
Development of a Robust Control Strategy for Fixed-Dose Combination Bilayer Tablets with Integrated Quality by Design, Statistical, and Process Analytical Technology Approach


Abstract
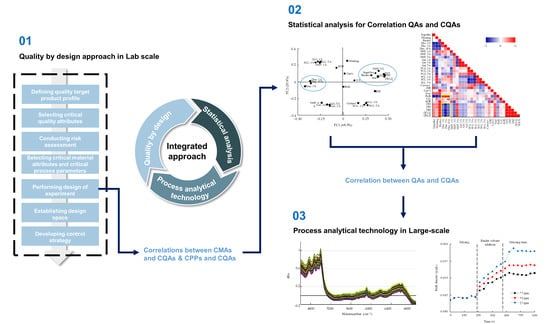
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Quality by Design Approach for Optimized Formulation
2.2.1. Design of Experiment for Metformin HCl Layer
2.2.2. Design of Experiment for Dapagliflozin l-Proline Layer
2.3. Quality by Design Approach for Optimized Process
2.3.1. Design of Experiment for the High-Shear Wet Granulation Process
2.3.2. Design of Experiment for the Roller Compaction Process
2.4. Preparation of Granules and Bilayer Tablet
2.4.1. Preparation of Metformin HCl Granules
2.4.2. Preparation of Dapagliflozin l-Proline Granules
2.4.3. Preparation of Bilayer Tablet
2.5. Measurement of CQAs
2.5.1. Assay and Content Uniformity (C.U.)
2.5.2. Hardness
2.5.3. Friability
2.5.4. In Vitro Dissolution Test
2.6. Measurement of QAs
2.6.1. Measurement of Granule Intrinsic Dissolution Rate
2.6.2. Measurement of Granule Properties
2.6.3. Measurement of Tablet Swelling Property
2.6.4. Measurement of Tablet Weight Gain and Tablet Mass Loss
2.6.5. Measurement of Tablet Gel Strength
2.6.6. Measurement of Tablet Contact Angle
2.7. HPLC Analysis Method
2.8. Multivariate Analysis
2.9. Process Analytical Technology Using Near-Infrared Spectrometer
2.9.1. Development of Calibration Models
2.9.2. In-Line NIR Monitoring during the Process
2.10. In Vivo Pharmacokinetic Study
2.10.1. LC–MS/MS Analysis Method
2.10.2. Study Design
2.10.3. Data Analysis
3. Results and Discussion
3.1. Initial Risk Assessment
3.2. Statistical Analysis of DoE for Metformin HCl Layer
3.2.1. Effect of Formulation Variables on Physical Properties of Metformin HCl Layer
3.2.2. Effect of Process Parameters on Physical Properties of Metformin HCl Layer
3.3. Statistical Analysis of DoE for Dapagliflozin l-Proline Layer
3.3.1. Effect of Formulation Variables on Physical Properties of Dapagliflozin l-Proline Layer
3.3.2. Effect of Process Parameters on Physical Properties of Dapagliflozin l-Proline Layer
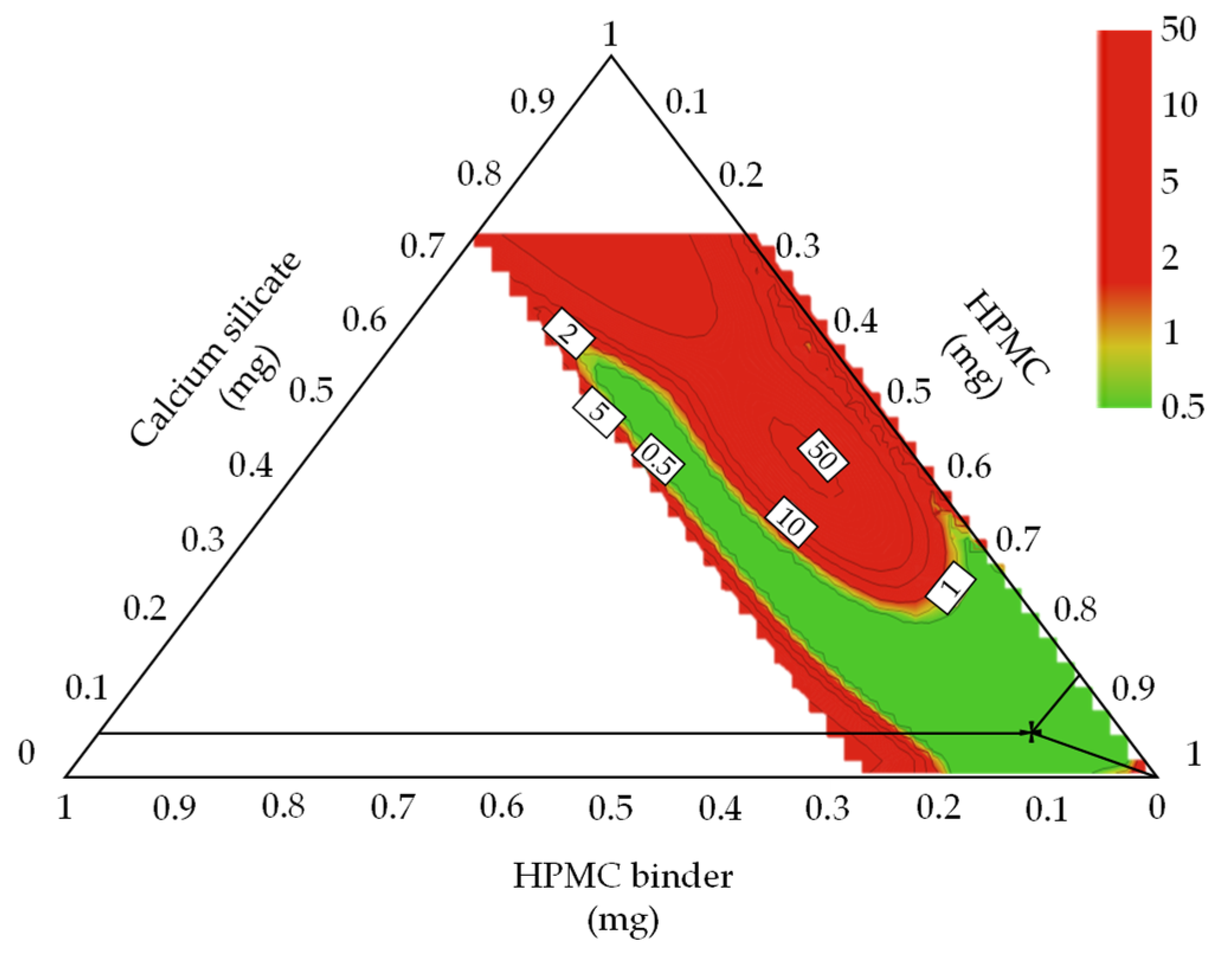
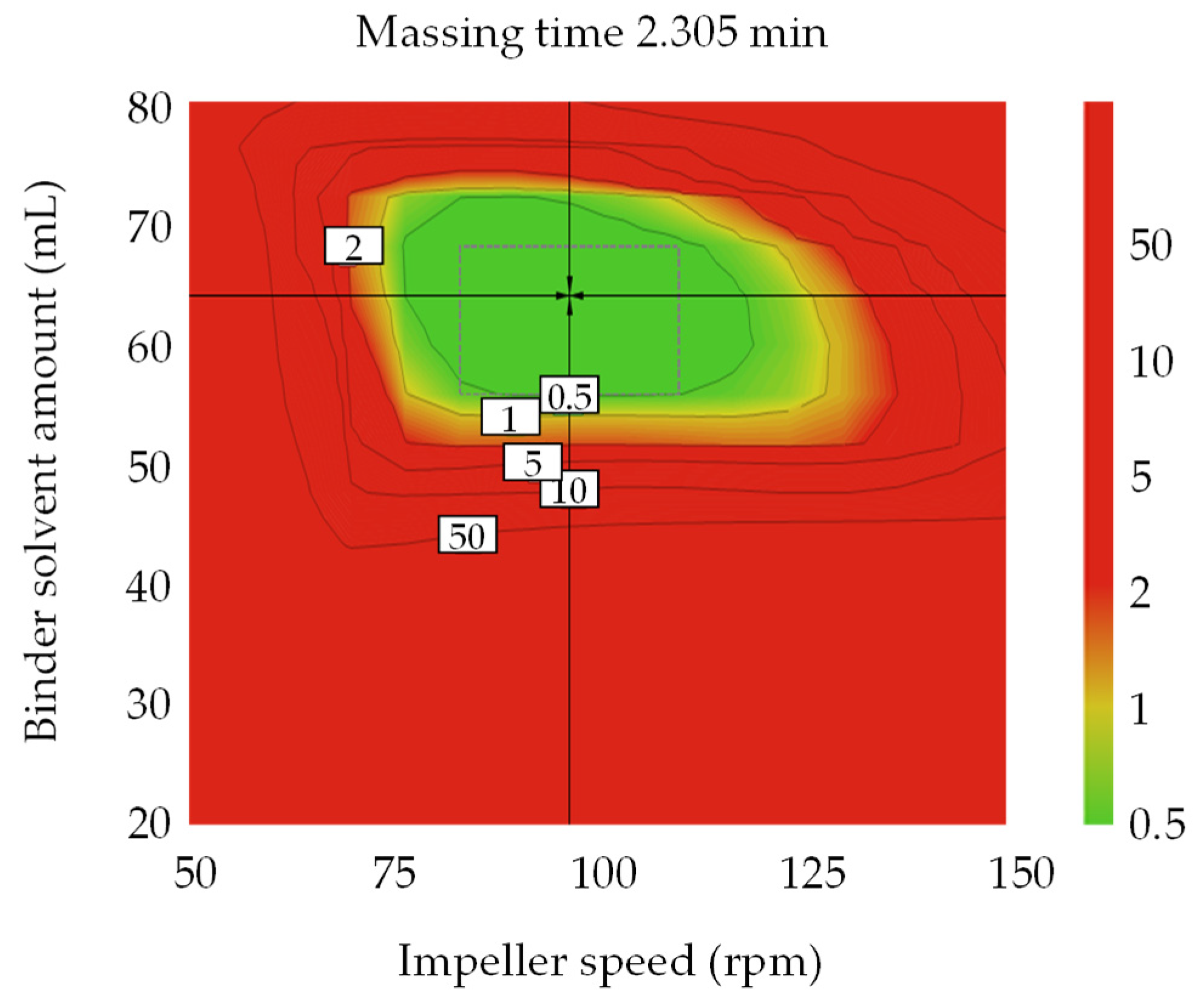
3.4. Optimal Settings and Robust Design Space
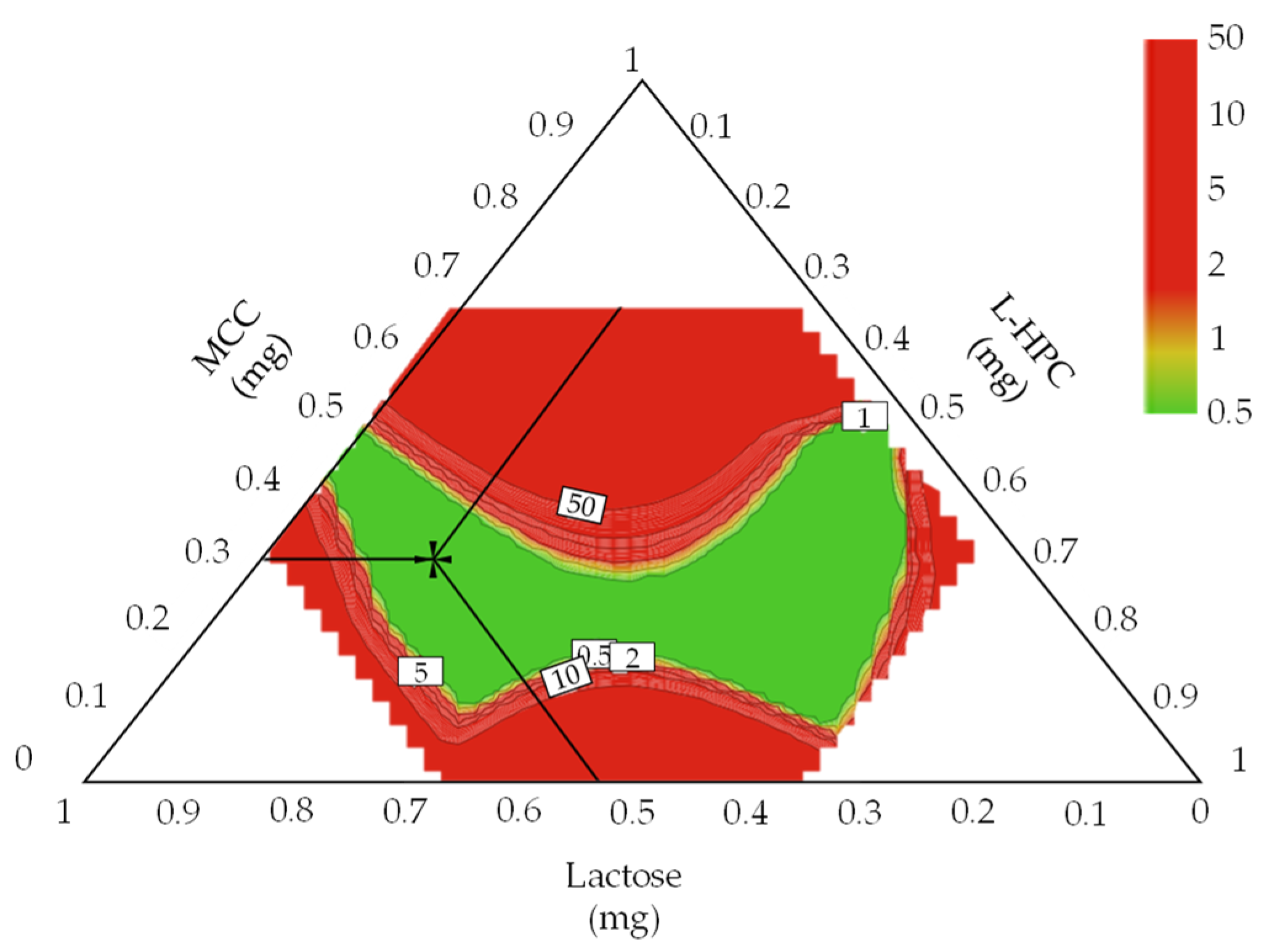
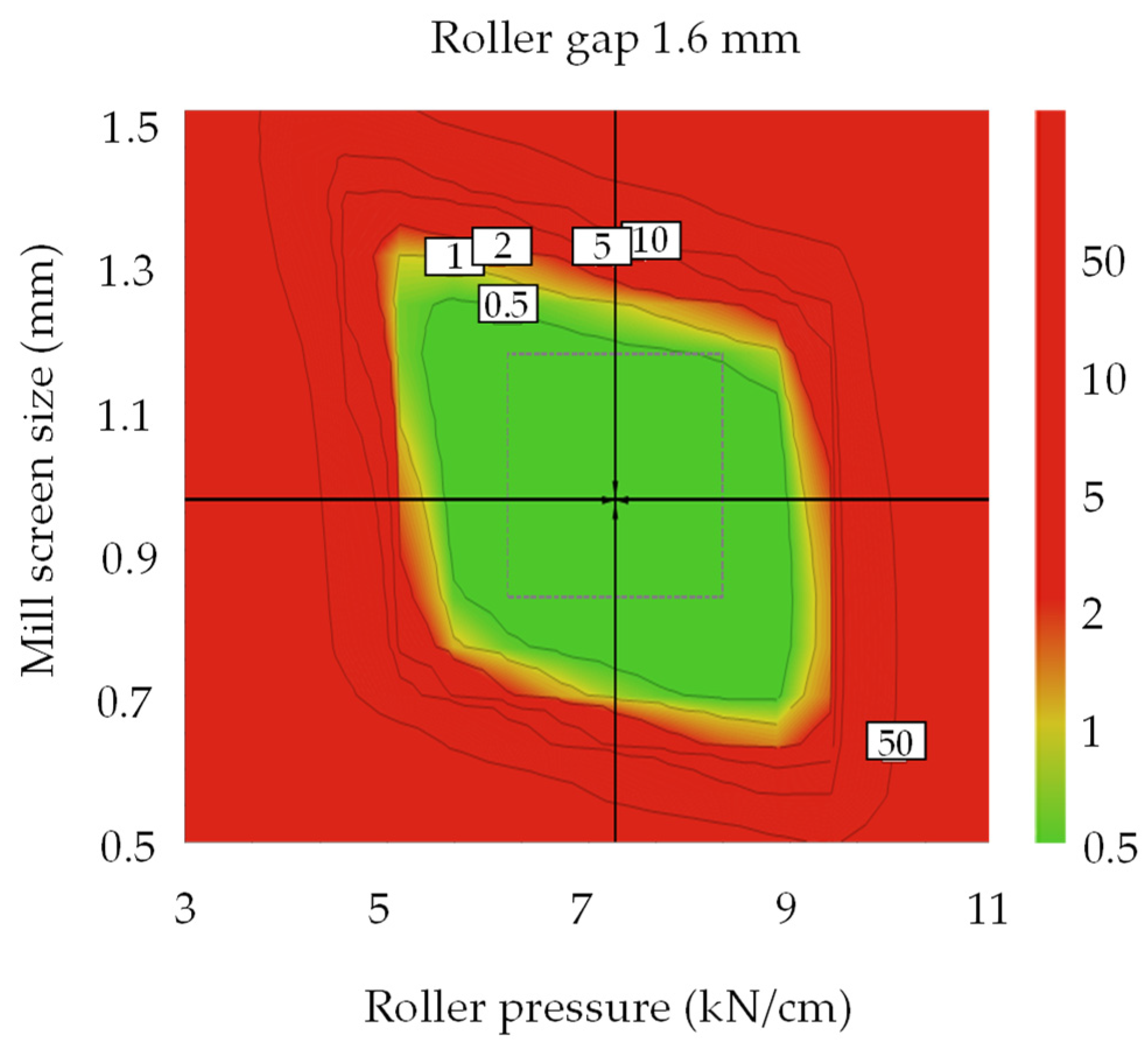
3.4.1. Optimal Settings of Metformin HCl Layer and the Robust Design Spaces
3.4.2. Optimal Settings of Dapagliflozin l-Proline Layer and the Robust Design Spaces
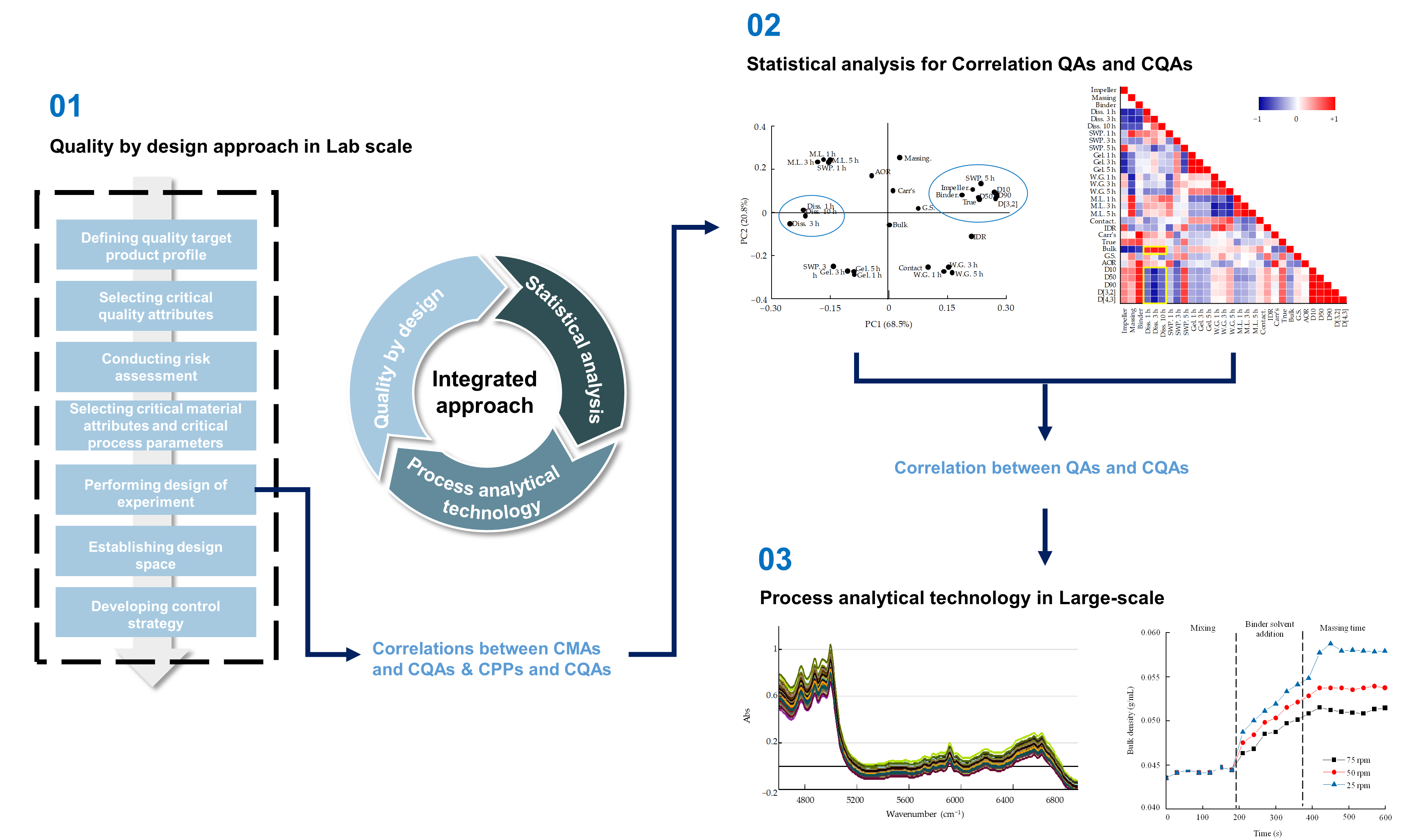
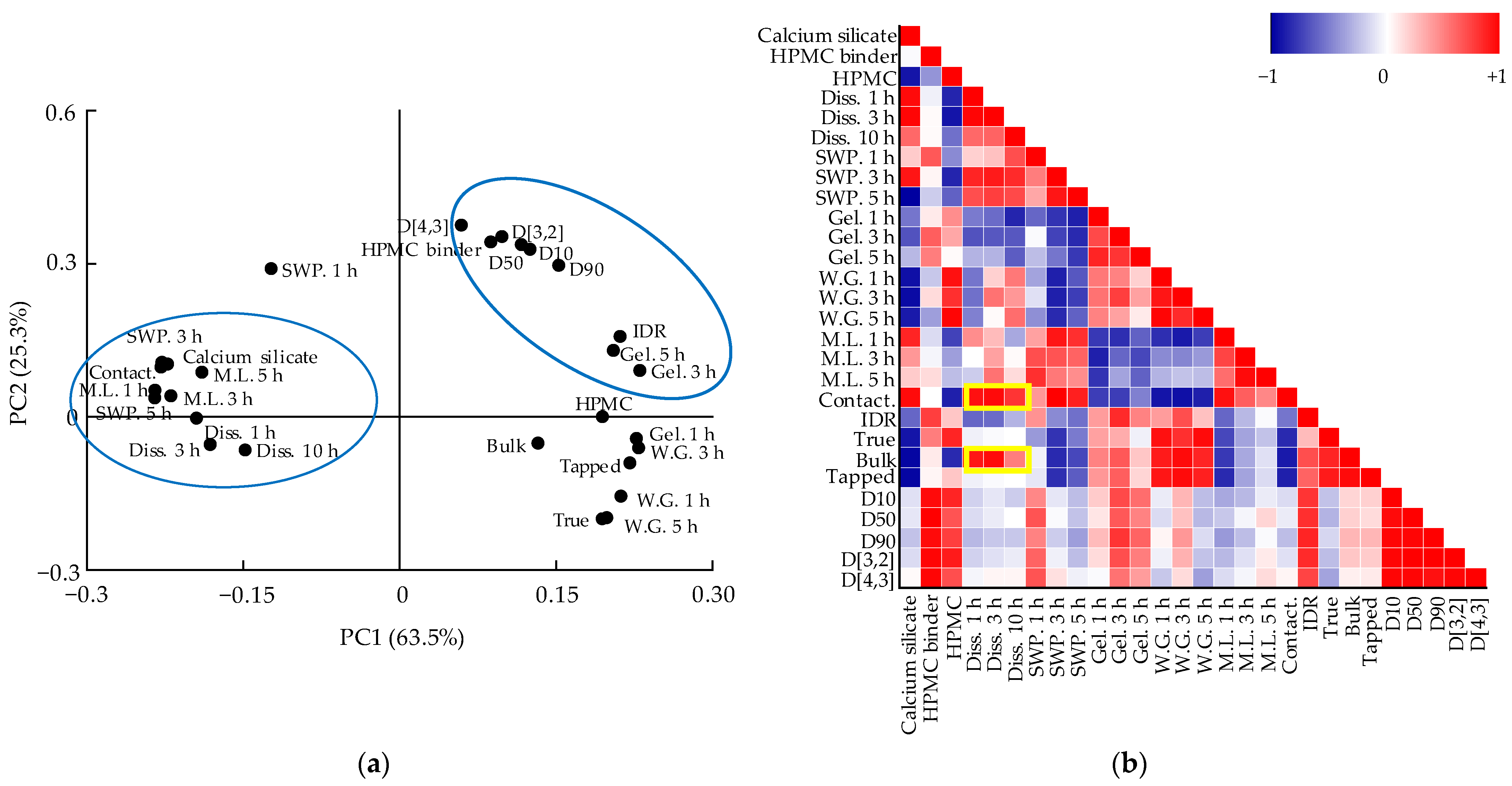
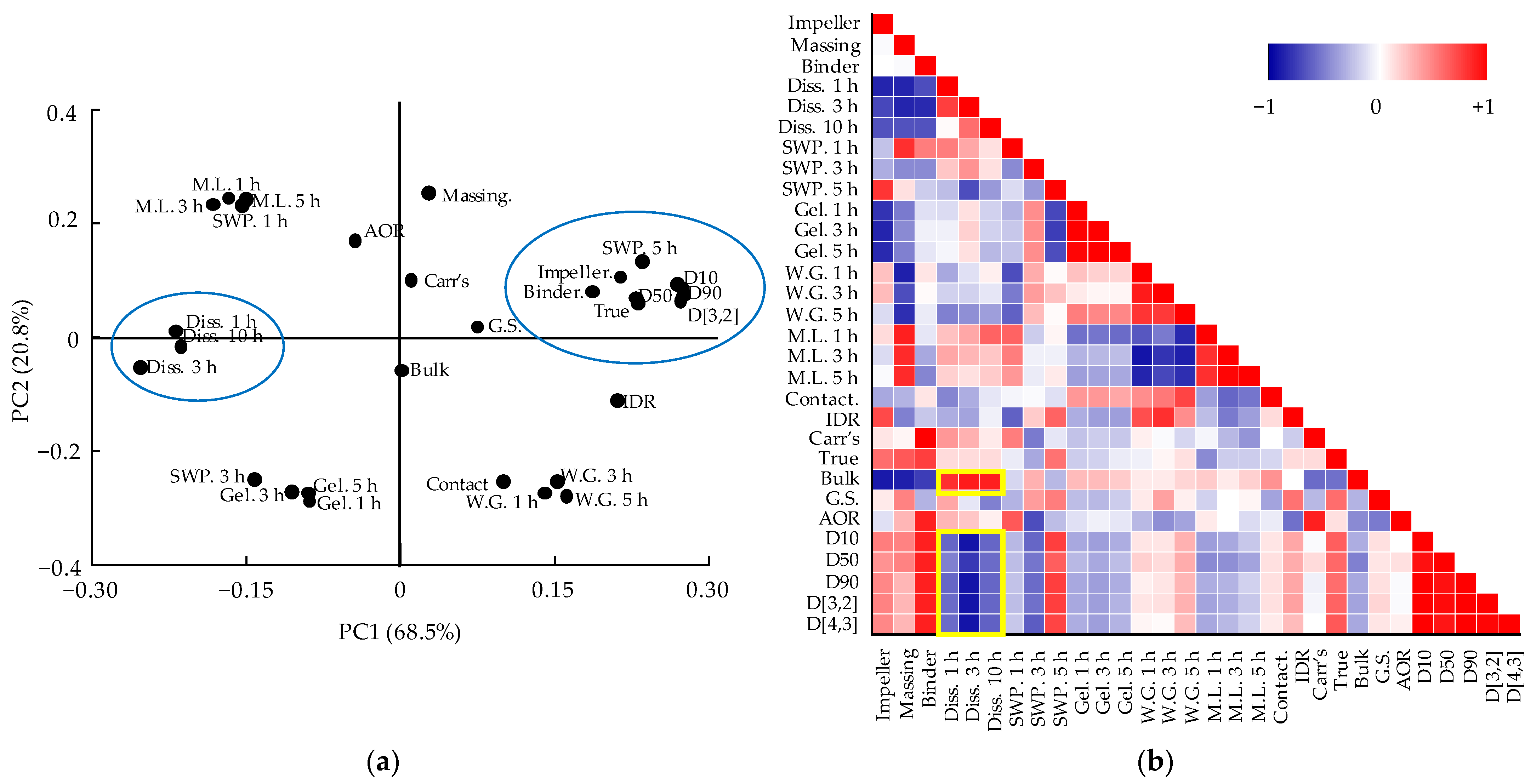
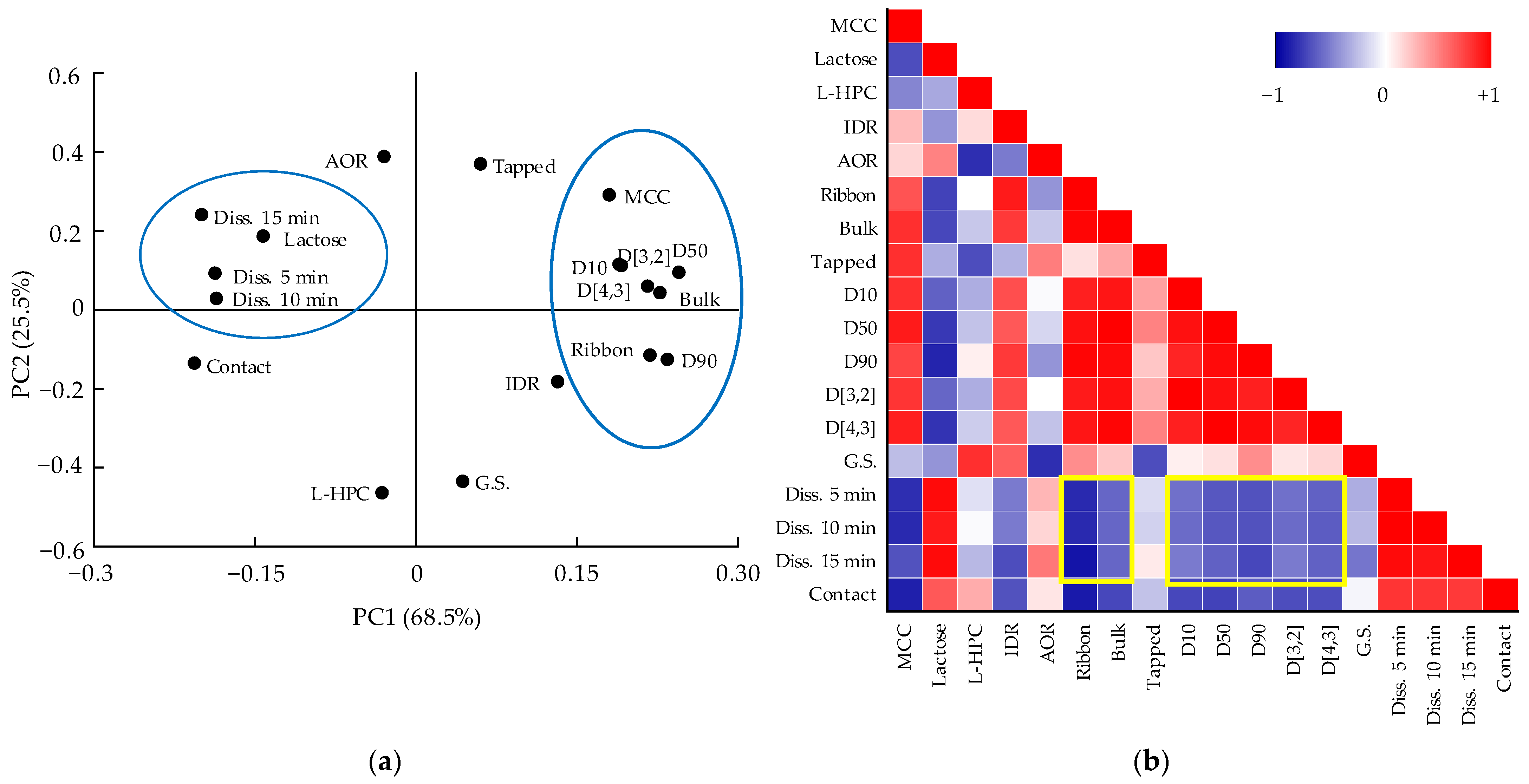
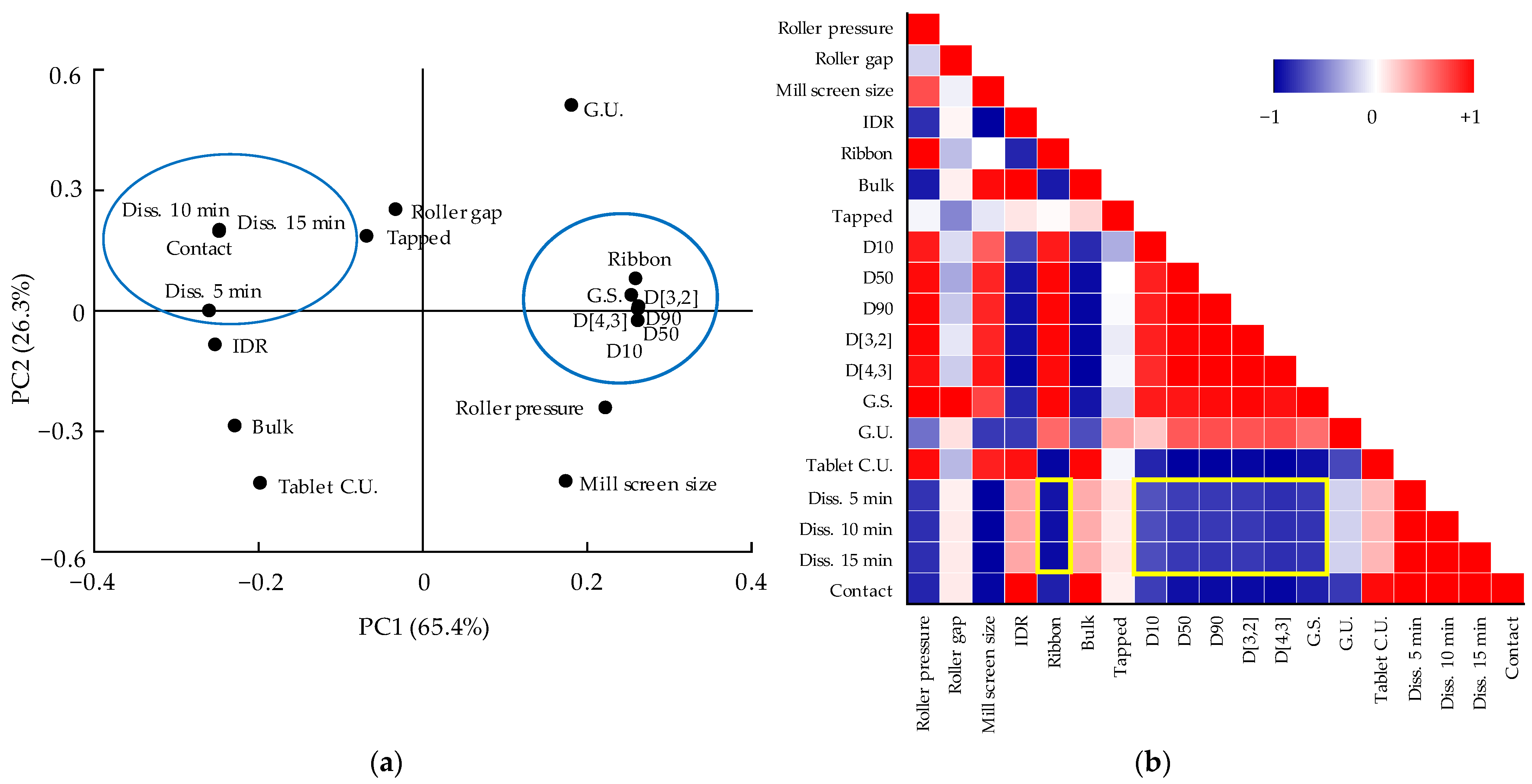
3.5. Multivariate Analysis for Correlations between QAs and CQAs
3.5.1. Correlation between QAs and CQAs of Metformin HCl Layer
3.5.2. Correlation between QAs and CQAs of Dapagliflozin l-Proline Layer
3.6. Process Analytical Technology Using Near-Infrared Spectroscopy for Monitoring Intermediate Product
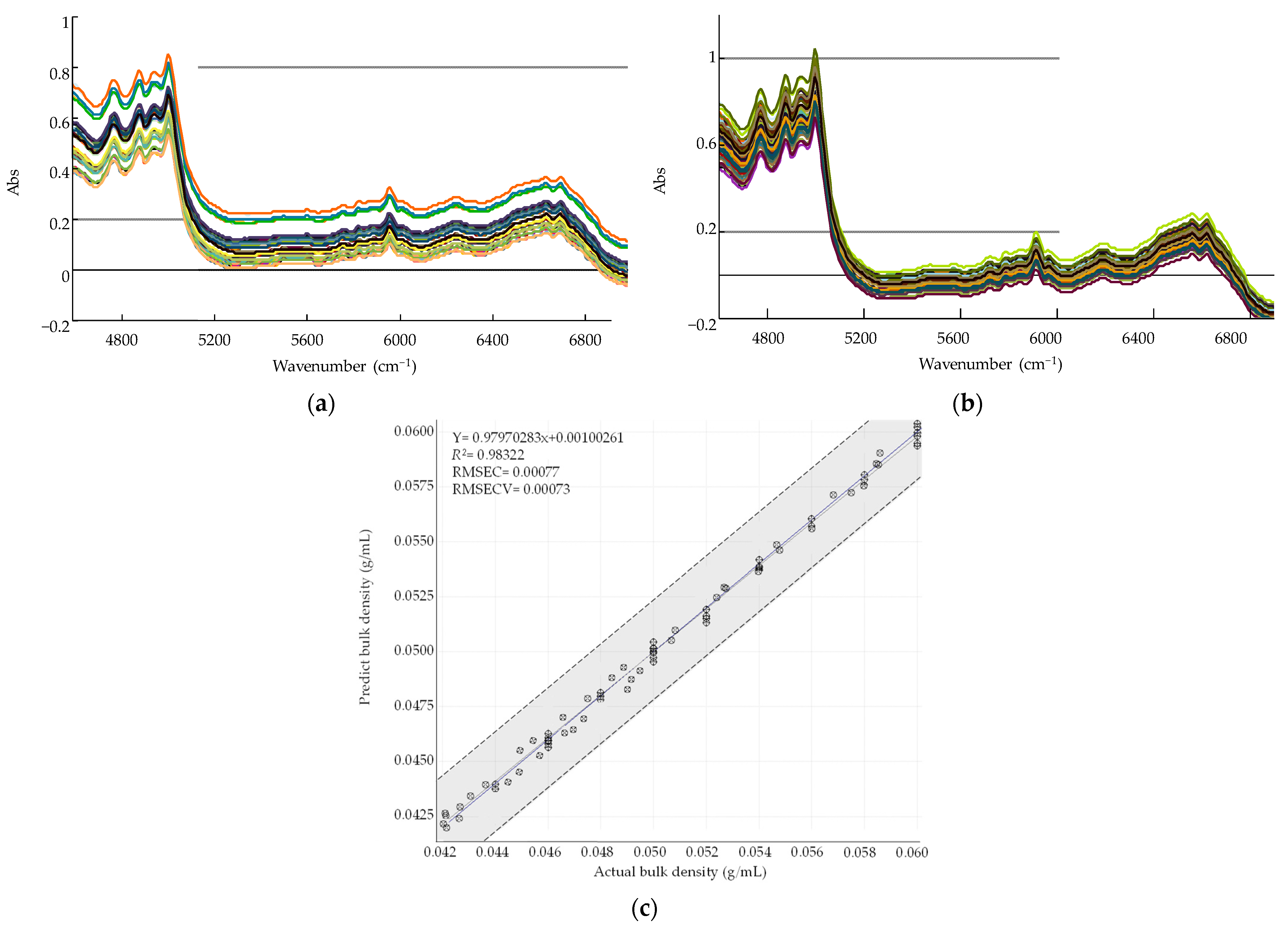
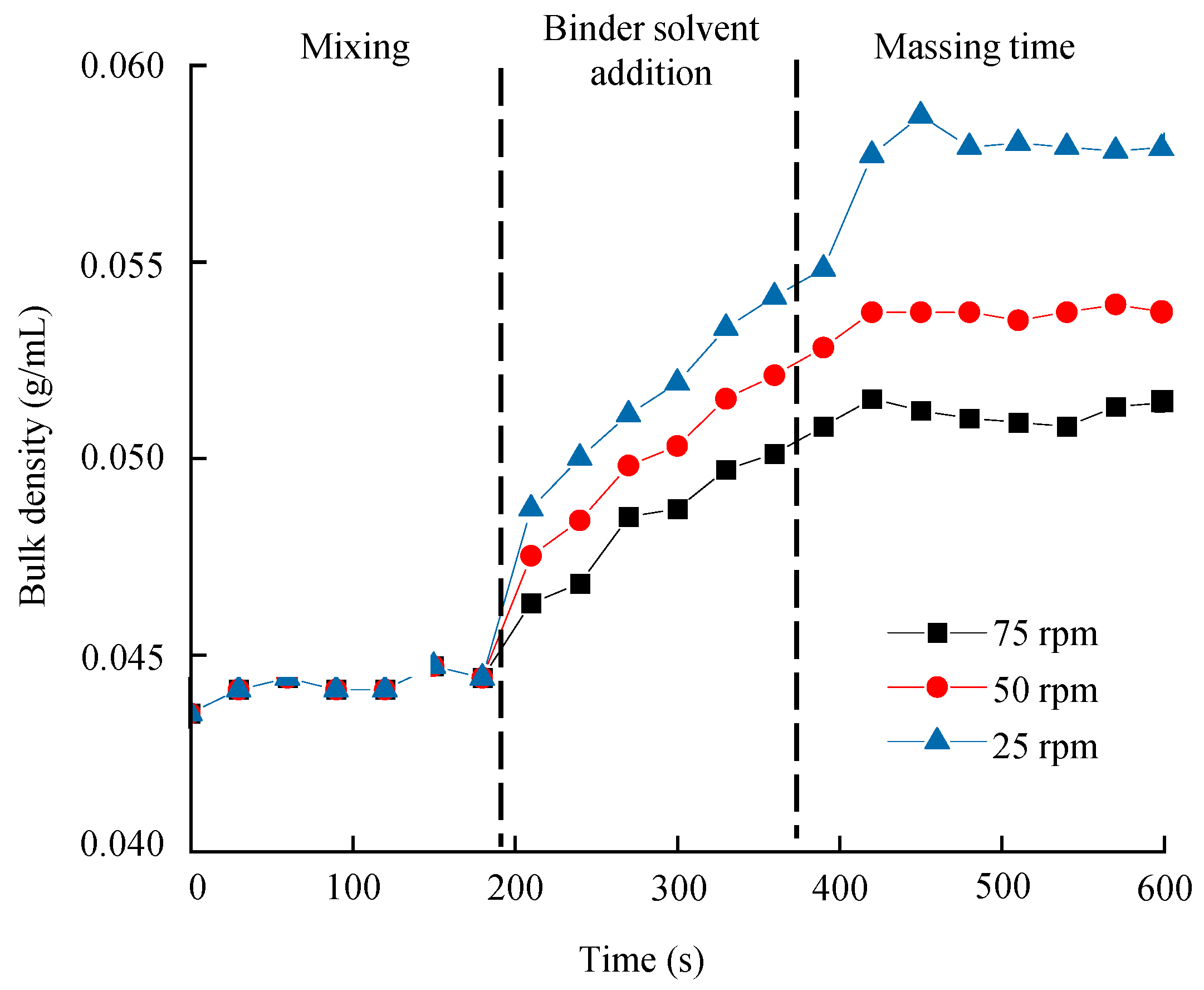
3.6.1. Process Analytical Technology in the High-Shear Wet Granulation Process
Development of a PLS Calibration Model for Bulk Density
Monitoring Granule Bulk Density in the Large-Scale High-Shear Wet Granulation Process
3.6.2. Process Analytical Technology in the Roller Compaction Process
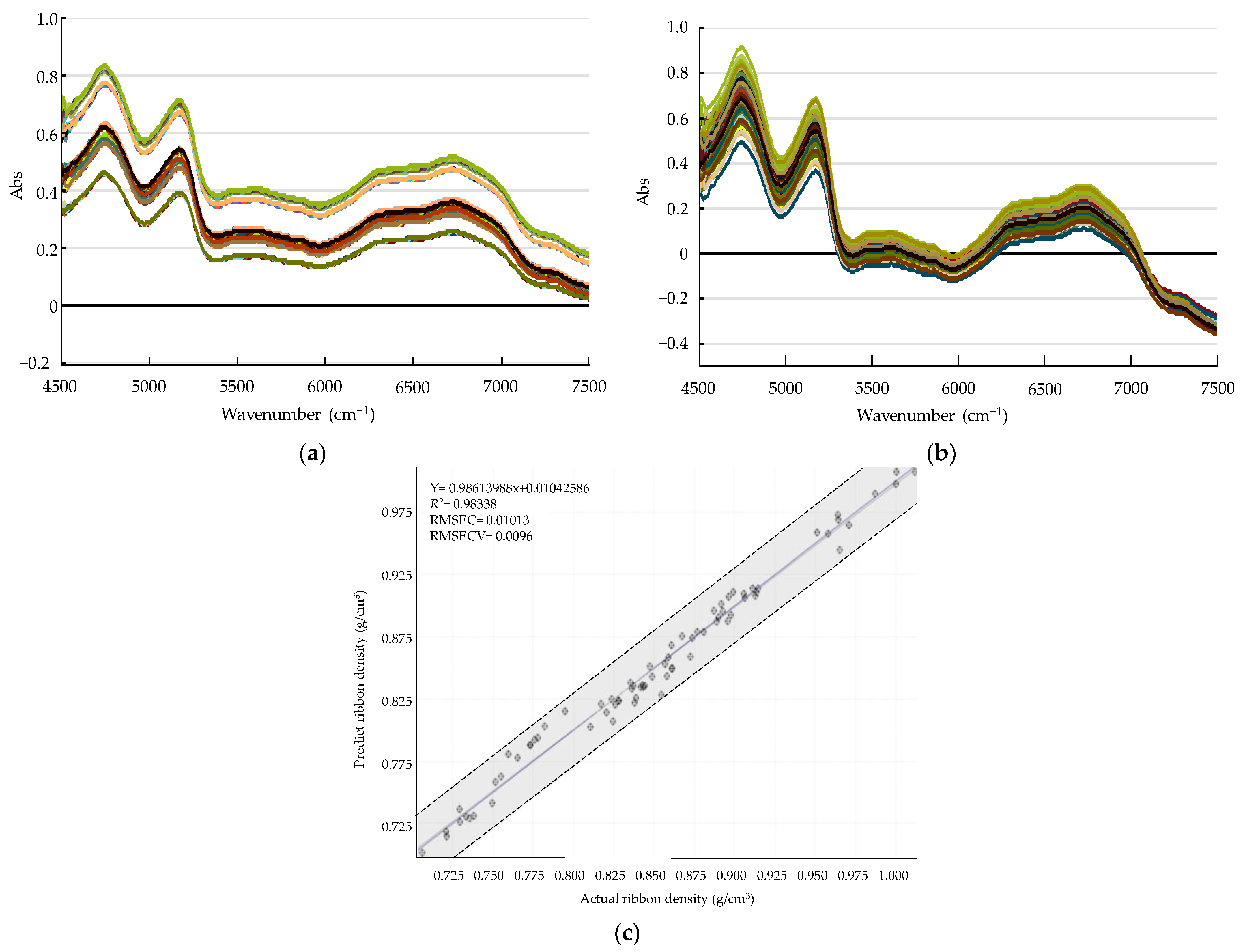
Development of a PLS Calibration Model for Ribbon Density
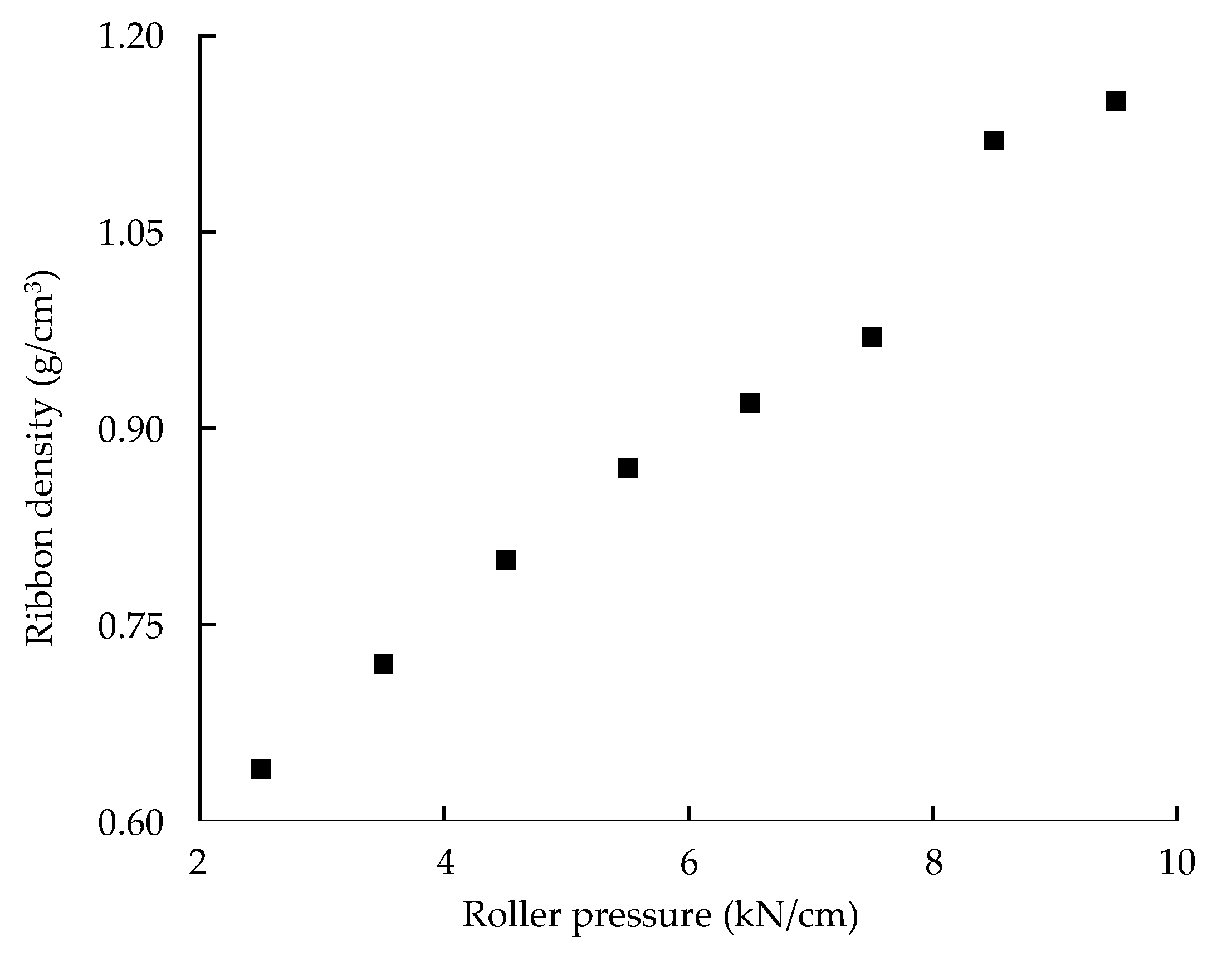
Monitoring Ribbon Density in the Large-Scale Roller Compaction Process
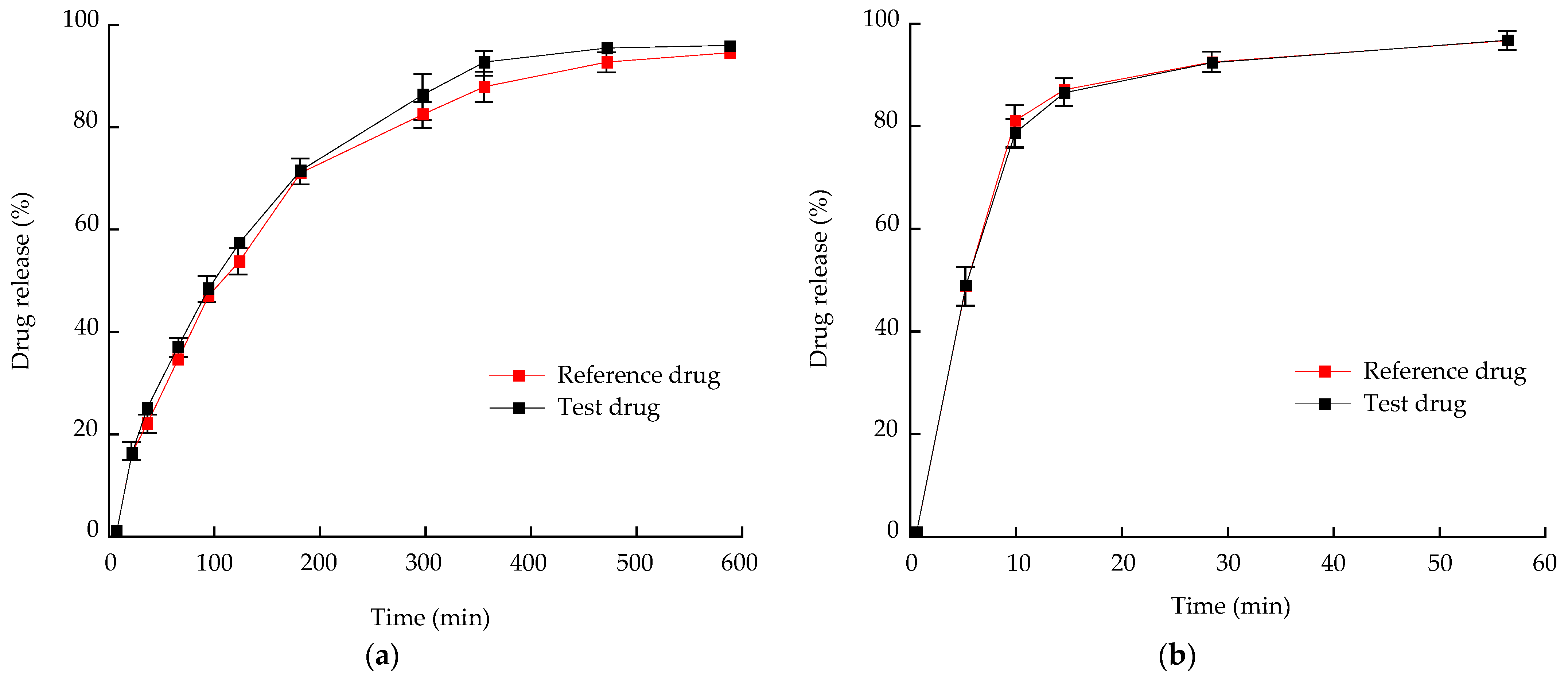
3.7. In Vitro Dissolution and Stability Test of Optimized Bilayer Tablet
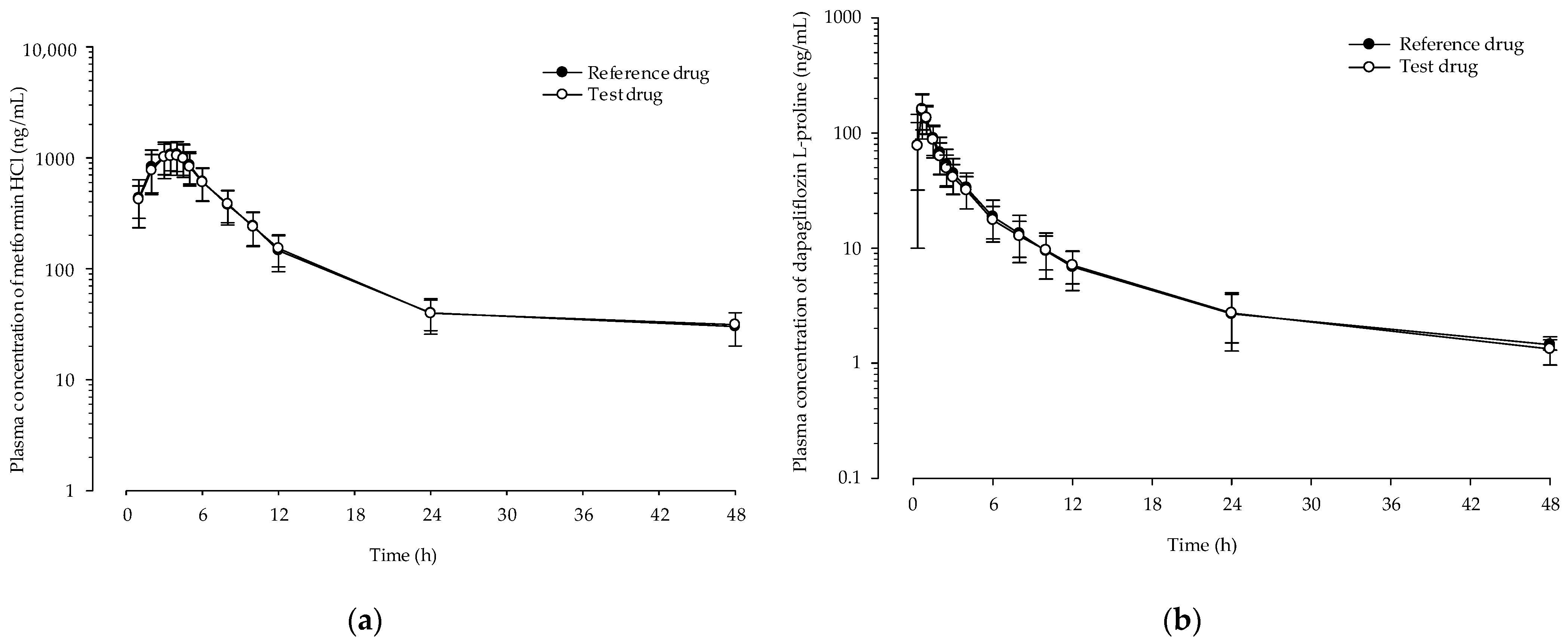
3.8. In Vivo Pharmacokinetic (PK) Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Del Prato, S.; Felton, A.M.; Munro, N.; Nesto, R.; Zimmet, P.; Zinman, B. Improving glucose management: Ten steps to get more patients with type 2 diabetes to glycaemic goal: Recommendations from the Global Partnership for Effective Diabetes Management. Int. J. Clin. Pract. 2005, 59, 1345–1355. [Google Scholar] [CrossRef]
- Kuecker, C.M.; Vivian, E.M. Patient considerations in type 2 diabetes–role of combination dapagliflozin–metformin XR. Diabetes Metab. Syndr. Obes. Targets Ther. 2016, 9, 25. [Google Scholar]
- Khomitskaya, Y.; Tikhonova, N.; Gudkov, K.; Erofeeva, S.; Holmes, V.; Dayton, B.; Davies, N.; Boulton, D.W.; Tang, W. Bioequivalence of Dapagliflozin/Metformin Extended-release Fixed-combination Drug Product and Single-component Dapagliflozin and Metformin Extended-release Tablets in Healthy Russian Subjects. Clin. Ther. 2018, 40, 550–561. [Google Scholar] [CrossRef]
- Wang, J.-S.; Huang, C.-N.; Hung, Y.-J.; Kwok, C.-F.; Sun, J.-H.; Pei, D.; Yang, C.-Y.; Chen, C.-C.; Lin, C.-L.; Sheu, W.H.-H. Acarbose plus metformin fixed-dose combination outperforms acarbose monotherapy for type 2 diabetes. Diabetes Res. Clin. Pract. 2013, 102, 16–24. [Google Scholar] [CrossRef]
- Bailey, C.; Day, C. Fixed-dose single tablet antidiabetic combinations. Diabetes Obes. Metab. 2009, 11, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Blonde, L.; Wogen, J.; Kreilick, C.; Seymour, A.A. Greater reductions in A1C in type 2 diabetic patients new to therapy with glyburide/metformin tablets as compared to glyburide co-administered with metformin. Diabetes Obes. Metab. 2003, 5, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Abebe, A.; Akseli, I.; Sprockel, O.; Kottala, N.; Cuitiño, A.M. Review of bilayer tablet technology. Int. J. Pharm. 2014, 461, 549–558. [Google Scholar] [CrossRef]
- Limin, Z.; John, F.; Hui, Z.; Harshad, P.; Scott, J. Dissolution Method Development for Fixed-Dose Combination Drug Products–Challenges and Strategies; American Pharmaceutical Review: Monroe, WA, USA, 2015. [Google Scholar]
- Food, U.; Administration, D. Guidance for Industry: Q8 (R2) Pharmaceutical Development; Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2009. [Google Scholar]
- Juran, J.; Godfrey, A.B. Quality Handbook; McGraw-Hill: New York, NY, USA, 1999; p. 173. [Google Scholar]
- Rathore, A.S.; Winkle, H. Quality by design for biopharmaceuticals. Nat. Biotechnol. 2009, 27, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Pramod, K.; Tahir, M.A.; Charoo, N.A.; Ansari, S.H.; Ali, J. Pharmaceutical product development: A quality by design approach. Int. J. Pharm. Investig. 2016, 6, 129. [Google Scholar]
- Björn, I.N.; Jansson, A.; Karlsson, M.; Folestad, S.; Rasmuson, A. Empirical to mechanistic modelling in high shear granulation. Chem. Eng. Sci. 2005, 60, 3795–3803. [Google Scholar] [CrossRef]
- Dumarey, M.; Talwar, S.; Yahyah, M.; Peterson, J. Empirical modelling to support scale up of primary pharmaceutical processes. In Computer Aided Chemical Engineerin; Elsevier: Amsterdam, The Netherlands, 2016; Volume 38, pp. 2241–2246. [Google Scholar]
- Lawrence, X.Y. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2008, 25, 781–791. [Google Scholar]
- Lawrence, X.Y.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar]
- Committee for Medicinal Products for Human Use. Guideline on Real Time Release Testing (formerly Guideline on Parametric Release); Techinal Report; European Medicines Agency: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Lendrem, D.; Owen, M.; Godbert, S. DOE (design of experiments) in development chemistry: Potential obstacles. Org. Process Res. Dev. 2001, 5, 324–327. [Google Scholar] [CrossRef]
- Huang, J.; Kaul, G.; Cai, C.; Chatlapalli, R.; Hernandez-Abad, P.; Ghosh, K.; Nagi, A. Quality by design case study: An integrated multivariate approach to drug product and process development. Int. J. Pharm. 2009, 382, 23–32. [Google Scholar] [CrossRef]
- Bowden, G.D.; Pichler, B.J.; Maurer, A. A Design of Experiments (DoE) Approach Accelerates the Optimization of Copper-Mediated 18 F-Fluorination Reactions of Arylstannanes. Sci. Rep. 2019, 9, 11370. [Google Scholar]
- Ferreira, A.P.; Tobyn, M. Multivariate analysis in the pharmaceutical industry: Enabling process understanding and improvement in the PAT and QbD era. Pharm. Dev. Technol. 2015, 20, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Han, J.K.; Shin, B.S.; Choi, D.H. Comprehensive study of intermediate and critical quality attributes for process control of high-shear wet granulation using multivariate analysis and the quality by design approach. Pharmaceutics 2019, 11, 252. [Google Scholar] [CrossRef] [Green Version]
- Jeong, G.; Bak, J.; Yoo, B. Physical and rheological properties of xanthan gum agglomerated in fluidized bed: Effect of HPMC as a binder. Int. J. Biol. Macromol. 2019, 121, 424–428. [Google Scholar] [CrossRef]
- Choi, D.H.; Kim, N.A.; Chu, K.R.; Jung, Y.J.; Yoon, J.H.; Jeong, S.H. Material properties and compressibility using Heckel and Kawakita equation with commonly used pharmaceutical excipients. J. Pharm. Investig. 2010, 40, 237–244. [Google Scholar]
- Yeom, S.B.; Choi, D.H. Scale-up strategy in quality by design approach for pharmaceutical blending process with discrete element method simulation. Pharmaceutics 2019, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.H.; Lim, J.Y.; Shin, S.; Choi, W.J.; Jeong, S.H.; Lee, S. A novel experimental design method to optimize hydrophilic matrix formulations with drug release profiles and mechanical properties. J. Pharm. Sci. 2014, 103, 3083–3094. [Google Scholar] [CrossRef]
- Jamzad, S.; Tutunji, L.; Fassihi, R. Analysis of macromolecular changes and drug release from hydrophilic matrix systems. Int. J. Pharm. 2005, 292, 75–85. [Google Scholar] [CrossRef]
- Jo, S.B.; Kim, H.K.; Lee, H.N.; Kim, Y.-J.; Dev Patel, K.; Campbell Knowles, J.; Lee, J.-H.; Song, M. Physical properties and biofunctionalities of bioactive root canal sealers in vitro. Nanomaterials 2020, 10, 1750. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L.J. Principal component analysis. In Wiley Interdisciplinary Reviews: Computational Statistics; Wiley: Hoboken, NJ, USA, 2010; pp. 433–459. [Google Scholar]
- Hauke, J.; Kossowski, T. Comparison of values of Pearson’s and Spearman’s correlation coefficients on the same sets of data. Quaest. Geogr. 2011, 30, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Shikata, F.; Kimura, S.; Hattori, Y.; Otsuka, M. Real-time monitoring of granule properties during high shear wet granulation by near-infrared spectroscopy with a chemometrics approach. RSC Adv. 2017, 7, 38307–38317. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Quality by Design for ANDAs: An Example for Immediate-Release Dosage Forms; U.S. Department of Health and Human Service FDA: Rockville, MD, USA, 2012.
- Desai, S.R. Quality by Design-based Formulation and Evaluation of Fast Dissolving Tablet of Aspirin. Asian J. Pharm. 2018, 12, S92–S101. [Google Scholar]
- Zakeri-Milani, P.; Barzegar-Jalali, M.; Azimi, M.; Valizadeh, H. Biopharmaceutical classification of drugs using intrinsic dissolution rate (IDR) and rat intestinal permeability. Eur. J. Pharm. Biopharm. 2009, 73, 102–106. [Google Scholar] [CrossRef]
- Issa, M.G.; Ferraz, H.G. Intrinsic dissolution as a tool for evaluating drug solubility in accordance with the biopharmaceutics classification system. Dissolution Technol. 2011, 18, 6–13. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Sui, Y.; She, Z.; Zhai, W.; Wang, C.; Deng, Y. Effect of particle size on solubility, dissolution rate, and oral bioavailability: Evaluation using coenzyme Q10 as naked nanocrystals. Int. J. Nanomed. 2012, 7, 5733. [Google Scholar]
- Van den Ban, S.; Goodwin, D.J. The impact of granule density on tabletting and pharmaceutical product performance. Pharm. Res. 2017, 34, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Chatlapalli, R.; Rohera, B.D. Physical characterization of HPMC and HEC and investigation of their use as pelletization aids. Int. J. Pharm. 1998, 161, 179–193. [Google Scholar] [CrossRef]
- Wang, L.; Chen, K.; Wen, H.; Ouyang, D.; Li, X.; Gao, Y.; Pan, W.; Yang, X. Design and evaluation of hydrophilic matrix system containing polyethylene oxides for the zero-order controlled delivery of water-insoluble drugs. AAPS PharmSciTech 2017, 18, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Lee, T.R. Contact angle and wetting properties. In Surface Science Techniques; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–34. [Google Scholar]
- Yang, B.; Wei, C.; Yang, Y.; Wang, Q.; Li, S. Evaluation about wettability, water absorption or swelling of excipients through various methods and the correlation between these parameters and tablet disintegration. Drug Dev. Ind. Pharm. 2018, 44, 1417–1425. [Google Scholar] [CrossRef]
- Razavykia, A.; Farahany, S.; Yusof, N.M. Evaluation of cutting force and surface roughness in the dry turning of Al–Mg2Si in-situ metal matrix composite inoculated with bismuth using DOE approach. Measurement 2015, 76, 170–182. [Google Scholar] [CrossRef]
- Khademi, A.; Ghorbani Renani, N.; Mofarrahi, M.; Rangraz Jeddi, A.; Mohd Yusof, N. The best location for speed bump installation using experimental design methodology. Promet Traffic Transp. 2013, 25, 565–574. [Google Scholar] [CrossRef]
- Brady, J.; Dürig, T.; Lee, P.I.; Li, J.X. Polymer Properties and Characterization. In Developing Solid Oral Dosage Forms; Qiu, Y., Chen, Y., Zhang, G.G.Z., Yu, L., Mantri, R.V., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 181–223. [Google Scholar]
- Jain, A.K.; Jain, S.K.; Yadav, A.; Agrawal, G.P. Controlled release calcium silicate based floating granular delivery system of ranitidine hydrochloride. Curr. Drug Deliv. 2006, 3, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Enayatifard, R.; Saeedi, M.; Akbari, J.; Tabatabaee, Y.H. Effect of hydroxypropyl methylcellulose and ethyl cellulose content on release profile and kinetics of diltiazem HCl from matrices. Trop. J. Pharm. Res. 2009, 8, 425–432. [Google Scholar] [CrossRef]
- Pabari, R.; Ramtoola, Z. Effect of a disintegration mechanism on wetting, water absorption, and disintegration time of orodispersible tablets. J. Young Pharm. 2012, 4, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhusudan Rao, Y.; Veni, J.K.; Jayasagar, G. Formulation and evaluation of diclofenac sodium using hydrophilic matrices. Drug Dev. Ind. Pharm. 2001, 27, 759–766. [Google Scholar] [CrossRef]
- Jain, K.K. Drug delivery systems-an overview. In Drug Delivery Systems; Humana Press: Passaic, NJ, USA, 2008; pp. 1–50. [Google Scholar]
- Alkan, M.; Yuksel, A. Granulation in a fluidized bed II Effect of binder amount on the final granules. Drug Dev. Ind. Pharm. 1986, 12, 1529–1543. [Google Scholar] [CrossRef]
- Nguyen, V.; Nguyen, Q.; Zhang, Y.; Lim, C.; Khoo, B.C. Effect of particle size on erosion characteristics. Wear 2016, 348, 126–137. [Google Scholar] [CrossRef]
- Goodwin, J.; Sage, W.; Tilly, G. Study of erosion by solid particles. Proc. Inst. Mech. 1969, 184, 279–292. [Google Scholar] [CrossRef]
- Thapa, P.; Choi, D.H.; Kim, M.S.; Jeong, S.H. Effects of granulation process variables on the physical properties of dosage forms by combination of experimental design and principal component analysis. Asian J. Pharm. Sci. 2019, 14, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Rahmanian, N.; Ghadiri, M.; Jia, X.; Stepanek, F. Characterisation of granule structure and strength made in a high shear granulator. Powder Technol. 2009, 192, 184–194. [Google Scholar] [CrossRef]
- Franceschinis, E.; Santomaso, A.; Benda, L.; Perissutti, B.; Voinovich, D.; Realdon, N. Influence of process variables on the properties of simvastatin self-emulsifying granules obtained through high shear wet granulation. Powder Technol. 2015, 274, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Shendurse, A.; Khedkar, C. Lactose. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 509–516. [Google Scholar]
- Bi, Y.; Sunada, H.; Yonezawa, Y.; Danjo, K. Evaluation of rapidly disintegrating tablets prepared by a direct compression method. Drug Dev. Ind. Pharm. 1999, 25, 571–581. [Google Scholar] [CrossRef]
- Zouai, O.; Thomas, C.; Pourcelot-Roubeau, Y. Microcrystalline cellulose: Investigation of porous structure of Avicel 102 from mercury porosimeter measurements. Drug Dev. Ind. Pharm. 1996, 22, 1253–1257. [Google Scholar] [CrossRef]
- Samal, P.K.; Newkirk, J.W. ASM Handbook: Powder Metallurgy; ASM International: Novelty, OH, USA, 2015; Volume 7, pp. 1–907. [Google Scholar]
- Souihi, N.; Josefson, M.; Tajarobi, P.; Gururajan, B.; Trygg, J. Design space estimation of the roller compaction process. Ind. Eng. Chem. Res 2013, 52, 12408–12419. [Google Scholar] [CrossRef]
- Al-Rabadi, G.J.; Gilbert, R.G.; Gidley, M.J. Effect of particle size on kinetics of starch digestion in milled barley and sorghum grains by porcine alpha-amylase. J. Cereal Sci. 2009, 50, 198–204. [Google Scholar] [CrossRef]
- Djuris, J.; Djuric, Z. Modeling in the quality by design environment: Regulatory requirements and recommendations for design space and control strategy appointment. Int. J. Pharm. 2017, 533, 346–356. [Google Scholar] [CrossRef]
- Chatterjee, S. Design space considerations. In Proceedings of the AAPS Annual Meeting, Chicago, IL, USA, 14 October 2012. [Google Scholar]
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and optimization of liquid chromatography analytical methods by using A QbD principles: Overview and recent advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Galbraith, S.; Ricart, B.; Stanton, C.; Smith-Goettler, B.; Verdi, L.; O’Connor, T.; Lee, S.; Yoon, S. Optimization of critical quality attributes in continuous twin-screw wet granulation via design space validated with pilot scale experimental data. Int. J. Pharm. 2017, 525, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Wikström, C.; Wold, S. Design of Experiments: Principles and Applications; MKS Umetrics AB: Umeå, Sweden, 2008. [Google Scholar]
- Zhang, Y.; Law, Y.; Chakrabarti, S. Physical properties and compact analysis of commonly used direct compression binders. AAPS PharmSciTech 2003, 4, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Heiman, J.; Tajarobi, F.; Gururajan, B.; Juppo, A.; Abrahmsén-Alami, S. Roller compaction of hydrophilic extended release tablets—combined effects of processing variables and drug/matrix former particle size. AAPS PharmSciTech 2015, 16, 267–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, S.I.; Narang, A.S.; LaMarche, K.; Subramanian, G.; Varia, S.A. Mechanistic basis for the effects of process parameters on quality attributes in high shear wet granulation. Int. J. Pharm. 2012, 439, 324–333. [Google Scholar] [CrossRef]
- Liu, B.; Wang, J.; Zeng, J.; Zhao, L.; Wang, Y.; Feng, Y.; Du, R. A review of high shear wet granulation for better process understanding, control and product development. Powder Technol. 2020, 381, 204–223. [Google Scholar] [CrossRef]
- Badawy, S.I.F.; Menning, M.M.; Gorko, M.A.; Gilbert, D.L. Effect of process parameters on compressibility of granulation manufactured in a high-shear mixer. Int. J. Pharm. 2000, 198, 51–61. [Google Scholar] [CrossRef]
- Crowley, M.E.; Hegarty, A.; McAuliffe, M.A.; O’Mahony, G.E.; Kiernan, L.; Hayes, K.; Crean, A.M. Near-infrared monitoring of roller compacted ribbon density: Investigating sources of variation contributing to noisy spectral data. Eur. J. Pharm. Sci. 2017, 102, 103–114. [Google Scholar] [CrossRef]
- Freitag, F.; Kleinebudde, P. How do roll compaction/dry granulation affect the tableting behaviour of inorganic materials? Comparison of four magnesium carbonates. Eur. J. Pharm. Sci. 2003, 19, 281–289. [Google Scholar] [CrossRef]
- Freeman, T.; Vom Bey, H.; Hanish, M.; Brockbank, K.; Armstrong, B. The influence of roller compaction processing variables on the rheological properties of granules. Asian J. Pharm. Sci. 2016, 11, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.W.; Flanner, H.H. Mathematical comparison of dissolution profiles. Pharm. Technol. 1996, 20, 64–74. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Metformin HCl | Dapagliflozin l-Proline | ||
|---|---|---|---|---|
| Reference (n = 32) | Test (n = 32) | Reference (n = 32) | Test (n = 32) | |
| t1/2 (h) | 5.22 ± 1.62 | 4.93 ± 2.46 | 8.75 ± 3.88 | 8.94 ± 3.77 |
| Cmax (ng/mL) | 1183.72 ± 378.92 | 1235.03 ± 319.71 | 170.83 ± 42.85 | 166.88 ± 53.28 |
| Tmax (h) | 3.41 ± 0.71 | 3.28 ± 0.94 | 0.72 ± 0.19 | 0.79 ± 0.23 |
| AUClast (ng·h/mL) | 7453.69 ± 2012.66 | 7435.50 ± 1824.21 | 475.38 ± 122.51 | 480.66 ± 147.52 |
| AUCinf (ng·h/mL) | 7798.04 ± 2028.24 | 7801.86 ± 1796.19 | 501.20 ± 129.25 | 509.59 ± 153.19 |
| Vd/F (L) | 1048.83 ± 552.35 | 949.16 ± 579.11 | 258.93 ± 95.36 | 261.84 ± 93.46 |
| CL/F (L/h) | 137.74 ± 40.81 | 135.25 ± 33.23 | 21.27 ± 5.42 | 21.30 ± 6.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chun, M.-H.; Kim, J.Y.; Park, E.-S.; Choi, D.H. Development of a Robust Control Strategy for Fixed-Dose Combination Bilayer Tablets with Integrated Quality by Design, Statistical, and Process Analytical Technology Approach. Pharmaceutics 2021, 13, 1443. https://doi.org/10.3390/pharmaceutics13091443
Chun M-H, Kim JY, Park E-S, Choi DH. Development of a Robust Control Strategy for Fixed-Dose Combination Bilayer Tablets with Integrated Quality by Design, Statistical, and Process Analytical Technology Approach. Pharmaceutics. 2021; 13(9):1443. https://doi.org/10.3390/pharmaceutics13091443
Chicago/Turabian StyleChun, Myung-Hee, Ji Yeon Kim, Eun-Seok Park, and Du Hyung Choi. 2021. "Development of a Robust Control Strategy for Fixed-Dose Combination Bilayer Tablets with Integrated Quality by Design, Statistical, and Process Analytical Technology Approach" Pharmaceutics 13, no. 9: 1443. https://doi.org/10.3390/pharmaceutics13091443