
Safety Testing of an Antisense Oligonucleotide Intended for Pediatric Indications in the Juvenile Göttingen Minipig, including an Evaluation of the Ontogeny of Key Nucleases

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
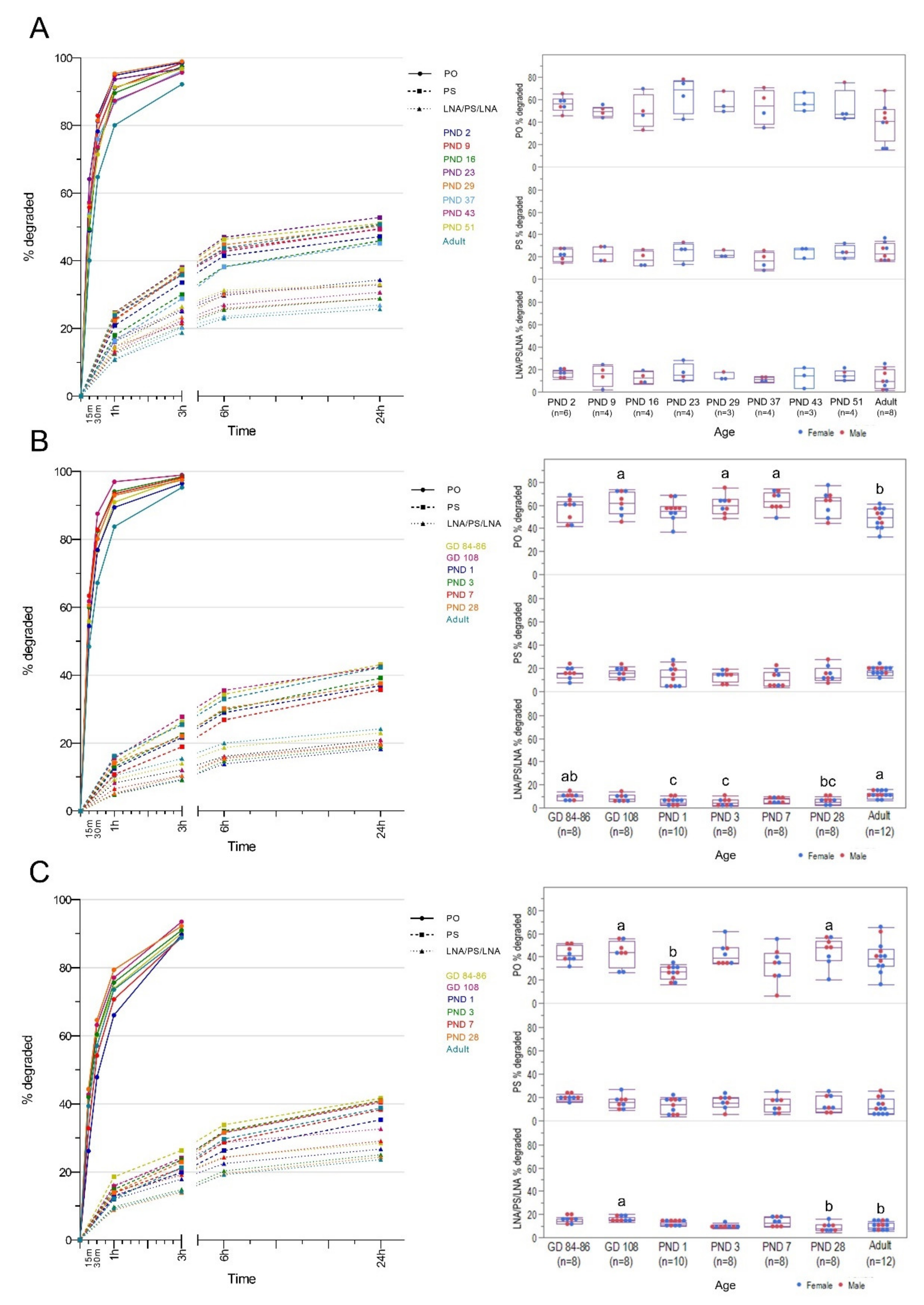
2. Materials and Methods
2.1. Antisense Oligonucleotides
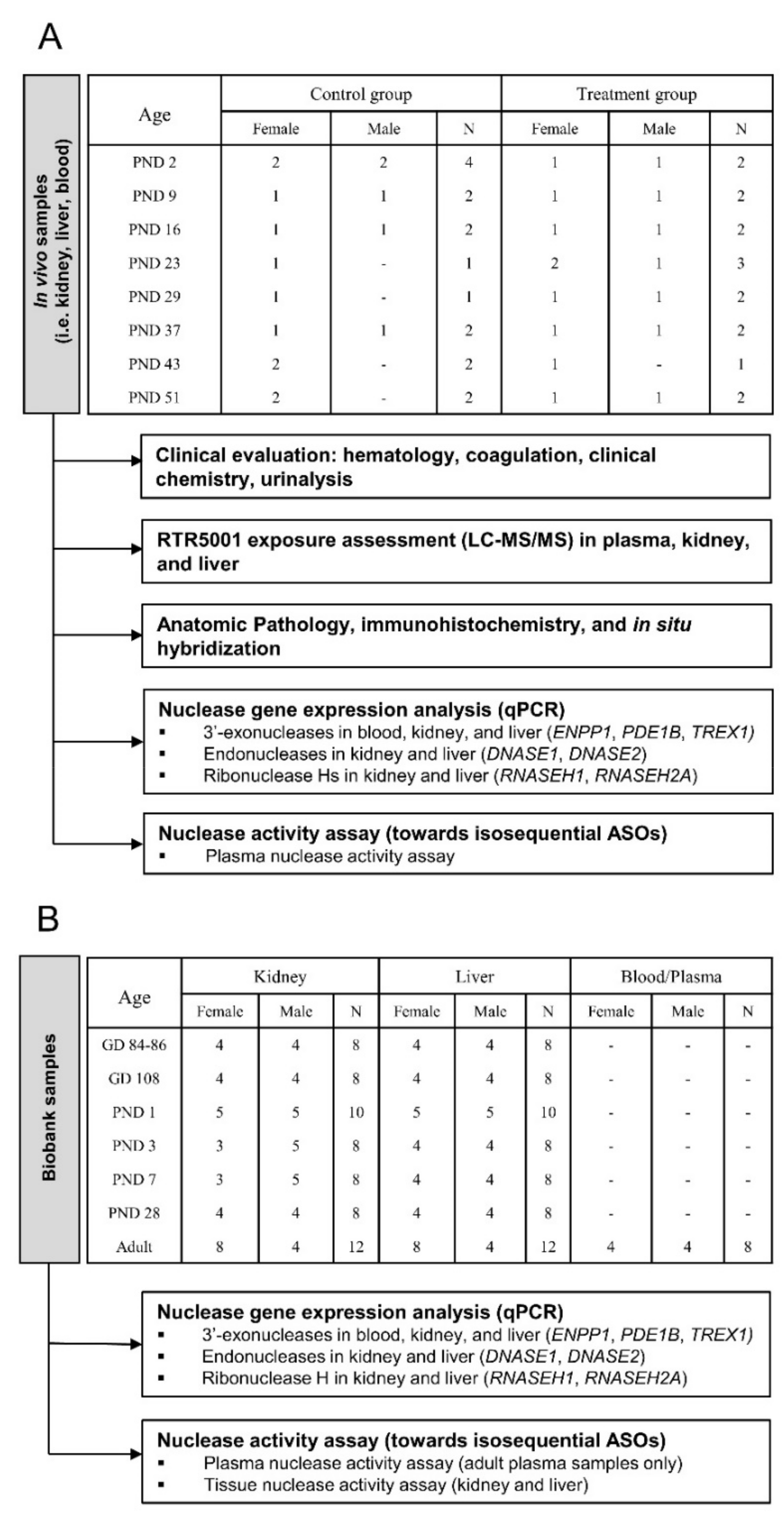
2.2. Study Design
2.3. In Vivo Study
2.3.1. In-Life Monitoring
2.3.2. Tissue Sampling
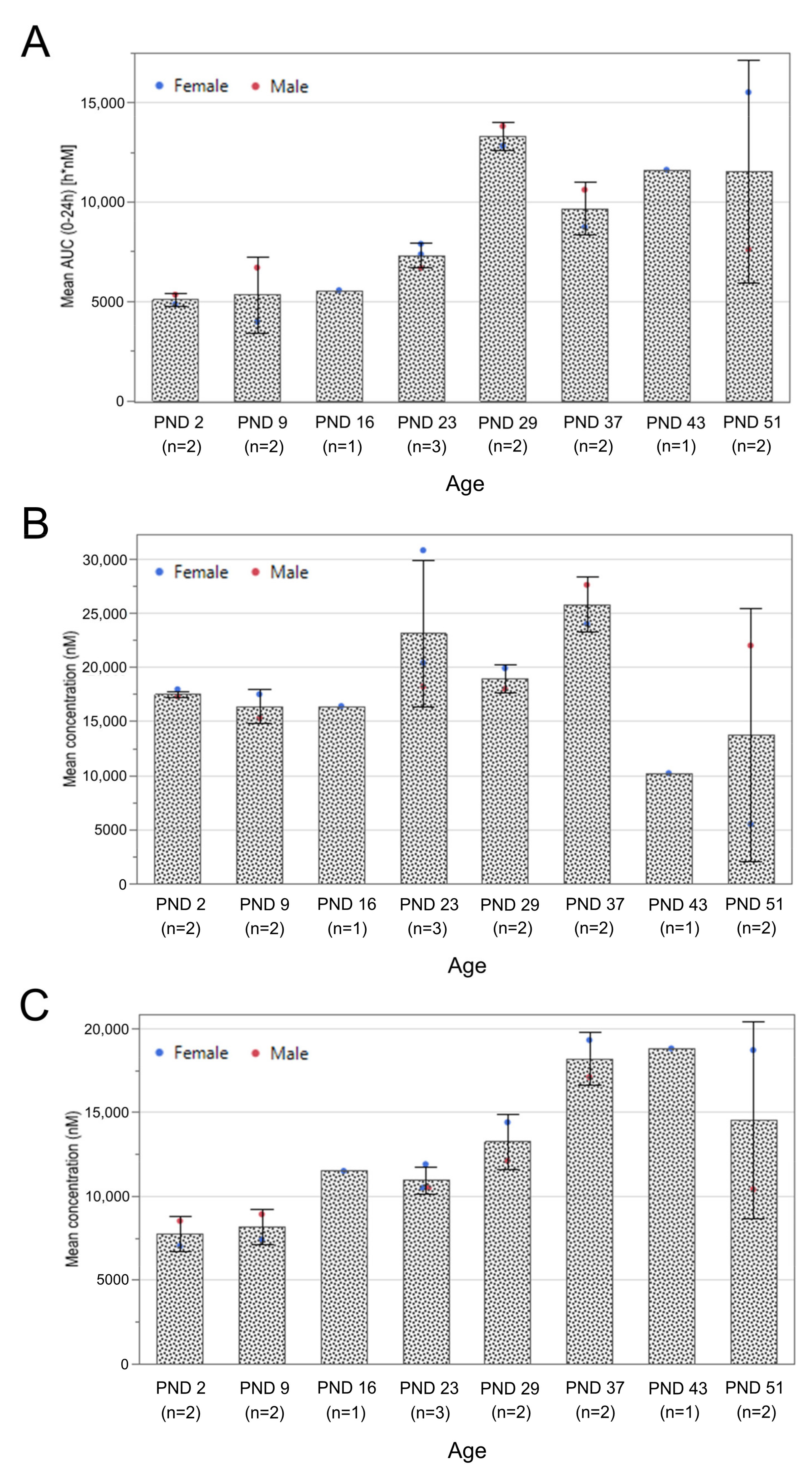
2.3.3. Exposure Assessment
2.3.4. Clinical Pathology
2.3.5. Necropsy, Anatomic Pathology, Immunohistochemistry, In Situ Hybridization
2.4. Biobank Study
2.5. Gene Expression
2.6. Plasma Nuclease Activity
2.7. Tissue Nuclease Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Name | Acc. Number | Rationale of Inclusion | Primer (5′–3′) | Amplicon Length | E% (B) | E% (K) | E% (L) |
|---|---|---|---|---|---|---|---|---|
| ENPP1 | Ectonucleotide Pyrophosphatase/ Phosphodiesterase 1 | XM_021087933 | human orthologue identified as the plasma 3′-exonuclease responsible for PS degradation [20] | (f) CAGATCATGGCATGGAACAAGGCA (r) TGGTTTGGTTCCTGGCAAGAAAG | 135 | 101 | 103 | 97 |
| PDE1B | Phosphodiesterase 1B | XM_003126207 | bovine orthologue was reported to show a similar 3′-exonuclease activity on PS oligonucleotide to that of the human plasma [57] | (f) GACTCGGCACAACCTCATCA (r) CAGTGGACCGTCTGGGTAAC | 147 | 94 | 100 | 93 |
| TREX1 | Three prime repair exonuclease 1 | XM_021070628 | major and most abundant 3′-exonuclease in mammalian cells [58,59] | (f) CCTGCCTGCTGTTCGGCTC (r) GGCTCTCCAGGGCACATCTAT | 175 | 94 | 106 | 98 |
| DNASE1 | Deoxyribonuclease 1 | NM_213991 | well-characterized role in DNA degradation [60,61] | (f) GGGATCTGGAGGACATCATGCT (r) CGACCACGATCCTGTCATAGGC | 177 | - | 97 | 88 |
| DNASE2 | Deoxyribonuclease 2 | NM_214196 | well-characterized role in DNA degradation [60,61] | (f) GGAGGAGGTAGTCAAGGGCCA (r) GCCAGAGTACAGGTCGTCTCC | 133 | - | 90 | 97 |
| RNASEH1 | Ribonuclease H1 | NM_001243681 | demonstrated its role for ASO pharmacologic activity [52] | (f) GCCAGGCCATCCTTTAAATGTAGG (r) CCCAGCTAGTGATGCCATTGATGG | 170 | - | 98 | 96 |
| RNASEH2A | Ribonuclease H2 subunit A | NM_001244444 | isoform of ribonuclease H in mammalian cells, capable of degrading target RNA in cell lysates [8,53] | (f) TTTGTGGGCTGGGCATTGGA (r) ACAAACACCTGGGCCACTTTC | 158 | - | 101 | 89 |
2.8. Statistical Analysis
3. Results
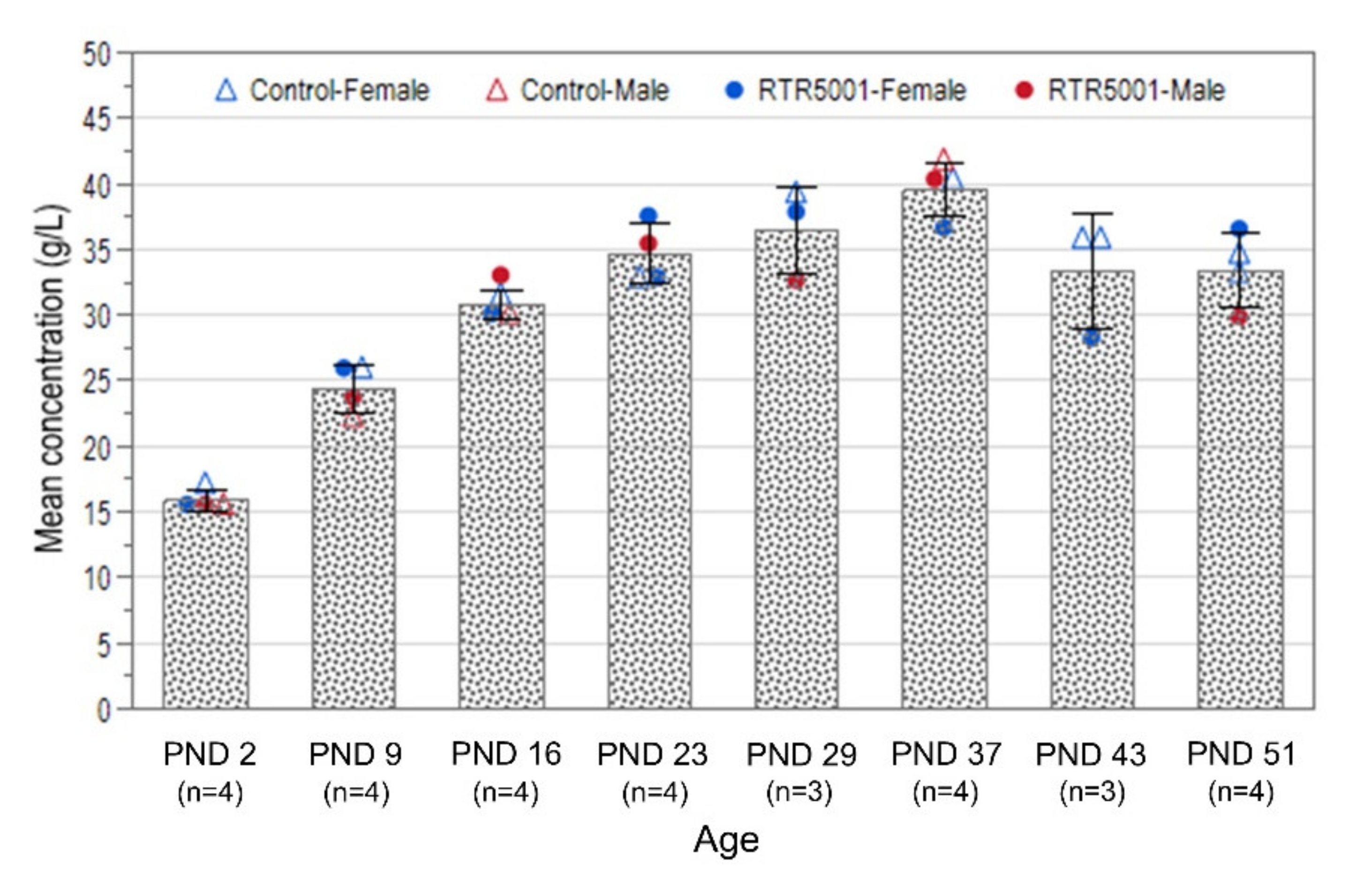
3.1. Exposure Assessment and Tissue Biodistribution
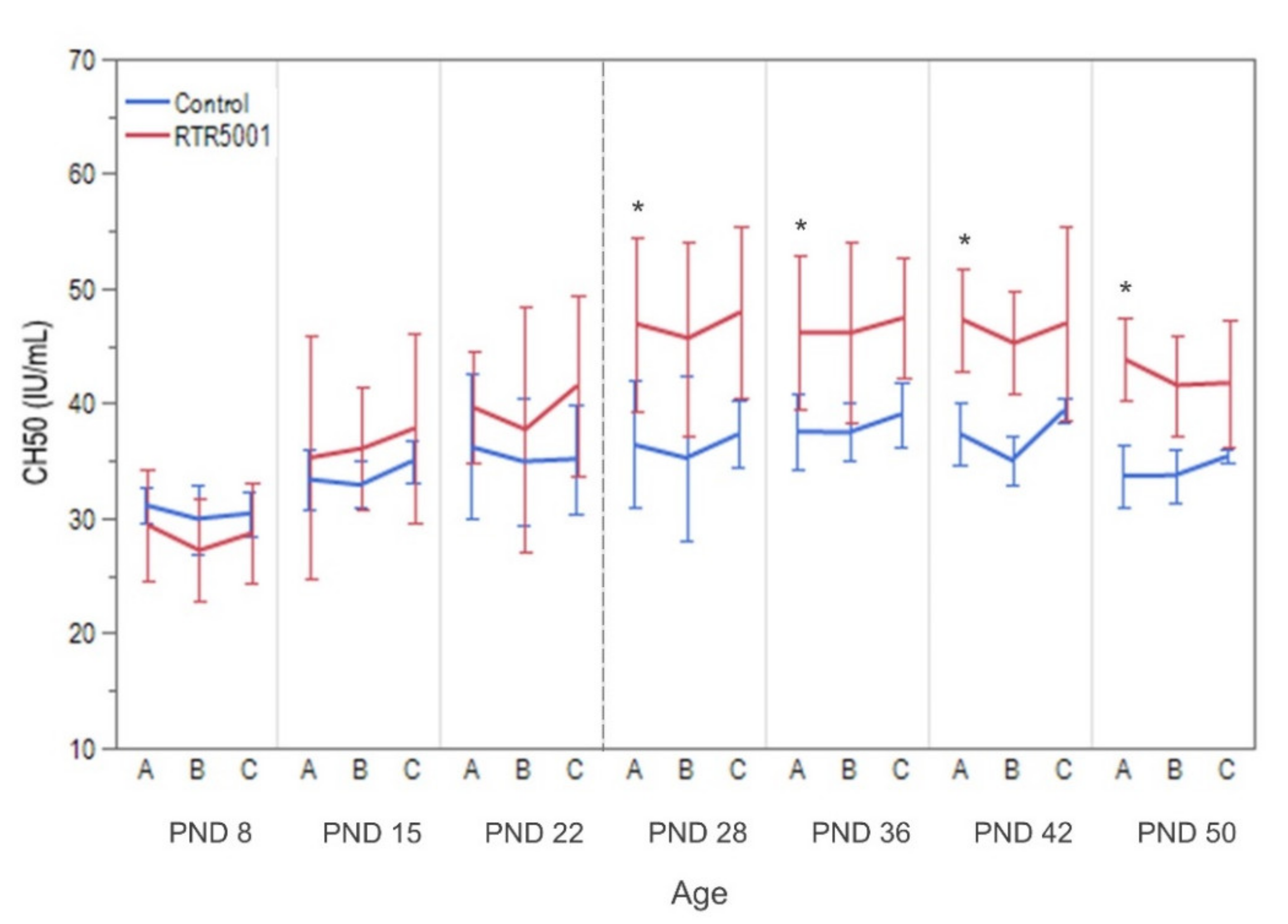
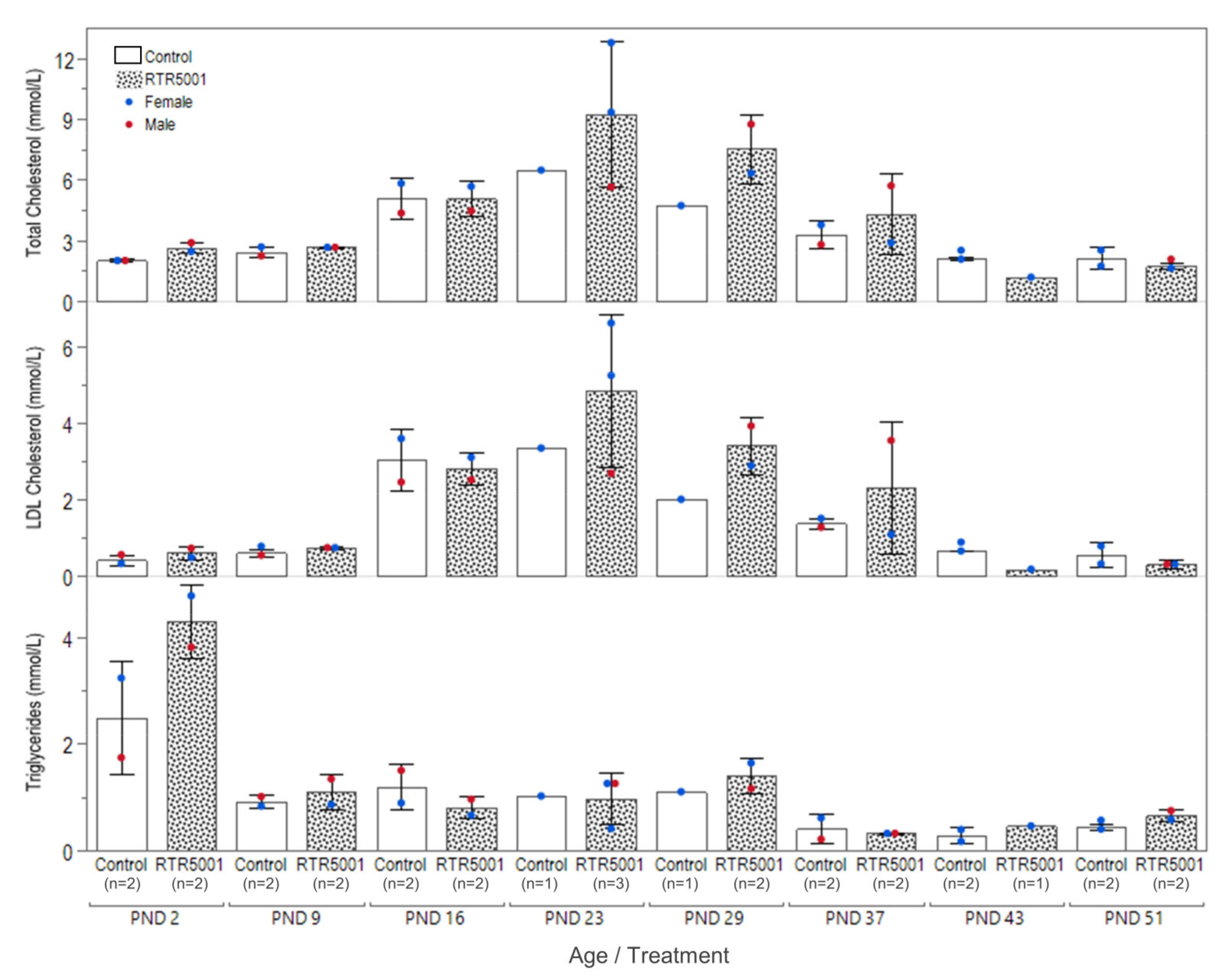
3.2. In-Life Observation and Clinical Pathology
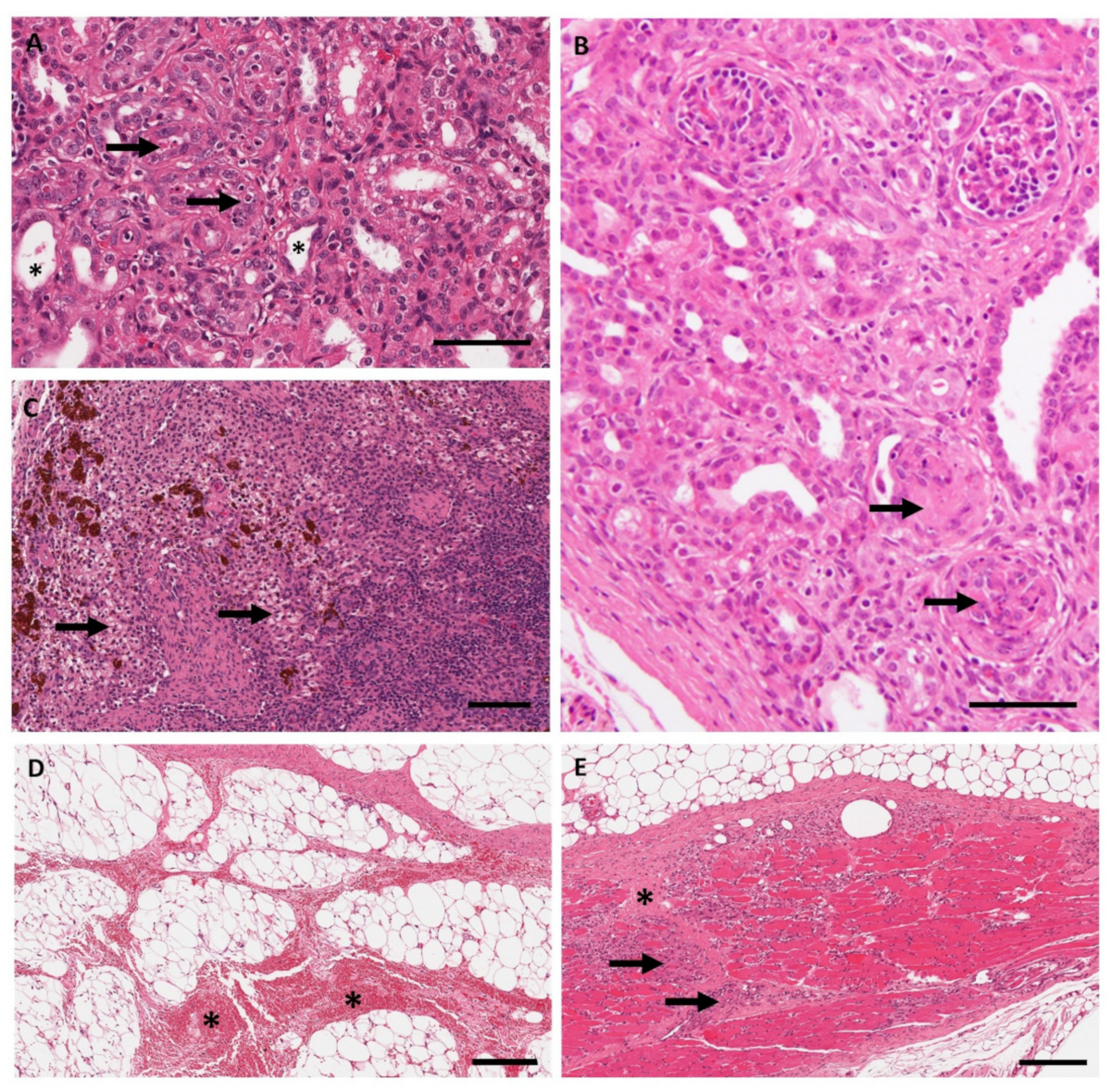
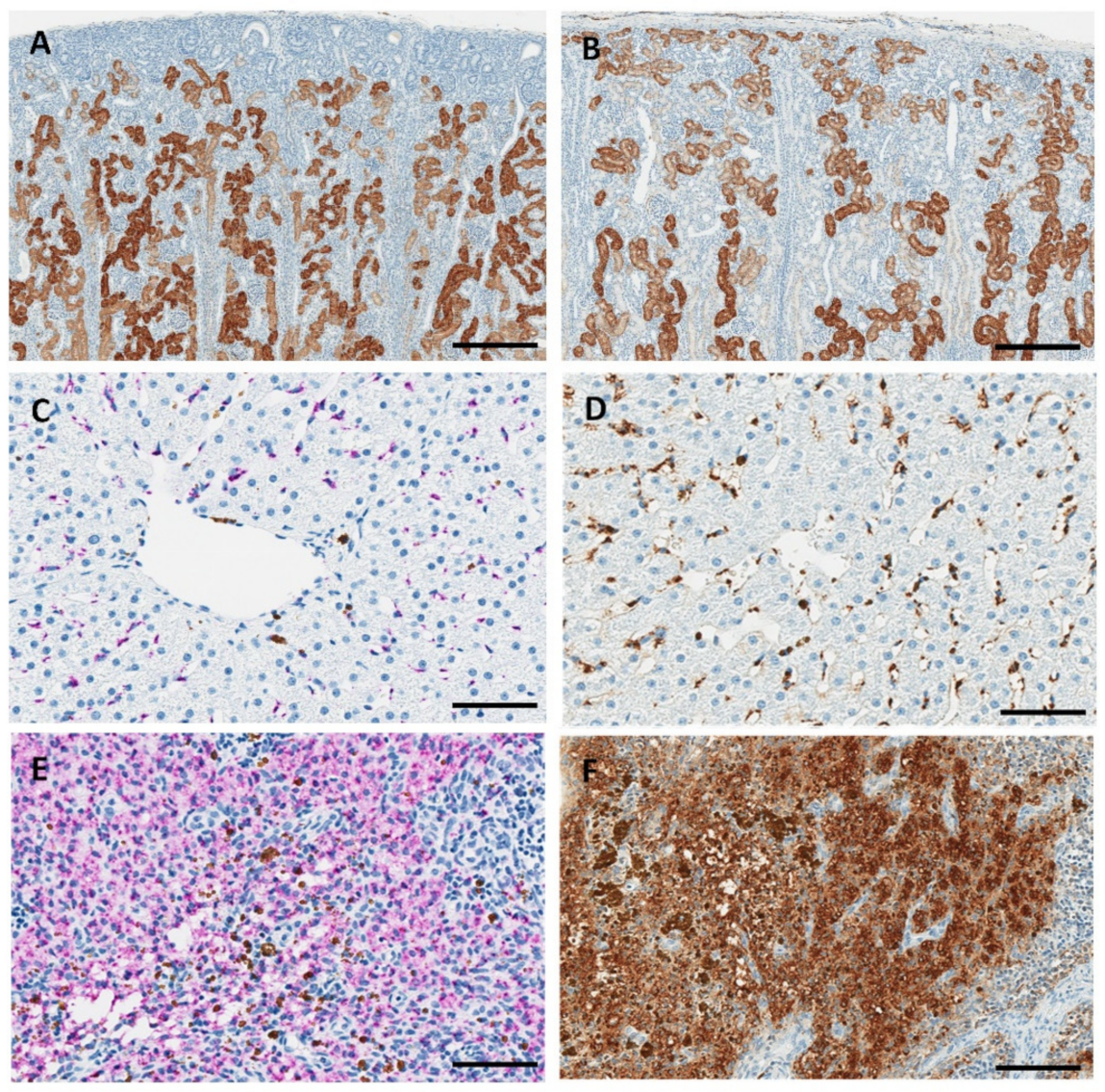
3.3. Anatomic Pathology, Immunohistochemistry, and In Situ Hybridization
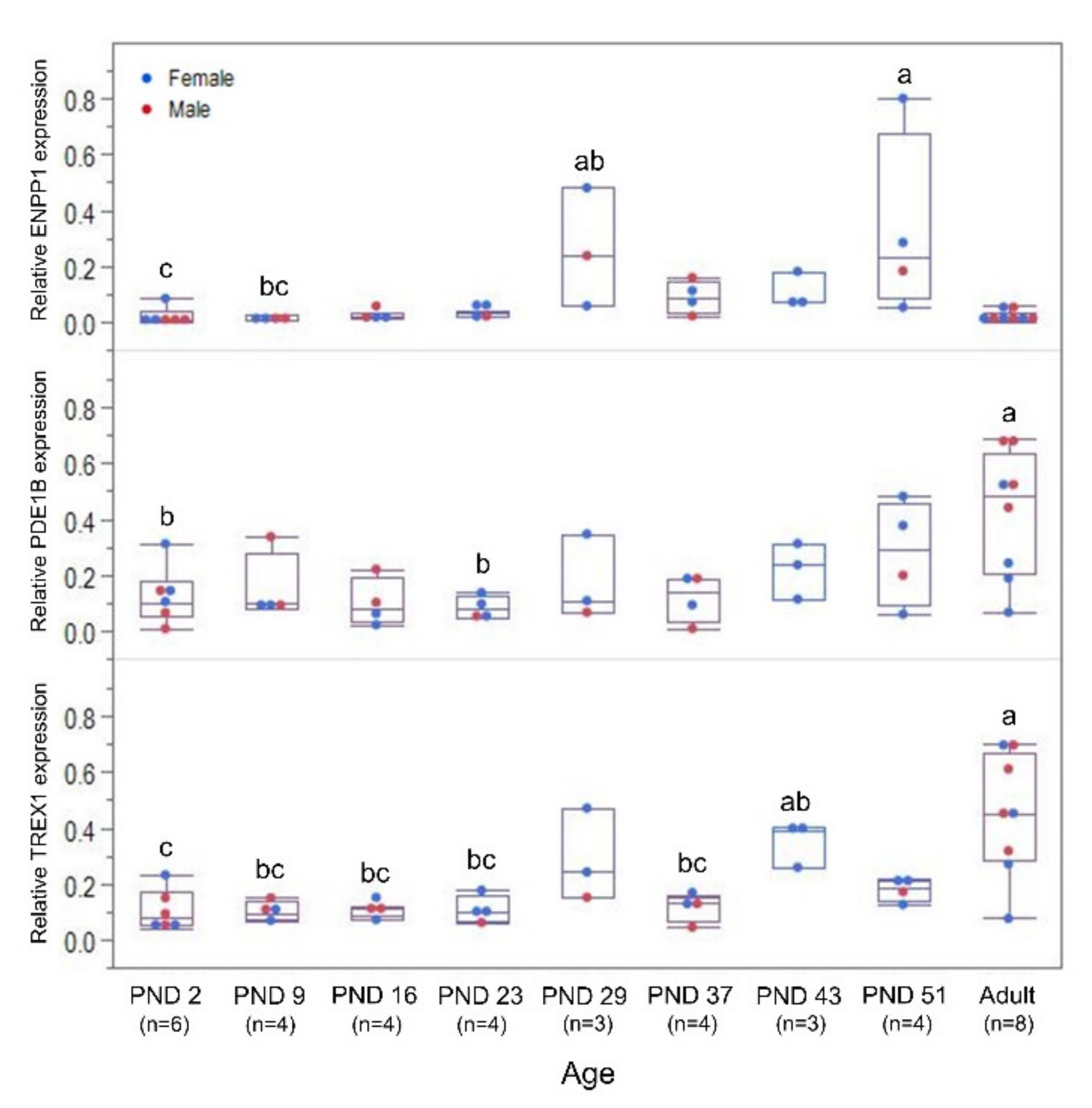
3.4. Ontogeny of Nuclease Gene Expression and Activity in the Blood
3.5. Ontogeny of Nuclease Gene Expression and Activity in the Kidney and Liver
3.6. Ontogeny of Ribonuclease H expression in the Kidney and Liver
4. Discussion
Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Mustonen, E.K.; Palomäki, T.; Pasanen, M. Oligonucleotide-based pharmaceuticals: Non-clinical and clinical safety signals and non-clinical testing strategies. Regul. Toxicol. Pharmacol. 2017, 90, 328–341. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent advances in oligonucleotide therapeutics in oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Rogge, M. Targeting RNA: A Transformative Therapeutic Strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Lima, W.; Wu, H.; Crooke, S.T. The RNase H mechanism. In Antisense Drug Technology: Principles, Strategies, and Applications, 2nd ed.; Crooke, S.T., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 47–74. [Google Scholar]
- Henry, S.P.; Johnson, M.; Zanardi, T.A.; Fey, R.; Auyeung, D.; Lappin, P.B.; Levin, A.A. Renal uptake and tolerability of a 2’-O-methoxyethyl modified antisense oligonucleotide (ISIS 113715) in monkey. Toxicology 2012, 301, 13–20. [Google Scholar] [CrossRef]
- Brown, D.A.; Kang, S.H.; Gryaznov, S.M.; DeDionisio, L.; Heidenreich, O.; Sullivan, S.; Xu, X.; Nerenberg, M.I. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 1994, 269, 26801–26805. [Google Scholar] [CrossRef]
- Geary, R.S.; Watanabe, T.A.; Truong, L.; Freier, S.; Lesnik, E.A.; Sioufi, N.B.; Sasmor, H.; Manoharan, M.; Levin, A.A. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 2001, 296, 890–897. [Google Scholar]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Frazier, K.S. Antisense Oligonucleotide Therapies:The Promise and the Challenges from a Toxicologic Pathologist’s Perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Z.; Geary, R.S.; Flaim, J.D.; Riley, G.C.; Tribble, D.L.; VanVliet, A.A.; Wedel, M.K. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2′-O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibe. Clin. Pharmacokinet. 2009, 48, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, F.; Yerino, P.; McCoy, C.; Parkinson, A.; Buckley, D.B.; Ogilvie, B.W. An assessment of the in vitro inhibition of cytochrome P450 enzymes, UDP-glucuronosyltransferases, and transporters by phosphodiester- or phosphorothioate-linked oligonucleotides. Drug Metab. Dispos. 2018, 46, 1066–1074. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.Z.; Kim, T.W.; Hong, A.; Watanabe, T.A.; Gaus, H.J.; Geary, R.S. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab. Dispos. 2007, 35, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffey, R.H.; Greig, M.J.; Gaus, H.J.; Liu, K.; Monteith, D.; Winniman, M.; Cummins, L.L. Characterization of oligonucleotide metabolism in vivo via liquid chromatography/electrospray tandem mass spectrometry with a quadrupole ion trap mass spectrometer. J. Mass Spectrom. 1997, 32, 305–313. [Google Scholar] [CrossRef]
- Yang, W. Nucleases: Diversity of structure, function and mechanism. Q. Rev. Biophys. 2011, 44, 1–93. [Google Scholar] [CrossRef]
- Eder, P.S.; Devine, R.J.; Dagle, J.M.; Walder, J.A. Substrate Specificity and Kinetics of Degradation of Antisense Oligonucleotides by a 3′ Exonuclease in Plasma. Antisense Res. Dev. 1991, 1, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, M.; Cieślak, M.; Stec, W.J.; Goding, J.W.; Koziołkiewicz, M. Nucleotide Pyrophosphatase/Phosphodiesterase 1 Is Responsible for Degradation of Antisense Phosphorothioate Oligonucleotides. Oligonucleotides 2007, 17, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geary, R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin. Drug Metab. Toxicol. 2009, 5, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.K. The Medicinal Chemistry of Antisense Oligonucleotides. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 39–69. [Google Scholar]
- Krishnan, A.V.; Mishra, D. Antisense Oligonucleotides: A Unique Treatment Approach. Indian Pediatr. 2020, 57, 165–171. [Google Scholar] [CrossRef]
- Laxton, C.; Brady, K.; Moschos, S.; Turnpenny, P.; Rawal, J.; Pryde, D.C.; Sidders, B.; Corbau, R.; Pickford, C.; Murray, E.J. Selection, optimization, and pharmacokinetic properties of a novel, potent antiviral locked nucleic acid-based antisense oligomer targeting hepatitis C virus internal ribosome entry site. Antimicrob. Agents Chemother. 2011, 55, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Kuespert, S.; Heydn, R.; Peters, S.; Wirkert, E.; Meyer, A.L.; Siebörger, M.; Johannesen, S.; Aigner, L.; Bogdahn, U.; Bruun, T.H. Antisense oligonucleotide in LNA-gapmer design targeting TGFBR2—A key single gene target for safe and effective inhibition of TGFβ signaling. Int. J. Mol. Sci. 2020, 21, 1952. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Fisker, N.; Asselin, M.C.; Lindholm, M.; Rosenbohm, C.; Ørum, H.; Elmén, J.; Seidah, N.G.; Straarup, E.M. A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression In Vitro and In Vivo. PLoS ONE 2010, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Abewe, H.; Deshmukh, S.; Mukim, A.; Beliakova-Bethell, N. Use of GapmeRs for gene expression knockdowns in human primary resting CD4+ T cells. J. Immunol. Methods 2020, 476, e112674. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- European Medicine Agency; Committee for Medicinal Products for Human Use; International Council for Harmonisation of Technical Requirements for Pharmaceuticas for Human Use. ICH Guideline M3 (R2) on Non-Clinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals, Step 5, EMA/CHMP/ICH/286/95; European Medicines Agency: London, UK, 11 February 2013; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-marketing-authorisation_en.pdf (accessed on 11 June 2021).
- Braendli-Baiocco, A.; Festag, M.; Erichsen, K.D.; Persson, R.; Mihatsch, M.J.; Fisker, N.; Funk, J.; Mohr, S.; Constien, R.; Ploix, C.; et al. The minipig is a suitable non-rodent model in the safety assessment of single stranded oligonucleotides. Toxicol. Sci. 2017, 157, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Heckel, T.; Schmucki, R.; Berrera, M.; Ringshandl, S.; Badi, L.; Steiner, G.; Ravon, M.; Küng, E.; Kuhn, B.; Kratochwil, N.A.; et al. Functional analysis and transcriptional output of the Göttingen minipig genome. BMC Genomics 2015, 16, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Vamathevan, J.J.; Hall, M.D.; Hasan, S.; Woollard, P.M.; Xu, M.; Yang, Y.; Li, X.; Wang, X.; Kenny, S.; Brown, J.R.; et al. Minipig and beagle animal model genomes aid species selection in pharmaceutical discovery and development. Toxicol. Appl. Pharmacol. 2013, 270, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Finkel, R.; Mercuri, E.; Muntoni, F. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc. Heal. 2018, 2, 600–609. [Google Scholar] [CrossRef]
- Abreu, N.J.; Waldrop, M.A. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr. Pulmonol. 2021, 56, 710–720. [Google Scholar] [CrossRef]
- Aoki, Y.; Wood, M.J.A. Emerging Oligonucleotide Therapeutics for Rare Neuromuscular Diseases. J. Neuromuscul. Dis. 2021, 1–16. [Google Scholar] [CrossRef]
- Osredkar, D.; Jílková, M.; Butenko, T.; Loboda, T.; Golli, T.; Fuchsová, P.; Rohlenová, M.; Haberlova, J. Children and young adults with spinal muscular atrophy treated with nusinersen. Eur. J. Paediatr. Neurol. 2021, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P. Pharmacotherapy of duchenne muscular dystrophy. Handb. Exp. Pharmacol. 2020, 261, 25–37. [Google Scholar] [CrossRef]
- Martinovich, K.M.; Shaw, N.C.; Kicic, A.; Schultz, A.; Fletcher, S.; Wilton, S.D.; Stick, S.M. The potential of antisense oligonucleotide therapies for inherited childhood lung diseases. Mol. Cell. Pediatr. 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafi, M.R.; Amanat, M.; Garshasbi, M.; Kameli, R.; Nilipour, Y.; Heidari, M.; Rezaei, Z.; Tavasoli, A.R. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies. Expert Rev. Neurother. 2020, 20, 65–84. [Google Scholar] [CrossRef]
- Oren, Y.S.; Irony-Tur Sinai, M.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C.; et al. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- European Medicine Agency; Committee for Medicinal Products for Human Use; International Council for Harmonisation of Technical Requirements for Pharmaceuticas for Human Use. ICH Guideline S11 on Noncliniclinical Safety Testing in Support of Development of Pediatric Pharmaceuticals, Step 5, EMA/CHMP/ICH/616110/2018; European Medicines Agency: Amsterdam, The Netherlands, 31 March 2020; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-s11-nonclinical-safety-testing-support-development-paediatric-pharmaceuticals-step-5_en.pdf (accessed on 11 June 2021).
- Barrow, P.C.; Barbellion, S.; Stadler, J. Preclinical evaluation of juvenile toxicity. Methods Mol. Biol. 2011, 691, 17–35. [Google Scholar] [CrossRef]
- Ayuso, M.; Buyssens, L.; Stroe, M.; Valenzuela, A.; Allegaert, K.; Smits, A.; Annaert, P.; Mulder, A.; Carpentier, S.; Van Ginneken, C.; et al. The neonatal and juvenile pig in pediatric drug discovery and development. Pharmaceutics 2021, 13, 44. [Google Scholar] [CrossRef]
- Bode, G.; Clausing, P.; Gervais, F.; Loegsted, J.; Luft, J.; Nogues, V.; Sims, J. The utility of the minipig as an animal model in regulatory toxicology. J. Pharmacol. Toxicol. Methods 2010, 62, 196–220. [Google Scholar] [CrossRef]
- Forster, R.; Bode, G.; Ellegaard, L.; van der Laan, J.W. The RETHINK project on minipigs in the toxicity testing of new medicines and chemicals: Conclusions and recommendations. J. Pharmacol. Toxicol. Methods 2010, 62, 236–242. [Google Scholar] [CrossRef]
- Lindholm, M.W.; Elmén, J.; Fisker, N.; Hansen, H.F.; Persson, R.; Møller, M.R.; Rosenbohm, C.; Ørum, H.; Straarup, E.M.; Koch, T. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol. Ther. 2012, 20, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.L.; Xiao, L.L.; Tang, Z.H.; Jiang, Z.S.; Liu, M.H. Role of PCSK9 in lipid metabolism and atherosclerosis. Biomed. Pharmacother. 2018, 104, 36–44. [Google Scholar] [CrossRef]
- Van Peer, E.; Downes, N.; Casteleyn, C.; Van Ginneken, C.; Weeren, A.; Van Cruchten, S. Organ data from the developing Göttingen minipig: First steps towards a juvenile PBPK model. J. Pharmacokinet. Pharmacodyn. 2016, 43, 179–190. [Google Scholar] [CrossRef]
- Geary, R.S.; Yu, R.Z.; Watanabe, T.; Henry, S.P.; Hardee, G.E.; Chappell, A.; Matson, J.; Sasmor, H.; Cummins, L.; Levin, A.A. Pharmacokinetics of a tumor necrosis factor-α phosphorothioate 2′-O-(2-methoxyethyl) modified antisense oligonucleotide: Comparison across species. Drug Metab. Dispos. 2003, 31, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, H.; Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: Effect on DNA-dependent RNA polymerase. Science 1969, 166, 393–395. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten Asbroek, A.L.M.A.; Van Groenigen, M.; Nooij, M.; Baas, F. The involvement of human ribonucleases H1 and H2 in the variation of response of cells to antisense phosphorothioate oligonucleotides. Eur. J. Biochem. 2002, 269, 583–592. [Google Scholar] [CrossRef]
- Nygard, A.B.; Jørgensen, C.B.; Cirera, S.; Fredholm, M. Selection of reference genes for gene expression studies in pig tissues using SYBR green qPCR. BMC Mol. Biol. 2007, 8, 67. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Ho, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides. Proc. Natl. Acad. Sci. USA. 2000, 97, 5633–5638. [Google Scholar] [CrossRef] [Green Version]
- Gilar, M.; Belenky, A.; Smisek, D.L.; Bourque, A.; Cohen, A.S. Kinetics of phosphorothioate oligonucleotide metabolism in biological fluids. Nucleic Acids Res. 1997, 25, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Mazur, D.J.; Perrino, F.W. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′→5′ exonucleases. J. Biol. Chem. 1999, 274, 19655–19660. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, D.; Spitzer, D.; Kothari, P.H.; Shaikh, A.; Liszewski, M.K.; Richards, A.; Atkinson, J.P. New roles for the major human 3′–5′ exonuclease TREX1 in human disease. Cell Cycle 2010, 7, 1718–1725. [Google Scholar] [CrossRef] [Green Version]
- Kishi, K.; Yasuda, T.; Ikehara, Y.; Sawazaki, K.; Sato, W.; Ida, R. Human serum deoxyribonuclease I (DNase I) polymorphism: Pattern similarities among isozymes from serum, urine, kidney, liver, and pancreas. Am. J. Hum. Genet. 1990, 47, 121–126. [Google Scholar]
- Evans, C.J.; Aguilera, R.J. DNase II: Genes, enzymes and function. Gene 2003, 322, 1–15. [Google Scholar] [CrossRef]
- Grossi, A.B.; Zeltner, A.; Christoffersen, C.; Søndergaard, A.C. Reference data of clinical chemistry and hematology in juvenile Göttingen Minipigs. Toxicol. Lett. 2016, 258, s234. [Google Scholar] [CrossRef]
- Seely, J.C. A brief review of kidney development, maturation, developmental abnormalities, and drug toxicity: Juvenile animal relevancy. J. Toxicol. Pathol. 2017, 30, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szudzik, M.; Starzyński, R.R.; Jończy, A.; Mazgaj, R.; Lenartowicz, M.; Lipiński, P. Erratum: Iron supplementation in suckling piglets: An ostensibly easy therapy of neonatal iron deficiency anemia. Pharmaceuticals 2019, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, K. Paediatric pharmacokinetics and drug doses. Aust. Prescr. 2016, 39, 208–210. [Google Scholar] [CrossRef] [Green Version]
- Van Donge, T.; Evers, K.; Koch, G.; van den Anker, J.; Pfister, M. Clinical pharmacology and pharmacometrics to better understand physiological changes during pregnancy and neonatal life. Handb. Exp. Pharmacol. 2020, 261, 325–337. [Google Scholar] [CrossRef]
- Bueters, R.; Bael, A.; Gasthuys, E.; Chen, C.; Schreuder, M.F.; Frazier, K.S. Ontogeny and Cross-species Comparison of Pathways Involved in Drug Absorption, Distribution, Metabolism, and Excretion in Neonates (Review): Kidney. Drug Metab. Dispos. 2020, 48, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V.A. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Basiri, B.; Hassan, C.; Punt, C.; van der Hage, E.; den Besten, C.; Bartlett, M.G. Metabolite Profiling of the Antisense Oligonucleotide Eluforsen Using Liquid Chromatography-Mass Spectrometry. Mol. Ther. Nucleic Acid 2019, 17, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Yu, R.Z.; Gaus, H.; Grundy, J.S.; Geary, R.S. In Vitro Metabolic Stabilities and Metabolism Antisense Oligonucleotides in Preincubated Rat or Human Whole Liver Homogenates. Oligonucleotides 2010, 20, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Crooke, R.M.; Graham, M.J.; Martin, M.J.; Lemonidis, K.M.; Wyrzykiewiecz, T.A.D.; Cummins, L.L. Metabolism of Antisense Oligonucleotides in Rat Liver Homogenates. J. Pharmacol. Exp. Ther. 2000, 292, 140–149. [Google Scholar]
- Sands, H.; Gorey-Feret, L.J.; Cocuzza, A.J.; Hobbs, F.W.; Chidester, D.; Trainor, G.L. Biodistribution and metabolism of internally 3H-labeled oligonucleotides. I. Comparison of a phosphodiester and a phosphorothioate. Mol. Pharmacol. 1994, 45, 932–943. [Google Scholar]
- Walder, R.Y.; Walder, J.A. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1988, 85, 5011–5015. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Hu, X.; Chang, S.; Chang, Y.; Bian, W.; Hu, R.; Wang, J.; Zhu, Q.; Qiu, J. Advances of Antisense Oligonucleotide Technology in the Treatment of Hereditary Neurodegenerative Diseases. Evidence-based Complement. Altern. Med. 2021, 2021, 6678422. [Google Scholar] [CrossRef]
- Chen, S.; Le, B.T.; Chakravarthy, M.; Kosbar, T.R.; Veedu, R.N. Systematic evaluation of 2′-Fluoro modified chimeric antisense oligonucleotide-mediated exon skipping in vitro. Sci. Rep. 2019, 9, 6078. [Google Scholar] [CrossRef] [PubMed]
- Dirin, M.; Winkler, J. Tissue Distribution, Metabolism, and Clearance. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 137–151. [Google Scholar]
- Levin, A.A. A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta 1999, 1489, 69–84. [Google Scholar] [CrossRef]
- Gaus, H.J.; Owens, S.R.; Winniman, M.; Cooper, S.; Cummins, L.L. On-Line HPLC Electrospray Mass Spectrometry of Phosphorothioate Oligonucleotide Metabolites. Anal. Chem. 1997, 69, 313–319. [Google Scholar] [CrossRef]
- Shaw, J.; Kent, K.; Bird, J.; Fishback, J.; Froehler, B. Modified deoxyoligonucleotides stable to exonuclease degradation in serum. Nucleic Acids Res. 1991, 19, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Østergaard, M.E.; De Hoyos, C.L.; Wan, W.B.; Shen, W.; Low, A.; Berdeja, A.; Vasquez, G.; Murray, S.; Migawa, M.T.; Liang, X.H.; et al. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020, 48, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Belli, S.I.; van Driel, I.R.; Goding, J.W. Identification and characterization of a soluble form of the plasma cell membrane glycoprotein PC-1 (5′-nucleotide phosphodiesterase). Eur. J. Biochem. 1993, 217, 421–428. [Google Scholar] [CrossRef]
- Loughney, K.; Ferguson, K. Identification and Quantification of PDE Isoenzymes and Subtypes by Molecular Biological Methods. In Phosphodiesterase Inhibitors; Schudt, C., Dent, G., Rabe, K., Eds.; Academic Press Ltd: San Diego, CA, USA, 1996; pp. 1–14. [Google Scholar]
- Wang, D. Discrepancy between mRNA and protein abundance: Insight from information retrieval process in computers. Comput. Biol. Chem. 2008, 32, 462–468. [Google Scholar] [CrossRef] [Green Version]
- Bruckmueller, H.; Martin, P.; Kähler, M.; Haenisch, S.; Ostrowski, M.; Drozdzik, M.; Siegmund, W.; Cascorbi, I.; Oswald, S. Clinically Relevant Multidrug Transporters Are Regulated by microRNAs along the Human Intestine. Mol. Pharm. 2017, 14, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Zapalska-Sozoniuk, M.; Chrobak, L.; Kowalczyk, K.; Kankofer, M. Is it useful to use several “omics” for obtaining valuable results? Mol. Biol. Rep. 2019, 46, 3597–3606. [Google Scholar] [CrossRef] [Green Version]
- Kamaliddin, C.; Guillochon, E.; Salnot, V.; Rombaut, D.; Huguet, S.; Guillonneau, F.; Houzé, S.; Cot, M.; Deloron, P.; Argy, N.; et al. Comprehensive Analysis of Transcript and Protein Relative Abundance during Blood Stages of Plasmodium falciparum Infection. J. Proteome Res. 2021, 20, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.Z.; Geary, R.S.; Levin, A.A. Pharmacokinetics and Pharmacodynamics of Antisense Oligonucleotides. In Pharmacokinetics and Pharmacodynamics of Biotech Drugs: Principles and Case Studies in Drug Development; Meibohm, B., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 93–115. [Google Scholar]
- Lundin, K.E.; Hansen, B.R.; Persson, R.; Bramsen, J.B.; Koch, T.; Wengel, J.; Smith, C.I.E. Biological Activity and Biotechnological Aspects of Locked Nucleic Acids. Adv. Genet. 2013, 82, 47–107. [Google Scholar] [CrossRef]
- Post, N.; Yu, R.; Greenlee, S.; Gaus, H.; Hurh, E.; Matson, J.; Wang, Y. Metabolism and disposition of volanesorsen, a 29-O-(2 methoxyethyl) antisense oligonucleotide, across species. Drug Metab. Dispos. 2019, 47, 1164–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillman, L.G.; Geary, R.S.; Hardee, G.E. Oral delivery of antisense oligonucleotides in man. J. Pharm. Sci. 2008, 97, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Geary, R.S. Clinical pharmacological properties of mipomersen ( Kynamro ), a second generation antisense inhibitor of apolipoprotein B. Br. J. Clin. Pharmacol. 2013, 76, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.Z.; Grundy, J.S.; Geary, R.S. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2013, 9, 169–182. [Google Scholar] [CrossRef]
- Schmidt, K.; Prakash, T.P.; Donner, A.J.; Kinberger, G.A.; Gaus, H.J.; Low, A.; Østergaard, M.E.; Bell, M.; Swayze, E.E.; Seth, P.P. Characterizing the effect of GalNAc and phosphorothioate backbone on binding of antisense oligonucleotides to the asialoglycoprotein receptor. Nucleic Acids Res. 2017, 45, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.M.; Donner, A.J.; Blank, E.E.; Egger, A.W.; Kellar, B.M.; Østergaard, M.E.; Seth, P.P.; Harris, E.N. Stabilin-1 and Stabilin-2 are specific receptors for the cellular internalization of phosphorothioate-modified antisense oligonucleotides (ASOs) in the liver. Nucleic Acids Res. 2016, 44, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Hvam, M.L.; Cai, Y.; Dagnæs-Hansen, F.; Nielsen, J.S.; Wengel, J.; Kjems, J.; Howard, K.A. Fatty Acid-Modified Gapmer Antisense Oligonucleotide and Serum Albumin Constructs for Pharmacokinetic Modulation. Mol. Ther. 2017, 25, 1710–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migawa, M.T.; Shen, W.; Wan, W.B.; Vasquez, G.; Oestergaard, M.E.; Low, A.; De Hoyos, C.L.; Gupta, R.; Murray, S.; Tanowitz, M.; et al. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res. 2019, 47, 5465–5479. [Google Scholar] [CrossRef]
- Srinivasan, S.K.; Tewary, H.K.; Iversen, P.L. Characterization of Binding Sites, Extent of Binding, and Drug Interactions of Oligonucleotides with Albumin. Antisense Res. Dev. 1995, 5, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.A.; Geary, R.S.; Levin, A.A. Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides 2006, 16, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Wartlick, H.; Spänkuch-Schmitt, B.; Strebhardt, K.; Kreuter, J.; Langer, K. Tumour cell delivery of antisense oligonuclceotides by human serum albumin nanoparticles. J. Control. Release 2004, 96, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Gaus, H.J.; Gupta, R.; Chappell, A.E.; Østergaard, M.E.; Swayze, E.E.; Seth, P.P. Characterization of the interactions of chemically-modified therapeutic nucleic acids with plasma proteins using a fluorescence polarization assay. Nucleic Acids Res. 2019, 47, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.; Schneider, A.; Hall, J. Pharmacokinetics and Pharmacodynamics of Antisense Oligonucleotides. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 107–136. [Google Scholar]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Rum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010, 327, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Weidolf, L.; Björkbom, A.; Dahlén, A.; Elebring, M.; Gennemark, P.; Hölttä, M.; Janzén, D.; Li, X.Q.; Andersson, S. Distribution and biotransformation of therapeutic antisense oligonucleotides and conjugates. Drug Discov. Today 2021. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic Administration of PRO051 in Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef]
- Levin, A.A.; Yu, R.Z.; Geary, R.S. Basic principles of the pharmacokinetics of antisense oligonucleotide drugs. In Antisense Drug Technology: Principles, Strategies, and Applications, 2nd ed.; Crooke, S.T., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 183–216. [Google Scholar]
- Straarup, E.M.; Fisker, N.; Hedtjärn, M.; Lindholm, M.W.; Rosenbohm, C.; Aarup, V.; Hansen, H.F.; Ørum, H.; Hansen, J.B.R.; Koch, T. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010, 38, 7100–7111. [Google Scholar] [CrossRef] [Green Version]
- Hagedorn, P.H.; Persson, R.; Funder, E.D.; Albæk, N.; Diemer, S.L.; Hansen, D.J.; Møller, M.R.; Papargyri, N.; Christiansen, H.; Hansen, B.R.; et al. Locked nucleic acid: Modality, diversity, and drug discovery. Drug Discov. Today 2018, 23, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Dai, G.; Marcucci, G.; Liu, Z.; Hoyt, D.; Blum, W.; Chan, K.K. A specific picomolar hybridization-based ELISA assay for the determination of phosphorothioate oligonucleotides in plasma and cellular matrices. Pharm. Res. 2006, 23, 1251–1264. [Google Scholar] [CrossRef]
- Deverre, J.R.; Boutet, V.; Boquet, D.; Ezan, E.; Grassi, J.; Grognet, J.M. A competitive enzyme hybridization assay for plasma determination of phosphodiester and phosphorothioate antisense oligonucleotides. Nucleic Acids Res. 1997, 25, 3584–3589. [Google Scholar] [CrossRef] [Green Version]
- Andersson, S.; Antonsson, M.; Elebring, M.; Jansson-Löfmark, R.; Weidolf, L. Drug metabolism and pharmacokinetic strategies for oligonucleotide- and mRNA-based drug development. Drug Discov. Today 2018, 23, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.S. Species Differences in Renal Development and Associated Developmental Nephrotoxicity. Birth Defects Res. 2017, 109, 1243–1256. [Google Scholar] [CrossRef]
- Hildebrandt-Eriksen, E.S.; Aarup, V.; Persson, R.; Hansen, H.F.; Munk, M.E.; Ørum, H. A locked nucleic acid oligonucleotide targeting microRNA 122 is well-tolerated in cynomolgus monkeys. Nucleic Acid Ther. 2012, 22, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Koch, T.; Ørum, H. Locked nucleic acid. In Antisense Drug Technology: Principles, Strategies, and Applications, 2nd ed.; Crooke, S.T., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 519–564. [Google Scholar]
- Graham, M.J.; Crooke, S.T.; Monteith, D.K.; Cooper, S.R.; Lemonidis, K.M.; Stecker, K.I.M.K.; Martin, M.J.; Crooke, R.M. In Vivo Distribution and Metabolism of a Phosphorothioate Oligonucleotide within Rat Liver after Intravenous Administration. J. Pharmacol. Exp. 1998, 286, 447–458. [Google Scholar]
- Altmann, K.H.; Dean, N.M.; Fabbro, D.; Freier, S.M.; Geiger, T.; Hanera, R.; Hiisken, D.A.; Martina, P.; Monia, P.B.; Miiller, M.; et al. Second generation of antisense oligonucleotides: From nuclease resistance to biological efficacy in animals. Chim. Int. J. Chem. 1996, 50, 168–176. [Google Scholar]
- Kim, J.; El Zahar, N.M.; Bartlett, M.G. In vitro metabolism of 2′-ribose unmodified and modified phosphorothioate oligonucleotide therapeutics using liquid chromatography mass spectrometry. Biomed. Chromatogr. 2020, 34, e4839. [Google Scholar] [CrossRef]
- Romero-Palomo, F.; Festag, M.; Lenz, B.; Schadt, S.; Brink, A.; Kipar, A.; Steinhuber, B.; Husser, C.; Koller, E.; Sewing, S.; et al. Safety, Tissue Distribution, and Metabolism of LNA-Containing Antisense Oligonucleotides in Rats. Toxicol. Pathol. 2021, 49, 1174–1192. [Google Scholar] [CrossRef]
- Kilanowska, A.; Studzińska, S. In vivo and in vitro studies of antisense oligonucleotides—a review. RSC Adv. 2020, 10, 34501–34516. [Google Scholar] [CrossRef]
- White, P.J.; Anastasopoulos, F.; Pouton, C.W.; Boyd, B.J. Overcoming biological barriers to in vivo efficacy of antisense oligonucleotides. Expert Rev. Mol. Med. 2009, 11, 1–19. [Google Scholar] [CrossRef]
- Yu, R.Z.; Lemonidis, K.M.; Graham, M.J.; Matson, J.E.; Crooke, R.M.; Tribble, D.L.; Wedel, M.K.; Levin, A.A.; Geary, R.S. Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem. Pharmacol. 2009, 77, 910–919. [Google Scholar] [CrossRef]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416-1–1004161-39. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Yu, R.Z.; Siwkowski, A.; Levin, A.A. Pharmacokinetic/Pharmacodynamic Properties of Phosphorothioate 2′-O-(2-Methoxyethyl)-Modified Antisense Oligonucleotides in Animals and Man. In Antisense Drug Technology: Principles, Strategies, and Applications, 2nd ed.; Crooke, S.T., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 305–326. [Google Scholar]
- Henry, S.P.; Jagels, M.A.; Hugli, T.E.; Manalili, S.; Geary, R.S.; Giclas, P.C.; Levin, A.A. Mechanism of alternative complement pathway dysregulation by a phosphorothioate oligonucleotide in monkey and human serum. Nucleic Acid Ther. 2014, 24, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Engelhardt, J.A.; Hung, G.; Yee, J.; Kikkawa, R.; Matson, J.; Tayefeh, B.; Machemer, T.; Giclas, P.C.; Henry, S.P. Effects of Repeated Complement Activation Associated with Chronic Treatment of Cynomolgus Monkeys with 2′-O-Methoxyethyl Modified Antisense Oligonucleotide. Nucleic Acid Ther. 2016, 26, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Henry, S.P.; Seguin, R.; Cavagnaro, J.; Berman, C.; Tepper, J.; Kornbrust, D. Considerations for the Characterization and Interpretation of Results Related to Alternative Complement Activation in Monkeys Associated with Oligonucleotide-Based Therapeutics. Nucleic Acid Ther. 2016, 26, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.; Den Besten, C. Preclinical and Clinical Drug-metabolism, Pharmacokinetics and Safety of Therapeutic Oligonucleotides. In RSC Drug Discovery Series: Advances in Nucleic Acid Therapeutics; Agrawal, S., Gait, M., Eds.; Royal Society of Chemistry: London, UK, 2019; pp. 474–517. [Google Scholar]
- Mcgreal, E.P.; Hearne, K.; Spiller, O.B. Immunobiology Off to a slow start: Under-development of the complement system in term newborns is more substantial following premature birth. Immunobiology 2012, 217, 176–186. [Google Scholar] [CrossRef]
- Seguin, R. Class-Related Proinflammatory Effects. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 227–242. [Google Scholar]
- Aartsma-Rus, A.; Jackson, A.L.; Levin, A.A. Mechanisms of Oligonucleotide Actions. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 1–38. [Google Scholar]
- Geary, R.S.; Wancewicz, E.; Matson, J.; Pearce, M.; Siwkowski, A.; Swayze, E.; Bennett, F. Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 2′-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN. Biochem. Pharmacol. 2009, 78, 284–291. [Google Scholar] [CrossRef]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Sun, H.; Shen, W.; Crooke, S.T. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015, 43, 2927–2945. [Google Scholar] [CrossRef] [Green Version]
- Pollak, A.J.; Hickman, J.H.; Liang, X.H.; Crooke, S.T. Gapmer Antisense Oligonucleotides Targeting 5S Ribosomal RNA Can Reduce Mature 5S Ribosomal RNA by Two Mechanisms. Nucleic Acid Ther. 2020, 30, 312–324. [Google Scholar] [CrossRef]
- Vickers, T.A.; Crooke, S.T. Antisense oligonucleotides capable of promoting specific target mRNA reduction via competing RNase H1-dependent and independent mechanisms. PLoS ONE 2014, 9, e108625. [Google Scholar] [CrossRef] [Green Version]
- Vickers, T.A.; Sabripour, M.; Crooke, S.T. U1 adaptors result in reduction of multiple pre-mRNA species principally by sequestering U1snRNP. Nucleic Acids Res. 2011, 39, e71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Allen, N.; Wang, S.; Vickers, T.A.; Shen, W.; Hsu, C.W.; Crooke, S.T. COPII vesicles can affect the activity of antisense oligonucleotides by facilitating the release of oligonucleotides from endocytic pathways. Nucleic Acids Res. 2018, 46, 10225–10245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, M.; Macgregor, R.D.; Hunt, C.A. Predictions of Effect for Intracellular Antisense Oligodeoxyribonucleotides from a Kinetic Model. Antisense Res. Dev. 1993, 3, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Spiller, D.G.; Giles, R.V.; Broughton, C.M.; Grzybowski, J.; Ruddell, C.J.; Tidd, D.M.; Clark, R.E. The influence of target protein half-life on the effectiveness of antisense oligonucleotide analog-mediated biologic responses. Antisense Nucleic Acid Drug Dev. 1998, 8, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Poelgeest, E.P.; Swart, R.M.; Betjes, M.G.H.; Moerland, M.; Weening, J.J.; Tessier, Y.; Hodges, M.R.; Levin, A.A.; Burggraaf, J. Acute kidney injury during therapy with an antisense oligonucleotide directed against PCSK9. Am. J. Kidney Dis. 2013, 62, 796–800. [Google Scholar] [CrossRef]
- Van Poelgeest, E.P.; Hodges, M.R.; Moerland, M.; Tessier, Y.; Levin, A.A.; Persson, R.; Lindholm, M.W.; Dumong Erichsen, K.; Ørum, H.; Cohen, A.F.; et al. Antisense-mediated reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9): A first-in-human randomized, placebo-controlled trial. Br. J. Clin. Pharmacol. 2015, 80, 1350–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]










| Parameters | Age | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PND 2 (n = 2) | PND 9 (n = 2) | PND 16 (n = 1) | PND 23 (n = 3) | PND 29 (n = 2) | PND 37 (n = 2) | PND 43 (n = 1) | PND 51 (n = 2) | Adult (4–6 mos) (n = 3) | |
| Hematology | Minimal to mild ↑ WBC, lymphocytes | Minimal to mild ↑ WBC, neutrophils, lymphocytes | Minimal to mild ↑ WBC, neutrophils | ||||||
| Clinical chemistry | Mild ↑ AST | Minimal to mild ↑ fibrinogen | Mild ↑ AST | Minimal to mild ↑ fibrinogen | Mild ↑ AST; minimal ↓ CHOL, LDL, minimal to mild ↑ fibrinogen | Minimal ↓ CHOL, LDL | ↓ CHOL, LDL, trig; ↑ crea in 1 of 3, BUN | ||
| Urinalysis | ↑ Na/Crea, Ca/Crea | ||||||||
| Total complement activity | Not detectable | ↑ CH50 | ↑ CH50 | ↑ CH50 | ↑ CH50 | No data | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenzuela, A.; Tardiveau, C.; Ayuso, M.; Buyssens, L.; Bars, C.; Van Ginneken, C.; Fant, P.; Leconte, I.; Braendli-Baiocco, A.; Parrott, N.; et al. Safety Testing of an Antisense Oligonucleotide Intended for Pediatric Indications in the Juvenile Göttingen Minipig, including an Evaluation of the Ontogeny of Key Nucleases. Pharmaceutics 2021, 13, 1442. https://doi.org/10.3390/pharmaceutics13091442
Valenzuela A, Tardiveau C, Ayuso M, Buyssens L, Bars C, Van Ginneken C, Fant P, Leconte I, Braendli-Baiocco A, Parrott N, et al. Safety Testing of an Antisense Oligonucleotide Intended for Pediatric Indications in the Juvenile Göttingen Minipig, including an Evaluation of the Ontogeny of Key Nucleases. Pharmaceutics. 2021; 13(9):1442. https://doi.org/10.3390/pharmaceutics13091442
Chicago/Turabian StyleValenzuela, Allan, Claire Tardiveau, Miriam Ayuso, Laura Buyssens, Chloe Bars, Chris Van Ginneken, Pierluigi Fant, Isabelle Leconte, Annamaria Braendli-Baiocco, Neil Parrott, and et al. 2021. "Safety Testing of an Antisense Oligonucleotide Intended for Pediatric Indications in the Juvenile Göttingen Minipig, including an Evaluation of the Ontogeny of Key Nucleases" Pharmaceutics 13, no. 9: 1442. https://doi.org/10.3390/pharmaceutics13091442