 271, 997–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the current state of Amber force field modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lipska, A.G.; Samsonov, S.A.; Murarka, R.K. Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems. Biomolecules 2021, 11, 1347. https://doi.org/10.3390/biom11091347
Liwo A, Czaplewski C, Sieradzan AK, Lipska AG, Samsonov SA, Murarka RK. Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems. Biomolecules. 2021; 11(9):1347. https://doi.org/10.3390/biom11091347
Chicago/Turabian StyleLiwo, Adam, Cezary Czaplewski, Adam K. Sieradzan, Agnieszka G. Lipska, Sergey A. Samsonov, and Rajesh K. Murarka. 2021. "Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems" Biomolecules 11, no. 9: 1347. https://doi.org/10.3390/biom11091347