
Emergence and Spread of a B.1.1.28-Derived P.6 Lineage with Q675H and Q677H Spike Mutations in Uruguay

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. SARS-CoV-2 Samples
2.3. Genome Sequencing
2.4. SARS-CoV-2 Lineage Assignment
2.5. Phylogenetic and Phylogeographic Analysis of B.1.1.28
2.6. Phylogenetic and Phylogeographic Analysis of P.2
2.7. Lineage Prevalence of Available Uruguayan Samples
2.8. Determination of Prevalence of Q675H + Q677H
2.9. Structural Representation of the Spike Protein
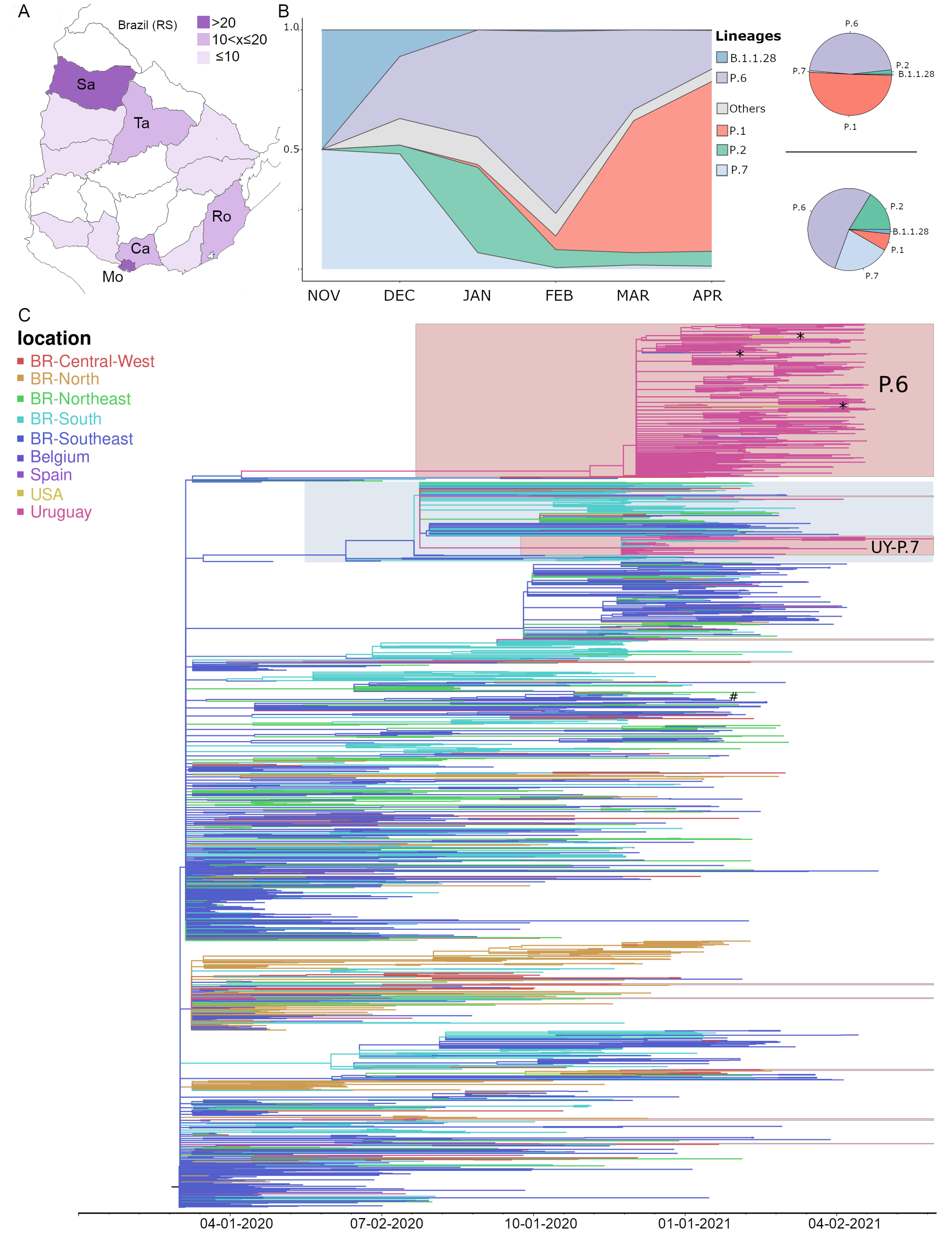
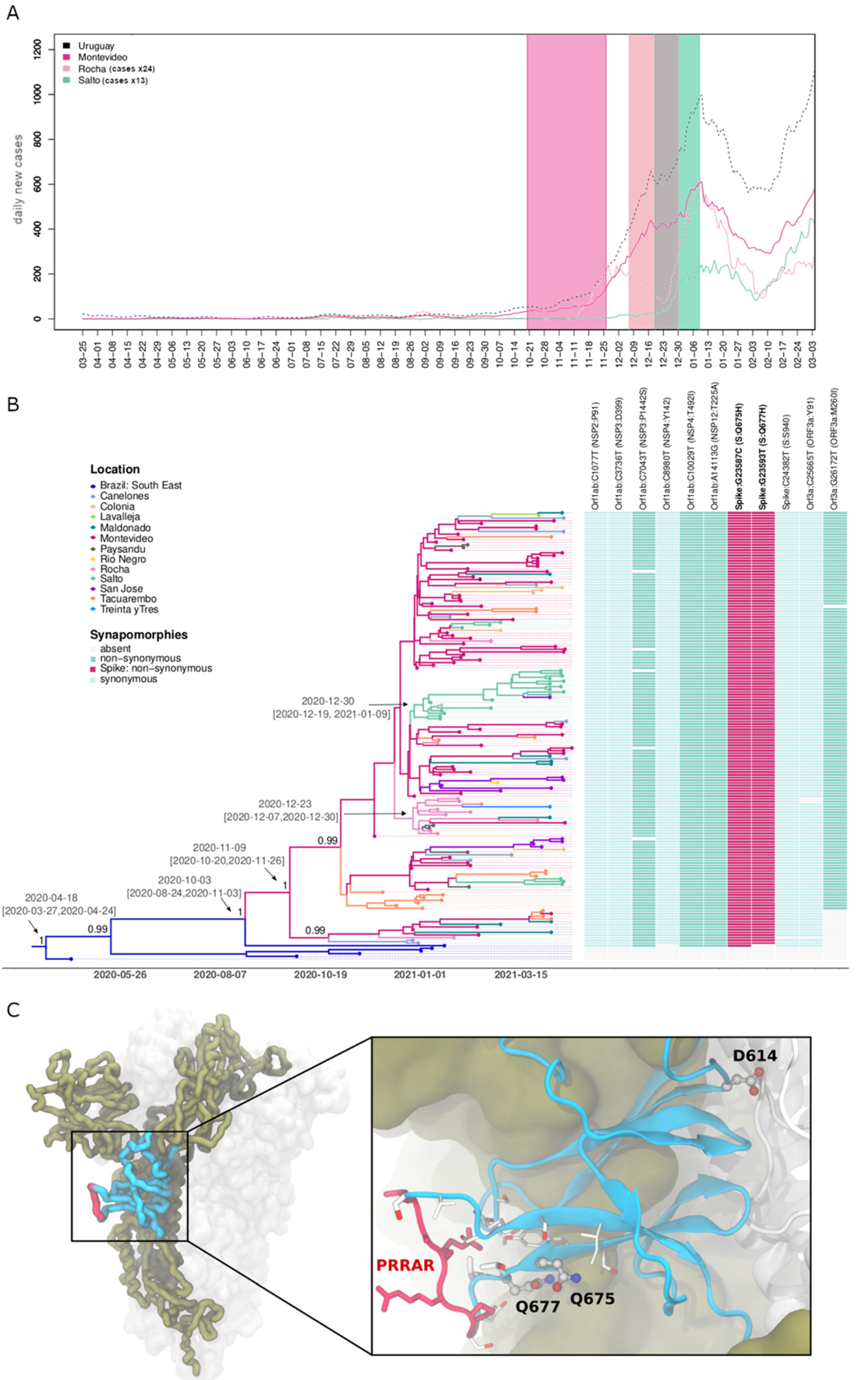
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC Coronavirus Disease 2019 (COVID-19). Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 17 August 2021).
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens 2021, 10, 633. [Google Scholar] [CrossRef]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and Epidemic Spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Resende, P.C.; Delatorre, E.; Gräf, T.; Mir, D.; Motta, F.C.; Appolinario, L.R.; da Paixão, A.C.D.; da Fonseca Mendonça, A.C.; Ogrzewalska, M.; Caetano, B.; et al. Evolutionary Dynamics and Dissemination Pattern of the SARS-CoV-2 Lineage B.1.1.33 During the Early Pandemic Phase in Brazil. Front. Microbiol. 2021, 11, 3565. [Google Scholar] [CrossRef] [PubMed]
- Rede Genomica Fiocruz. Available online: http://www.genomahcov.fiocruz.br/ (accessed on 17 August 2021).
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 Variant in Travelers from Brazil to Japan. Emerg. Infect. Dis. 2021, 27, 1243. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Lima Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- Cov-Lineages. Available online: https://cov-lineages.org/lineage.html?lineage=P.1 (accessed on 17 August 2021).
- Cov-Lineages. Available online: https://cov-lineages.org/lineage.html?lineage=P.2 (accessed on 17 August 2021).
- Voloch, C.M.; da Silva Francisco, R.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; de C Guimarães, A.P.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic Characterization of a Novel SARS-CoV-2 Lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Lamarca, A.P.; de Almeida, L.G.P.; da S. Francisco, R.; Lima, L.F.A.; Scortecci, K.C.; Perez, V.P.; Brustolini, O.J.; Sousa, E.S.S.; Secco, D.A.; Santos, A.M.G.; et al. Genomic Surveillance of SARS-CoV-2 Tracks Early Interstate Transmission of P.1 Lineage and Diversification within P.2 Clade in Brazil. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.03.21.21253418v2 (accessed on 17 August 2021). [CrossRef]
- Franceschi, V.B.; Ferrareze, P.A.G.; Zimerman, R.A.; Cybis, G.B.; Thompson, C.E. Mutation hotspots and spatiotemporal distribution of SARS-CoV-2 lineages in Brazil, February 2020–2021. Virus Res. 2021, 304, 198532. [Google Scholar] [CrossRef] [PubMed]
- da Silva Francisco, R., Jr.; Lamarca, A.P.; de Almeida, L.G.P.; Cavalcante, L.; Machado, D.T.; Martins, Y.; Brustolini, O.; Gerber, A.L.; de C Guimarães, A.P.; Gonçalves, R.B.; et al. Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.07.20.21260890v1 (accessed on 17 August 2021). [CrossRef]
- Potential New B.1.1.28 Sub-Lineage with L452R in Brazil Issue #68·Cov-Lineages/Pango-Designation. Available online: https://github.com/cov-lineages/pango-designation/issues/68 (accessed on 17 August 2021).
- Tablizo, F.A.; Kim, K.M.; Lapid, C.M.; Castro, M.J.R.; Yangzon, M.S.L.; Maralit, B.A.; Ayes, M.E.C.; Cutiongco-de la Paz, E.M.; De Guzman, A.R.; Yap, J.M.C.; et al. Genome Sequencing and Analysis of an Emergent SARS-CoV-2 Variant Characterized by Multiple Spike Protein Mutations Detected from the Central Visayas Region of the Philippines. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.03.03.21252812v2 (accessed on 17 August 2021). [CrossRef]
- da Silva Francisco, R., Jr.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; de C Guimarães, A.P.; Antunes, A.K.E.; et al. Pervasive Transmission of E484K and Emergence of VUI-NP13L with Evidence of SARS-CoV-2 Co-Infection Events by Two Different Lineages in Rio Grande Do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, F.H.; Varela, A.P.M.; Prichula, J.; Comerlato, J.; Comerlato, C.B.; Roglio, V.S.; Pereira, G.F.M.; Moreno, F.; Seixas, A.; Wendland, E.M. Emergence of the Novel SARS-CoV-2 Lineage VUI-NP13L and Massive Spread of P.2 in South Brazil. Emerg. Microbes Infect. 2021, 10, 1431–1440. [Google Scholar] [CrossRef]
- Estadisticasuy. Available online: https://guiad-covid.github.io/estadisticasuy.html (accessed on 26 July 2021).
- Taylor, L. Why Uruguay Lost Control of COVID. Nature 2021, 595, 21. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, V.; Harkins, G.W.; Mabvakure, B.; Smidt, S.; Zappile, P.; Marier, C.; Maurano, M.T.; Perez, V.; Mazza, N.; Beloso, C.; et al. SARS-CoV-2 Genomic Characterization and Clinical Manifestation of the COVID-19 Outbreak in Uruguay. Emerg. Microbes Infect. 2021, 10, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Mir, D.; Rego, N.; Resende, P.C.; Tort, F.; Fernández-Calero, T.; Noya, V.; Brandes, M.; Possi, T.; Arleo, M.; Reyes, N.; et al. Recurrent Dissemination of SARS-CoV-2 Through the Uruguayan–Brazilian Border. Front. Microbiol. 2021, 12, 653986. [Google Scholar] [CrossRef]
- Fraser, C.; Riley, S.; Anderson, R.M.; Ferguson, N.M. Factors That Make an Infectious Disease Outbreak Controllable. Proc. Natl. Acad. Sci. USA 2004, 101, 6146–6151. [Google Scholar] [CrossRef] [Green Version]
- Grantz, K.H.; Lee, E.C.; McGowan, L.D.; Lee, K.H.; Metcalf, C.J.E.; Gurley, E.S.; Lessler, J. Maximizing and Evaluating the Impact of Test-Trace-Isolate Programs: A Modeling Study. PLoS Med. 2021, 18, e1003585. [Google Scholar] [CrossRef]
- Uruguay Medidas. Available online: https://medios.presidencia.gub.uy/tav_portal/2020/noticias/AH_204/Medidas%2016.12.2020.pdf (accessed on 16 December 2020).
- Rego, N.; Costábile, A.; Paz, M.; Salazar, C.; Perbolianachis, P.; Spangenberg, L.; Ferrés, I.; Arce, R.; Fajardo, A.; Arleo, M.; et al. Real-Time Genomic Surveillance for SARS-CoV-2 Variants of Concern, Uruguay. Emerg. Infect. Dis. 2021, 27. [Google Scholar] [CrossRef]
- Cov-Lineages. Available online: https://cov-lineages.org/lineage.html?lineage=P.6 (accessed on 17 August 2021).
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-NCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antiviral Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Quick, J. NCoV-2019 Sequencing Protocol v3 (LoCost). 2020. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye/metrics (accessed on 31 August 2021).
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore. BioRxiv 2020. [Google Scholar] [CrossRef]
- Resende, P. Long Reads Nanopore Sequencing to Recover SARS-CoV-2 Whole Genome. Protocols 2020. [Google Scholar] [CrossRef]
- Freed, N.; Silander, O. SARS-CoV2 Genome Sequencing Protocol (1200bp Amplicon “Midnight” Primer Set, Using Nanopore Rapid Kit) V5. Available online: https://www.protocols.io/view/sars-cov2-genome-sequencing-protocol-1200bp-amplic-btsrnnd6?version_warning=no (accessed on 31 August 2021).
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and Inexpensive Whole-Genome Sequencing of SARS-CoV-2 Using 1200 Bp Tiled Amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore Technologies. Available online: https://nanoporetech.com/ (accessed on 2 June 2021).
- GitHub-Replikation/PoreCov: SARS-CoV-2 Workflow for Nanopore Sequence Data. Available online: https://github.com/replikation/poreCov (accessed on 23 July 2021).
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational Strategies to Combat COVID-19: Useful Tools to Accelerate SARS-CoV-2 and Coronavirus Research. Brief. Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Loman, N.J.; Quick, J.; Simpson, J.T. A Complete Bacterial Genome Assembled de Novo Using Only Nanopore Sequencing Data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific Containers for Mobility of Compute. PLoS ONE 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.M.; Gamaarachchi, H.; Nguyen, T.; Gollon, A.; Tong, S.; Aquilina-Reid, C.; Bowen-James, R.; Deveson, I.W. InterARTIC: An Interactive Web Application for Whole-Genome Nanopore Sequencing Analysis of SARS-CoV-2 and Other Viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Hölzer, M. PoreCov-An Easy to Use, Fast, and Robust Workflow for SARS-CoV-2 Genome Reconstruction via Nanopore Sequencing. Front. Genet. 2021, 12, 711437. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- COG-UK. Available online: https://pangolin.cog-uk.io/ (accessed on 26 July 2021).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, veab064. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—from Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2. Virus Evol. 2020, 6. [Google Scholar] [CrossRef]
- Time Dependence of SARS-CoV-2 Substitution Rates—SARS-CoV-2 Coronavirus/NCoV-2019 Evolutionary History. Available online: https://virological.org/t/time-dependence-of-sars-cov-2-substitution-rates/542 (accessed on 23 July 2021).
- Ishikawa, S.A.; Zhukova, A.; Iwasaki, W.; Gascuel, O. A Fast Likelihood Method to Reconstruct and Visualize Ancestral Scenarios. Mol. Biol. Evol. 2019, 36, 2069–2085. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.A.R.; Suchard, M.A. Bayesian Analysis of Elapsed Times in Continuous-Time Markov Chains. Can. J. Stat. 2008, 36, 355–368. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; Maio, N.D.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 26 July 2021).
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- D. E. Shaw Research. Technical Data Molecular Dynamics Simulations Related to SARS-CoV-2. Available online: https://www.deshawresearch.com/downloads/download_trajectory_sarscov2.cgi/ (accessed on 26 July 2021).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lemmin, T.; Kalbermatter, D.; Harder, D.; Plattet, P.; Fotiadis, D. Structures and Dynamics of the Novel S1/S2 Protease Cleavage Site Loop of the SARS-CoV-2 Spike Glycoprotein. J. Struct. Biol. X 2020, 4, 100038. [Google Scholar] [CrossRef]
- Bagdonaite, I.; Thompson, A.J.; Wang, X.; Søgaard, M.; Fougeroux, C.; Frank, M.; Diedrich, J.K.; Yates, J.R.; Salanti, A.; Vakhrushev, S.Y.; et al. Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells. Viruses 2021, 13, 551. [Google Scholar] [CrossRef] [PubMed]
- Sanda, M.; Morrison, L.; Goldman, R. N- and O-Glycosylation of the SARS-CoV-2 Spike Protein. Anal. Chem. 2021, 93, 2003–2009. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Stalls, V.; Kopp, M.F.; Manne, K.; Li, D.; Wiehe, K.; et al. Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 2021. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Domman, D.B.; Snyder, D.J.; Oguntuyo, K.Y.; Diest, M.V.; Densmore, K.H.; Schwalm, K.C.; Femling, J.; Carroll, J.L.; Scott, R.S.; et al. Emergence in Late 2020 of Multiple Lineages of SARS-CoV-2 Spike Protein Variants Affecting Amino Acid Position 677. medRxiv 2021. [Google Scholar] [CrossRef]
- Detection of the Recurrent Substitution Q677H in the Spike Protein of SARS-CoV-2 in Cases Descended from the Lineage B.1.429—SARS-CoV-2 Coronavirus/NCoV-2019 Genomic Epidemiology. Available online: https://virological.org/t/detection-of-the-recurrent-substitution-q677h-in-the-spike-protein-of-sars-cov-2-in-cases-descended-from-the-lineage-b-1-429/660 (accessed on 22 July 2021).
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.D.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 Variant through Europe in the Summer of 2020. Nature 2021, 1–6. [Google Scholar] [CrossRef]
- Örd, M.; Faustova, I.; Loog, M. The Sequence at Spike S1/S2 Site Enables Cleavage by Furin and Phospho-Regulation in SARS-CoV2 but Not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 16944. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural Impact on SARS-CoV-2 Spike Protein by D614G Substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Chakraborty, S.; Nguyen, K.; Montefiori, D.C.; Korber, B.; Gnanakaran, S. The SARS-CoV-2 Spike Variant D614G Favors an Open Conformational State. Sci. Adv. 2021, 7, eabf3671. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The Effect of the D614G Substitution on the Structure of the Spike Glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Gobeil, S.M.-C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Mohammad, A.; Alshawaf, E.; Marafie, S.K.; Abu-Farha, M.; Abubaker, J.; Al-Mulla, F. Higher Binding Affinity of Furin for SARS-CoV-2 Spike (S) Protein D614G Mutant Could Be Associated with Higher SARS-CoV-2 Infectivity. Int. J. Infect. Dis. 2021, 103, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Chao, T.-L.; Li, C.-L.; Wang, S.-H.; Kao, H.-C.; Tsai, Y.-M.; Wang, H.-Y.; Hsieh, C.-L.; Lin, Y.-Y.; Chen, P.-J.; et al. D614G Substitution of SARS-CoV-2 Spike Protein Increases Syncytium Formation and Virus Titer via Enhanced Furin-Mediated Spike Cleavage. mBio 2021, 12, e00587-21. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, T.; Mueller, D.S.; Mark, A.E.; Young, P.R.; Kobe, B. The Role of Histidine Residues in Low-PH-Mediated Viral Membrane Fusion. Structure 2006, 14, 1481–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. bioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.08.12.456173v1 (accessed on 26 June 2021). [CrossRef]
- Maher, M.C.; Bartha, I.; Weaver, S.; di Iulio, J.; Ferri, E.; Soriaga, L.; Lempp, F.A.; Hie, B.L.; Bryson, B.; Berger, B.; et al. Predicting the Mutational Drivers of Future SARS-CoV-2 Variants of Concern. medRxiv 2021. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rego, N.; Salazar, C.; Paz, M.; Costábile, A.; Fajardo, A.; Ferrés, I.; Perbolianachis, P.; Fernández-Calero, T.; Noya, V.; Machado, M.R.; et al. Emergence and Spread of a B.1.1.28-Derived P.6 Lineage with Q675H and Q677H Spike Mutations in Uruguay. Viruses 2021, 13, 1801. https://doi.org/10.3390/v13091801
Rego N, Salazar C, Paz M, Costábile A, Fajardo A, Ferrés I, Perbolianachis P, Fernández-Calero T, Noya V, Machado MR, et al. Emergence and Spread of a B.1.1.28-Derived P.6 Lineage with Q675H and Q677H Spike Mutations in Uruguay. Viruses. 2021; 13(9):1801. https://doi.org/10.3390/v13091801
Chicago/Turabian StyleRego, Natalia, Cecilia Salazar, Mercedes Paz, Alicia Costábile, Alvaro Fajardo, Ignacio Ferrés, Paula Perbolianachis, Tamara Fernández-Calero, Veronica Noya, Matias R. Machado, and et al. 2021. "Emergence and Spread of a B.1.1.28-Derived P.6 Lineage with Q675H and Q677H Spike Mutations in Uruguay" Viruses 13, no. 9: 1801. https://doi.org/10.3390/v13091801