
Activation of Cryptic Antibiotic Biosynthetic Gene Clusters Guided by RNA-seq Data from Both Streptomyces ansochromogenes and ΔwblA

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
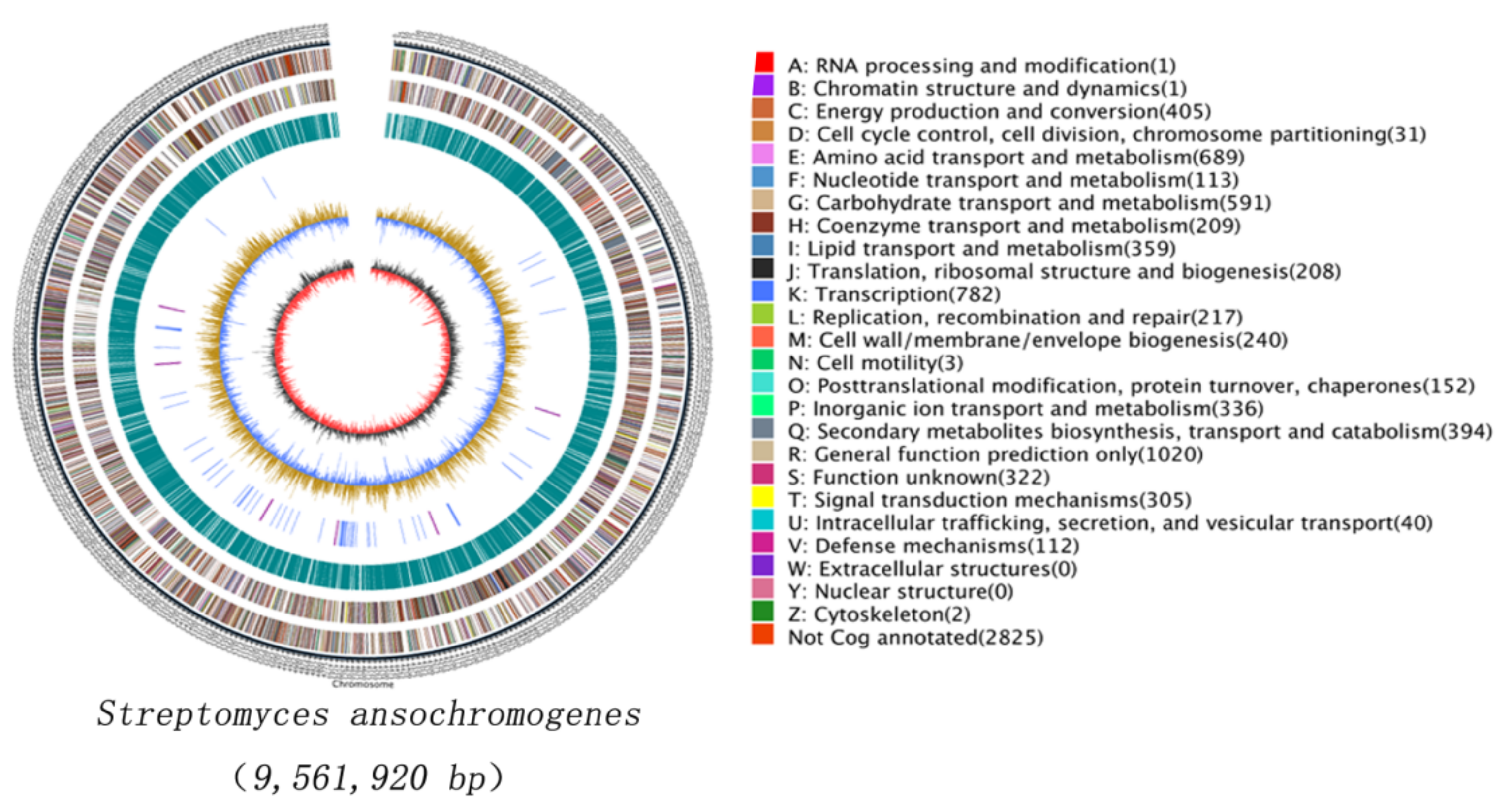
2.1. Sequencing and AntiSMASH Analysis of S. Ansochromogenes 7100 Genome
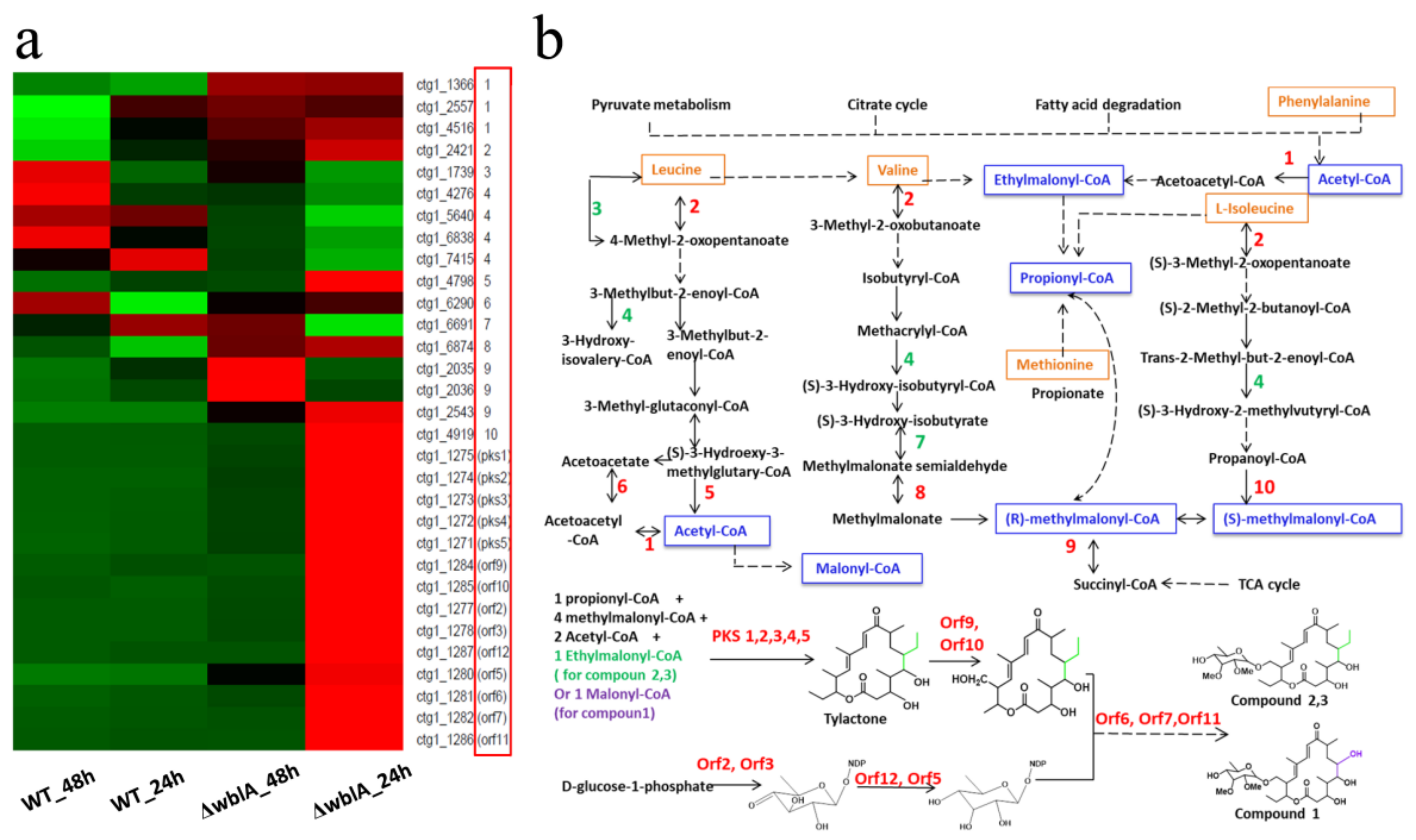
2.2. Genome-Wide Differentially Expressed Genes in ΔwblA
2.3. The Silent Nikkomycin Pathway and the Activated Tylosin Analogue Compounds (TACs) Biosynthetic Pathway in ΔwblA
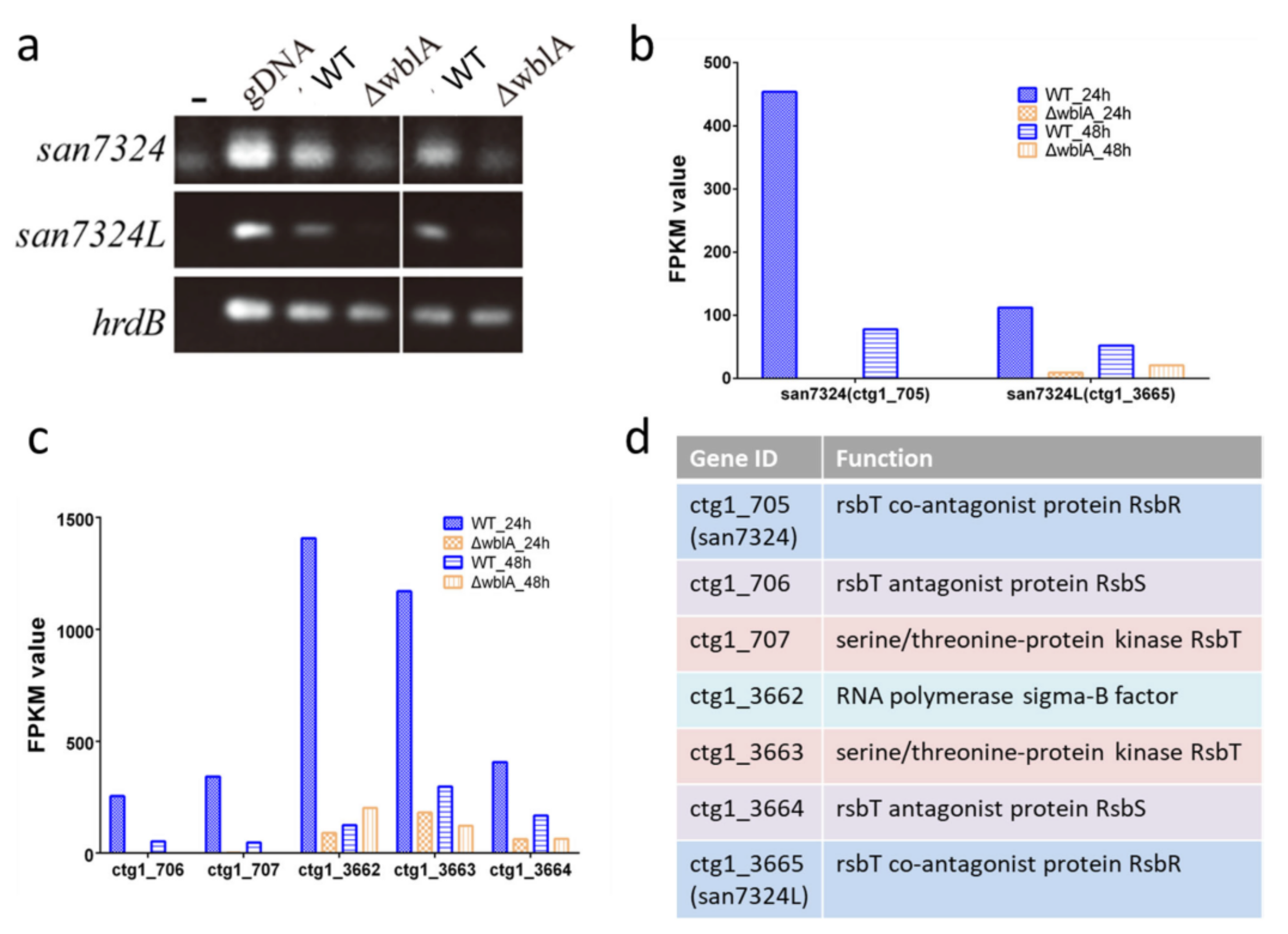
2.4. Production of an Anthracycline Antibiotic Oviedomycin Activated by Disrupting Genes san7324 plus san7324L
2.5. Understanding on Activation of Cryptic Antibiotic Biosynthetic Gene Clusters Guided by RNA-seq Data
3. Materials and Methods
3.1. Strains and Growth Condition
3.2. HPLC Analysis of Oviedomycin
3.3. PacBio Sequencing, Assembly and Gene Annotation
3.4. RNA Isolation, Library Construction and Sequencing
3.5. Quality Control and Reads Mapping to the Reference Genome
3.6. Quantification of Gene Expression Level and Differential Expression Analysis
3.7. RT-PCR Analysis
3.8. Data Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Li, J.; Tian, Z.; Xu, Y.; Zhang, J.; Liu, W.; Tan, H. Coordinative Modulation of Chlorothricin Biosynthesis by Binding of the Glycosylated Intermediates and End Product to a Responsive Regulator ChlF1. J. Biol. Chem. 2016, 291, 5406–5417. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, J.; Zheng, J.; Guan, H.; Liu, W.; Tan, H. Co-expression of a SARP Family Activator ChlF2 and a Type II Thioesterase ChlK Led to High Production of Chlorothricin in Streptomyces antibioticus DSM 40725. Front. Bioeng. Biotechnol. 2020, 8, 1013. [Google Scholar] [CrossRef]
- Liu, G.; Chater, K.F.; Chandra, G.; Niu, G.; Tan, H. Molecular Regulation of Antibiotic Biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 2013, 77, 112–143. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, P.; Challis, G. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Genet. 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Li, Y.; Tan, H. Biosynthesis and molecular regulation of secondary metabolites in microorganisms. Sci. China Life Sci. 2017, 60, 935–938. [Google Scholar] [CrossRef]
- Schroeckh, V.; Scherlach, K.; Nützmann, H.-W.; Shelest, E.; Schmidt-Heck, W.; Schuemann, J.; Martin, K.; Hertweck, C.; Brakhage, A.A. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 2009, 106, 14558–14563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhang, J.; Tian, Y.; Tan, H. Enhancement of salinomycin production by ribosome engineering in Streptomyces albus. Sci. China Life Sci. 2019, 62, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, J.A.; Kautsar, S.A.; Medema, M.H.; Linington, R.G. Microbial natural product databases: Moving forward in the multi-omics era. Nat. Prod. Rep. 2021, 38, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Kim, H.U.; Medema, M.H.; Weber, T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2019, 20, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Guo, Z.; Tang, W.; Chen, Y. Activation of three natural product biosynthetic gene clusters from Streptomyces lavendulae CGMCC 4.1386 by a reporter-guided strategy. Synth. Syst. Biotechnol. 2018, 3, 254–260. [Google Scholar] [CrossRef]
- Ji, Z.-Y.; Nie, Q.-Y.; Yin, Y.; Zhang, M.; Pan, H.-X.; Hou, X.-F.; Tang, G.-L. Activation and Characterization of Cryptic Gene Cluster: Two Series of Aromatic Polyketides Biosynthesized by Divergent Pathways. Angew. Chem. Int. Ed. 2019, 58, 18046–18054. [Google Scholar] [CrossRef]
- Liu, N.; Guan, H.; Niu, G.; Jiang, L.; Li, Y.; Zhang, J.; Li, J.; Tan, H. Molecular mechanism of mureidomycin biosynthesis activated by introduction of an exogenous regulatory gene ssaA into Streptomyces roseosporus. Sci. China Life Sci. 2021, 1–15. [Google Scholar] [CrossRef]
- Bush, M.J. The actinobacterial WhiB-like (Wbl) family of transcription factors. Mol. Microbiol. 2018, 110, 663–676. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, H.N.; Kim, P.; Lee, H.S.; Kim, E.S. Negative Role of wblA in Response to Oxidative Stress in Streptomyces coelicolor. J. Microbiol. Biotechnol. 2012, 22, 736–741. [Google Scholar] [CrossRef]
- Nah, H.-J.; Park, J.; Choi, S.; Kim, E.-S. WblA, a global regulator of antibiotic biosynthesis in Streptomyces. J. Ind. Microbiol. Biotechnol. 2021, 48. [Google Scholar] [CrossRef]
- Huang, X.; Ma, T.; Tian, J.; Shen, L.; Zuo, H.; Hu, C.; Liao, G. wblA, a pleiotropic regulatory gene modulating morphogenesis and daptomycin production in Streptomyces roseosporus. J. Appl. Microbiol. 2017, 123, 669–677. [Google Scholar] [CrossRef]
- Singh, A.; Guidry, L.; Narasimhulu, K.V.; Mai, D.; Trombley, J.; Redding, K.E.; Giles, G.; Lancaster, J.R.; Steyn, A.J.C. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe–4S] cluster and is essential for nutrient starvation survival. Proc. Natl. Acad. Sci. USA 2007, 104, 11562–11567. [Google Scholar] [CrossRef] [Green Version]
- Burian, J.; Yim, G.; Hsing, M.; Axerio-Cilies, P.; Cherkasov, A.; Spiegelman, G.B.; Thompson, C.J. The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res. 2013, 41, 10062–10076. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Alam, S.; Bajpai, R.; Kishan, K.R.; Agrawal, P. Redox biology of Mycobacterium tuberculosis H37Rv: Protein-protein interaction between GlgB and WhiB1 involves exchange of thiol-disulfide. BMC Biochem. 2009, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Wu, W.; Tian, Y.; Yang, H.; Song, Y.; Dong, K.; Huang, X.; Song, D. Studies on the differentiation and properties of stretptomyces ansochromogens. Acta Microbiol. Sin. 1994, 34, 398–402. [Google Scholar]
- Pan, Y.; Wang, L.; He, X.; Tian, Y.; Liu, G.; Tan, H. SabR enhances nikkomycin production via regulating the transcriptional level of sanG, a pathway-specific regulatory gene in Streptomyces ansochromogenes. BMC Microbiol. 2011, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Liao, G.; Zhang, J.; Tan, H. Identification of novel tylosin analogues generated by a wblA disruption mutant of Streptomyces ansochromogenes. Microb. Cell Factories 2015, 14, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palaniappan, K.; Chen, I.-M.A.; Chu, K.; Ratner, A.; Seshadri, R.; Kyrpides, N.; Ivanova, N.N.; Mouncey, N.J. IMG-ABC v.5.0: An update to the IMG/Atlas of Biosynthetic Gene Clusters Knowledgebase. Nucleic Acids Res. 2019, 48, D422–D430. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, G.; Tan, H. Nucleoside antibiotics: Biosynthesis, regulation, and biotechnology. Trends Microbiol. 2015, 23, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Moy, B.E.; Seshu, J. STAS Domain Only Proteins in Bacterial Gene Regulation. Front. Cell Infect. Microbiol. 2021, 11, 679982. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Xin, Y.-P.; Shan, Y.; Zhang, X.; Song, H.-H.; Fang, W.-H. Phosphorylation residue T175 in RsbR protein is required for efficient induction of sigma B factor and survival of Listeria monocytogenes under acidic stress. J. Zhejiang Univ. Sci. B 2019, 20, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Van der Steen, J.B.; Ávila-Pérez, M.; Knippert, D.; Vreugdenhil, A.; van Alphen, P.; Hellingwerf, K.J. Differentiation of Function among the RsbR Paralogs in the General Stress Response of Bacillus subtilis with Regard to Light Perception. J. Bacteriol. 2012, 194, 1708–1716. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, J.; Li, Y.; Cai, Y.; Tan, H. Effects of san7324 and san7324L disruption on morphological differentiation and nikkomycin production of Streptomyces ansochromogenes. Acta Microbiol. Sin. 2019, 59, 235–246. [Google Scholar]
- Xu, J.; Zhang, J.; Zhuo, J.; Li, Y.; Tian, Y.; Tan, H. Activation and mechanism of a cryptic oviedomycin gene cluster via the disruption of a global regulatory gene, adpA, in Streptomyces ansochromogenes. J. Biol. Chem. 2017, 292, 19708–19720. [Google Scholar] [CrossRef] [Green Version]
- Fowler-Goldsworthy, K.; Gust, B.; Mouz, S.; Chandra, G.; Findlay, K.C.; Chater, K.F. The actinobacteria-specific gene wblA controls major developmental transitions in Streptomyces coelicolor A3(2). Microbiology 2011, 157, 1312–1328. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.Y.Y.; Bush, M.; Crack, J.C.; Buttner, M.; Le Brun, N.E. Interaction of the Streptomyces Wbl protein WhiD with the principal sigma factor sigma(σHrdB) depends on the WhiD [4Fe–4S] cluster. J. Biol. Chem. 2020, 295, 9752–9765. [Google Scholar] [CrossRef] [Green Version]
- Zhiwu, Q.; Gui, S.; Dequan, L.; Pan, L.; Wang, S.; Ke, W.; Liang, D.; Ding, Y. A precise chloroplast genome of Nelumbo nucifera (Nelumbonaceae) evaluated with Sanger, Illumina MiSeq, and PacBio RS II sequencing platforms: Insight into the plastid evolution of basal eudicots. BMC Plant Biol. 2014, 14, 289. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Li, Y.; Guan, H.; Zhang, J.; Tan, H. Enhancement of neomycin production by engineering the entire biosynthetic gene cluster and feeding key precursors in Streptomyces fradiae CGMCC 4.576. Appl. Microbiol. Biotechnol. 2019, 103, 2263–2275. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Type | From (bp) | To (bp) | Most Similar Known Cluster | Similarity |
|---|---|---|---|---|---|
| Region 1 | NRPS, NRPS-like, betalactone, nucleoside | 118,096 | 202,793 | neopolyoxin C | 100% |
| Region 2 | lanthipeptide-class-ii, terpene | 287,202 | 318,846 | heronamide A/heronamide B/heronamide C/heronamide D/heronamide E/heronamide F | 8% |
| Region 3 | NRPS, T1PKS, terpene | 413,811 | 476,353 | eponemycin | 21% |
| Region 4 | ectoine | 579,945 | 589,601 | ectoine | 75% |
| Region 5 | NRPS-like | 681,454 | 723,406 | livipeptin | 100% |
| Region 6 | terpene, T1PKS, PKS-like, NRPS, RiPP-like | 941,834 | 1,090,502 | filipin | 38% |
| Region 7 | RiPP-like | 1,402,262 | 1,411,931 | - | |
| Region 8 | T2PKS, T1PKS | 1,481,390 | 1,632,620 | spiramycin | 46% |
| Region 9 | terpene | 1,658,532 | 1,682,854 | hopene | 92% |
| Region 10 | butyrolactone | 2,066,935 | 2,076,146 | lactonamycin | 5% |
| Region 11 | siderophore | 2,110,497 | 2,123,136 | - | - |
| Region 12 | terpene | 2,269,830 | 2,290,914 | ashimides | 16% |
| Region 13 | butyrolactone | 2,300,561 | 2,311,180 | prejadomycin/rabelomycin/gaudimycin C/gaudimycin D/UWM6/ gaudimycin A | 6% |
| Region 14 | RiPP-like | 2,317,869 | 2,327,714 | streptonigrin | 5% |
| Region 15 | butyrolactone | 2,408,057 | 2,418,502 | - | - |
| Region 16 | NRPS, NAPAA | 2,440,769 | 2,482,644 | stenothricin | 13% |
| Region 17 | siderophore | 2,636,536 | 2,648,477 | - | - |
| Region 18 | thioamitides, RiPP-like | 3,052,710 | 3,074,720 | A-503083 A/A-503083 B/A-503083 E/A-503083 F | 3% |
| Region 19 | terpene | 3,258,980 | 3,279,405 | albaflavenone | 100% |
| Region 20 | NRPS, RRE-containing | 3,416,101 | 3,487,684 | granaticin | 8% |
| Region 21 | T2PKS | 4,177,007 | 4,248,593 | oviedomycin | 95% |
| Region 22 | betalactone | 4,526,013 | 4,553,501 | divergolide A/divergolide B/divergolide C/divergolide D | 6% |
| Region 23 | terpene | 5,147,528 | 5,167,076 | mitomycin | 3% |
| Region 24 | siderophore | 5,623,772 | 5,635,544 | desferrioxamin B/desferrioxamine E | 83% |
| Region 25 | melanin, RRE-containing, phosphonate | 5,736,851 | 5,791,306 | fosfazinomycin A | 78% |
| Region 26 | ectoine | 6,302,336 | 6,311,857 | showdomycin | 52% |
| Region 27 | T2PKS | 6,332,547 | 6,405,060 | spore pigment | 83% |
| Region 28 | terpene | 6,423,141 | 6,443,139 | neocarazostatin A | 100% |
| Region 29 | ectoine | 6,762,458 | 6,772,862 | ectoine | 100% |
| Region 30 | NRPS-like, NRPS, terpene | 7,351,300 | 7,421,452 | enduracidin | 4% |
| Region 31 | T3PKS | 7,458,394 | 7,497,631 | herboxidiene | 7% |
| Region 32 | T1PKS, NRPS, PKS-like | 7,669,936 | 7,736,833 | LL-D49194α1 (LLD) | 3% |
| Region 33 | melanin | 7,963,719 | 7,974,099 | melanin | 57% |
| Region 34 | T1PKS | 8,145,007 | 8,259,932 | sceliphrolactam | 92% |
| Region 35 | T3PKS, lanthipeptide-class-i, T1PKS | 8,397,854 | 8,505,277 | A-47934 | 8% |
| Region 36 | lanthipeptide-class-i | 8,764,849 | 8,790,087 | primycin | 8% |
| Region 37 | butyrolactone | 8,824,305 | 8,835,291 | - | |
| Region 38 | lassopeptide | 8,842,116 | 8,864,247 | SSV-2083 | 36% |
| Region 39 | NRPS, lassopeptide | 8,883,560 | 8,950,374 | achromosin | 100% |
| Region 40 | terpene | 9,046,490 | 9,072,076 | carotenoid | 63% |
| Region 41 | lassopeptide | 9,413,607 | 9,436,052 | - | - |
| Gene ID | Step or Gene Name | KO_id in KEGG Pathway | Fold Change (ΔwblA_24 h/WT_24 h) | Fold Change (ΔwblA_48 h/WT_48 h) | Start | End | Strand | Function |  Up Up  Down Down |
|---|---|---|---|---|---|---|---|---|---|
| ctg1_1366 | step 1 | K00626 | 2.9 | 2.5 | 1701042 | 1702256 | − | acetyl-CoA C-acetyltransferase | |
| ctg1_2557 | step 1 | K00626 | 1.0 | 8.3 | 3127039 | 3128238 | − | acetyl-CoA C-acetyltransferase | |
| ctg1_4516 | step 1 | K00626 | 1.6 | 7.3 | 5304370 | 5305590 | − | acetyl-CoA C-acetyltransferase | |
| ctg1_2421 | step 2 | K00826 | 1.7 | 2.6 | 2966975 | 2968063 | − | branched-chain amino acid aminotransferase | |
| ctg1_1739 | step 3 | K00263 | 0.6 | 0.6 | 2144241 | 2145296 | + | leucine dehydrogenase | |
| ctg1_4276 | step 4 | K01692 | 0.1 | 0.2 | 5031893 | 5032639 | + | enoyl-CoA hydratase | |
| ctg1_5640 | step 4 | K01692 | 0.2 | 0.5 | 6578023 | 6578784 | − | enoyl-CoA hydratase | |
| ctg1_6838 | step 4 | K01692 | 0.1 | 0.3 | 7905240 | 7906040 | + | enoyl-CoA hydratase | |
| ctg1_7415 | step 4 | K01692 | 0.2 | 0.7 | 8560092 | 8560883 | + | enoyl-CoA hydratase | |
| ctg1_4798 | step 5 | K01640 | 6.5 | 3.9 | 5638643 | 5639584 | − | hydroxymethylglutaryl-CoA lyase | |
| ctg1_6290 | step 6 | K01907 | 3.2 | 0.7 | 7305337 | 7307313 | + | acetoacetyl-CoA synthetase | |
| ctg1_6691 | step 7 | K00020 | 0.4 | 1.3 | 7743630 | 7744517 | − | 3-hydroxyisobutyrate dehydrogenase | |
| ctg1_6874 | step 8 | K00128 | 7.7 | 2.1 | 7947480 | 7948802 | + | aldehyde dehydrogenase (NAD+) | |
| ctg1_2035 | step 9 | K01847 | 0.6 | 14.6 | 2497253 | 2499067 | + | methylmalonyl-CoA mutase | |
| ctg1_2036 | step 9 | K01847 | 1.0 | 12.8 | 2499067 | 2501241 | + | methylmalonyl-CoA mutase | |
| ctg1_2543 | step 9 | K01848 | 2.8 | 1.7 | 3114436 | 3116136 | − | methylmalonyl-CoA mutase, N-terminal domain | |
| ctg1_4919 | step 10 | K01965 | 15.9 | 2.0 | 5767295 | 5768551 | − | propionyl-CoA carboxylase alpha chain | |
| ctg1_1275 | PKS1 | * | 90.0 | 7.0 | 1574987 | 1588348 | − | Polyketone synthase | |
| ctg1_1274 | PKS2 | * | 27.4 | 3.7 | 1568736 | 1574990 | − | Polyketone synthase | |
| ctg1_1273 | PKS3 | * | 32.9 | 5.0 | 1557006 | 1568672 | − | Polyketone synthase | |
| ctg1_1272 | PKS4 | * | 37.7 | 8.4 | 1552212 | 1556942 | − | Polyketone synthase | |
| ctg1_1271 | PKS5 | * | 20.3 | 4.2 | 1545995 | 1552108 | − | Polyketone synthase | |
| ctg1_1284 | Orf9 | * | 14.4 | 1.7 | 1599380 | 1599619 | − | ferredoxin | |
| ctg1_1285 | Orf10 | * | 25.7 | 1.5 | 1599659 | 1600816 | − | cytochrome p450 | |
| ctg1_1277 | Orf2 | * | 72.2 | 5.3 | 1590321 | 1591208 | + | dTDP-glucose_synthase | |
| ctg1_1278 | Orf3 | * | 192.0 | 11.3 | 1591278 | 1592276 | + | dTDP-glucose 4,6-dehydratase | |
| ctg1_1287 | Orf12 | * | 39.2 | 4.3 | 1601980 | 1602597 | + | dTDP-4-dehydrorhamnose 3,5-epimerase | |
| ctg1_1280 | Orf5 | * | 2.8 | 1.6 | 1594851 | 1595702 | + | 4-ketoreductase_in_D-allose_pathway | |
| ctg1_1281 | Orf6 | * | 22.3 | 4.1 | 1595710 | 1597002 | − | D-allose_glycosyltransferase | |
| ctg1_1282 | Orf7 | * | 40.3 | 3.8 | 1597061 | 1598248 | − | 2’OH-methyltransferase | |
| ctg1_1286 | Orf11 | * | 40.5 | 3.5 | 1601199 | 1601969 | + | methyltransferase | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Yu, H.; Guan, H.; Li, J.; Zhang, J.; Xiang, H.; Li, J.; Tan, H. Activation of Cryptic Antibiotic Biosynthetic Gene Clusters Guided by RNA-seq Data from Both Streptomyces ansochromogenes and ΔwblA. Antibiotics 2021, 10, 1097. https://doi.org/10.3390/antibiotics10091097
Li Y, Yu H, Guan H, Li J, Zhang J, Xiang H, Li J, Tan H. Activation of Cryptic Antibiotic Biosynthetic Gene Clusters Guided by RNA-seq Data from Both Streptomyces ansochromogenes and ΔwblA. Antibiotics. 2021; 10(9):1097. https://doi.org/10.3390/antibiotics10091097
Chicago/Turabian StyleLi, Yue, Haiying Yu, Hanye Guan, Jingjing Li, Jihui Zhang, Hua Xiang, Jine Li, and Huarong Tan. 2021. "Activation of Cryptic Antibiotic Biosynthetic Gene Clusters Guided by RNA-seq Data from Both Streptomyces ansochromogenes and ΔwblA" Antibiotics 10, no. 9: 1097. https://doi.org/10.3390/antibiotics10091097