
Abstract
Soft materials, such as hydrogels, are used as scaffolds in tissue engineering and regenerative medicine to help tissues regenerate and heal. Recently, supramolecular hydrogels, based on non-covalent interactions, have grown in popularity, especially in the development of materials for biomedical use. Their potential lies in the dynamic, reversible and temporary nature of their crosslinks, which can make them responsive to stimuli, injectable and suitable for 3D printing. Such versatility and processability is important when developing new biomaterials for drug delivery or as implantable scaffolds. The behavior and properties of such hydrogels are different compared to those of chemically crosslinked hydrogels. In this review, we give an overview on supramolecular hydrogels which contain hyaluronic acid (HA) as one of the building blocks. HA is particularly interesting, due to its hydrophilicity, biofunctionality and ease of chemical modification. Specifically, we focus on HA-based hydrogels that make use of hydrogen bonding, hydrophobic interactions, electrostatic interactions, metal–ion coordination and guest–host interactions, and are intended for applications in the biomedical field, with potential for clinical translation.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Hydrogels are very popular soft materials used and investigated for a multitude of biomedical applications, such as contact lenses, drug delivery, tissue engineering and wound dressing [1]. They consist of hydrophilic polymer chains, which constitute a water absorbing 3D polymeric network. The polymer chains are held together by crosslinks, which can be either based on covalent bonds or physical interactions. Properties such as stiffness, mesh size, biodegradability, and swelling can be tuned by changing the polymer components or by using different methods of crosslinking [2]. Hydrogels are often considered biocompatible and can be tuned to have similar properties as the extracellular matrix (ECM) [3].
Chemically (covalently) crosslinked hydrogels have been widely explored, but the permanence of the bonds often results in elastic materials, which cannot handle repetitive mechanical stress [4]. Moreover, such gels are difficult to process (e.g. by extrusion) due to the permanent nature of the crosslinks. For the same reason, the hydrogel network structure is generally not reversible upon changes in the environment (e.g. temperature and pH). Therefore, the introduction of physical, dynamic bonds between polymer chains have gained interest recently. Such temporary bonds (supramolecular interactions) in hydrogels allow for the material's properties to be designed, fine-tuned and customized by means of a 'bottom-up' approach. In this way, viscoelastic and mechanical properties of the hydrogels are matched to those of biological tissues and ECM. Natural tissues are dynamic and do not exhibit perfectly elastic behavior, resulting in a type of material whose mechanical response is both time- and rate-dependent [5, 6]. Such viscoelastic properties can be replicated by making use of supramolecular (reversible) interactions, while at the same time bringing about plenty of other useful and highly sought-after properties. For example, supramolecular hydrogels have the ability to self-heal, which can lead to a longer life-time of the hydrogel [4]. Also, supramolecular gels often possess shear-thinning properties (decrease in viscosity upon increased shear) making them easily injectable and suitable for 3D bioprinting. These properties and tunable viscoelasticity cannot be easily achieved by covalently crosslinked hydrogels, without undergoing significant polymer and/or crosslinker degradation. Overall, compared to covalent hydrogels, supramolecular ones offer greater versatility and better biomimetic properties. There is a range of physical interactions that can be employed to design supramolecular hydrogels, such as hydrogen bonding, guest–host, hydrophobic, ionic, and metal coordination interactions. Most of these interactions are tunable and reversible, giving possibilities to form dynamic materials [7]. These dynamic, supramolecular hydrogels predominantly find their application as injectable drug delivery systems, or as biomaterial scaffolds to facilitate tissue engineering [2, 4].
Supramolecular hydrogels based on either naturally occurring or synthetic polymers have been widely investigated for potential biomedical applications, as reviewed elsewhere [8, 9]. In this work, we focus more closely to one particular type of biopolymer frequently used in hydrogel fabrication and very relevant for the biomedical field—hyaluronic acid (HA), a natural linear anionic polysaccharide, formed by the repeating units of N-acetyl-β-D-glucosamine and β-D-glucuronic acid [10]. HA is a glycosaminoglycan (GAG), present in the ECM of most tissues and most predominantly present in ocular and cartilage tissues. It is a very versatile and popular polymer to use in biomedical applications, due to its biocompatibility, biodegradability, bioactivity, non-immunogenicity, and non-thrombogenicity [10, 11]. It specifically interacts with the CD44 receptor, known to be overexpressed in different cell types during cancer development and progression [12].
One of the key parameters of HA is its molecular weight (Mw). It was found that in the human body it ranges between 4000 and 8 × 106 Da. Consequently, many of the structural and physicochemical properties of HA are highly dependent on its Mw. An increase of HA Mw or the concentration in aqueous solutions leads to an increase of viscosity and pseudo-gelling (structural organization towards an intermolecular polymer network). This is due to chain entanglement and extensive hydrogen bonds formed both intra- and inter-molecularly [13–15]. For example, such network formation is observed with 5 MDa HA at relatively low concentrations (0.1 mg ml−1) [16]. Being an anionic polysaccharide (carboxylate groups), HA's behavior in aqueous solutions is also dependent on the pH and salt concentrations, due to the effect of these parameters on the balance between repulsing and attracting forces of polymer chains. Moreover, rheological properties and viscoelastic behavior of the HA solutions in water are also dependent on polymer Mw [13]. Concisely, at Mw lower than 350 kDa HA molecules are efficiently dispersed in solution, whereas at Mw above 1600 kDa, a polymer network is formed (at 1.0%) [17]. For designing specifically crosslinked hydrogels based on HA, chemical modification of HA and/or use of crosslinkers is often necessary. Therefore, the Mw of HA, the degree of functionalization and the polymer concentration are all key parameters that determine final hydrogel properties. Introduced functionalities on HA can affect the porosity, mechanical strength, and other properties of the corresponding hydrogels to fine-tune the characteristics for the desired biomedical applications. The present review focuses on the research that has been done regarding HA-based hydrogels making use of supramolecular interactions (figure 1). We give readers a complete overview on the current status of HA-based supramolecular gels intended for biomedical applications, specifically hydrogels that are held together by hydrogen bonds, hydrophobic interactions, ionic interactions, metal ion coordination and inclusion complexes.
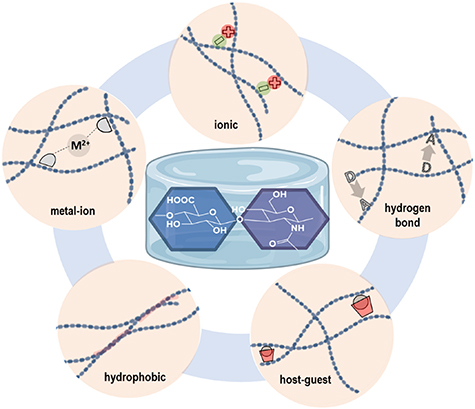
Figure 1. Schematic representation of the HA disaccharide (repeating unit) and different types of physical interactions employed for fabrication of supramolecular hydrogels.
Download figure:
Standard image High-resolution image2. Hydrogen bonding
Hydrogen bonding can be classified as a non-covalent physical interaction. Hydrogen bonding is a specific dipole–dipole interaction, which takes place when there is a donor (an electronegative atom to which hydrogen is attached) and an acceptor (usually a vicinal electronegative atom possessing a lone electron pair) [18]. This type of interaction is short-range and transient. In addition, hydrogen bonding is a highly directional interaction, given that it depends on the geometry of the molecules, and the maximum interaction energy is achieved when there is linearity between donor and acceptor [18]. This interaction is commonly found in biomacromolecules, such as proteins and DNA [19]. In fact, it is one of the most important forces responsible for the secondary and tertiary structures of proteins, as well as for Watson–Crick base-pairing in DNA. Therefore, it is no surprise that intermolecular hydrogen bonding motifs are frequently employed to design and construct reversible supramolecular hydrogels [20–22]. There is a vast range of hydrogen bonding units traditionally used to fabricate such hydrogels, including amides [23, 24], ureido-pyrimidinones [25–27] and ureas [28]. A single hydrogen bond interaction is often too weak to remain intact on relevant time scales due to thermal motions of the molecules. Therefore, in order to create stable and well-defined supramolecular systems, hydrogen bonds present in a single molecule should act cooperatively [29, 30]. A drawback of hydrogen bonding in water-based systems such as hydrogels, is the fact that functional groups responsible for hydrogen bonding can also extensively interact with surrounding water molecules, which in turn weakens the gel. However, it has been observed that by combining hydrogen bonds with hydrophobic interactions in particular, gelation and stability properties are improved, as hydrogen bonding functional groups existing within hydrophobic regions are shielded from the water molecules [26, 31, 32].
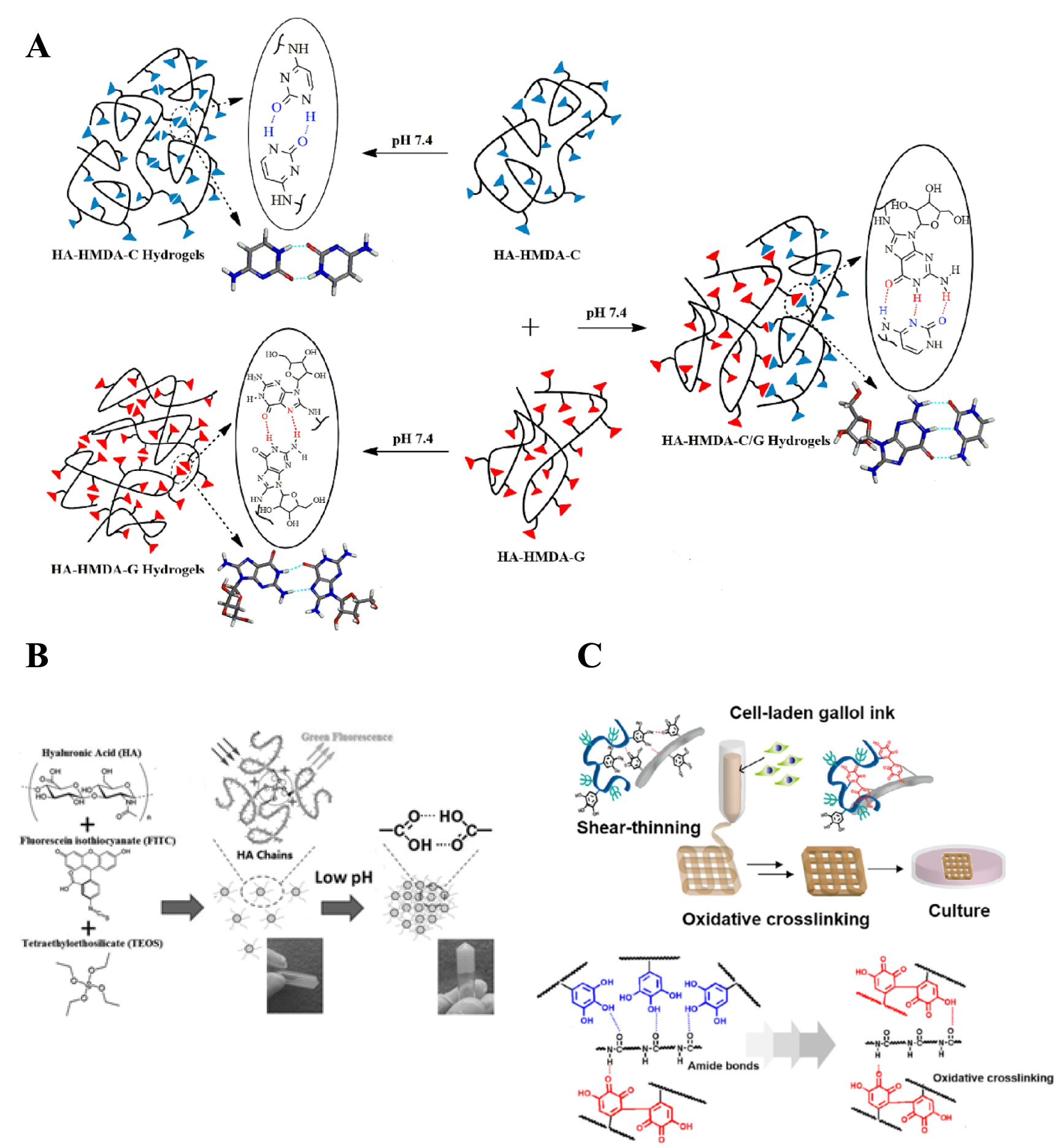
Several supramolecular hydrogels containing HA relying on hydrogen bonding have been reported for biomedical applications, as summarized in table 1. Ye et al reported on the design of an injectable supramolecular hydrogel formed via hydrogen bonding alone [33]. Hydrogen bond crosslinking was based on complementary base-pairing between cytosine and guanosine units (figure 2(A)). In particular, such functionalities were introduced on the HA polymer backbone (200 kDa), using a 1,6-hexamethylenediamine linker. Hydrogels were prepared by mixing the modified HA polymers in PBS at 37 °C. Although gels were formed based on cytosine–cytosine and guanosine–guanosine hydrogen bonds as well, the best candidate in terms of mechanical properties was formed upon mixing cytosine modified HA and guanosine modified HA at 10 wt% (1:1 ratio). The hydrogel was formed within 6 min after mixing and displayed a storage modulus (G') of 100 kPa. All prepared hydrogels were tested for protein delivery applications with bovine serum albumin (BSA) as a model protein. The protein was added to the polymer solution prior to the crosslinking and BSA release was monitored in time after hydrogel formation. All formulations displayed a burst release of 30%–50% in the 1st 12 h, while a sustained release was observed in the following days. Complete BSA release was reached after 1 week, due to the hydrogel dissolution. Therefore, the authors argue that these gels could be suitable platforms for short- and medium-term release (days) of therapeutic proteins. Moreover, such hydrogels exhibited self-healing properties, due to the transient and dynamic character of the physical interactions. For the same reason, these supramolecular hydrogels are also injectable. Depending on the composition and polymer concentration, hydrogels with tunable viscoelastic and injectability properties could be obtained.

Figure 2. (A) Schematic description of the cytosine- and guanosine-modified HA polymers and their assembly into a hydrogel via hydrogen bonding. Reprinted from [33], © 2016 Elsevier Ltd. All rights reserved. (B) Components used to fabricate silica-based nanoconjugates end-capped with HA and containing fluorescein isothiocyanate (FITC) for labeling purposes. At lower pH (6.8) hydrogen bonding in carboxyl dimers are responsible for transition from nanoconjugates solution into a supramolecular hydrogel. [34] John Wiley & Sons. © 2016, John Wiley and Sons. (C) Schematic representation of the 3D printing process of the cell-laden gallol modified HA and gelatin (top); mechanism for the formation of the supramolecular network due to hydrogen bonding and the covalent crosslinking caused by auto-oxidation of gallol moieties (bottom). Reprinted from [35], © 2018 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageTable 1. Examples of supramolecular hydrogels containing HA.
| Type of interactions | Additional components (functional groups) | Application | Figure | Reference |
|---|---|---|---|---|
| Hydrogen bonding | Cytosine, guanosine | Drug delivery | 2(A) | [33] |
| Hydrogen bonding | Silica | Drug delivery | 2(B) | [34] |
| Hydrogen bonding | PVA and Ag | Wound-dressing | — | [36] |
| Hydrogen bonding | Gallol–gelatin | Soft tissue engineering | 2(C) | [35] |
| Hydrogen bonding a | Fmoc-FF | Bioinks | — | [37] |
| Hydrogen bonding a | Fmoc-FFY | Hydrogel coating | — | [38] |
| Hydrophobic | PNIPAM and chitosan | Prevention of post-operative peritendinous adhesion | — | [39] |
| Hydrophobic | PNIPAM and chitosan | Prevention of post-operative peritoneal adhesion | 3(A) | [40] |
| Hydrophobic | PNIPAM and chitosan | Cell delivery and tissue engineering | 3(B) | [41] |
| Hydrophobic | PNIPAM | Cell and drug delivery | — | [42] |
| Hydrophobic | PNIPAM | Nucleus pulposus tissue engineering | — | [43] |
| Hydrophobic | PNIPAM | Nucleus pulposus tissue engineering | 3(C) | [44] |
| Hydrophobic | PNIPAM | Cartilage tissue engineering | — | [45] |
| Hydrophobic | PNIPAM | Drug delivery | 3(D) | [46] |
| Ionic | Carboxymethyl hexanyl chitosan | Drug delivery | 4(B) | [47] |
| Ionic | Chondroitin sulfate | Model CNS tissue for studying drug diffusion | 4(A) | [48] |
| Metal coordination | Doxycycline, Mg2+ | Drug delivery | — | [49] |
| Metal coordination | Zn2+, recombinant peptide and BDNF | Neural cell carrier | — | [50] |
| Metal coordination | Magnesium silicate nanoparticles | Drug delivery | — | [51] |
| Metal coordination | Ca2+ | Bioink for 3D bioprinting | 5(A) | [52] |
| Metal coordination | Ag+ | Wound healing | 5(B) | [53] |
| Inclusion complex | β-CD and 2-NAA-dextran | Tissue engineering scaffold | — | [54] |
| Inclusion complex | β-CD and adamantane | Drug delivery | 6(A) | [55] |
| Inclusion complex | β-CD, adamantane, and peptide tethers | Drug delivery | — | [56] |
| Inclusion complex | β-CD and adamantane | Bioink for 3D printing | 6(B) | [57] |
| Inclusion complex | β-CD and adamantane | Drug delivery | — | [58] |
| Inclusion complex | β-CD and adamantane | Drug delivery | 6(C) | [59] |
| Inclusion complex | β-CD and adamantane, gold nanorods | Drug delivery | — | [60] |
| Inclusion complex | CB[6], DAH, and SPM | 3D cellular engineering | — | [61] |
| Inclusion complex | CB[6] and DAH | Stem cell therapy | — | [62] |
| Inclusion complex | CB[6] and DAH | Cartilage tissue engineering | 7(A) | [63] |
| Inclusion complex | CB[8] and CF | Drug delivery | — | [64] |
| Inclusion complex | CB[8] and CF | Drug delivery | — | [65] |
| Inclusion complex | β-CD and azobenzene | Drug delivery | 7(B) | [66] |
| Inclusion complex | β-CD and azobenzene | Wound healing | — | [67] |
a Supramolecular hydrogels are made by hydrogen bonds, π–π stacking and hydrophobic interactions.
A system for the delivery of anticancer drugs from a HA-based supramolecular nanocomposite hydrogel was developed by Chen et al [34]. Specifically, nanoconjugates based on HA (10 kDa) and silica were fabricated by a base-catalyzed condensation reaction of tetraethoxysilane in the presence of cetyltrimethylammonium bromide as surfactant. This way, mesoporous silica nanoconjugates were obtained, end-capped with HA, with a pore size within the silica core of around 2 nm and diameter of the spherical nanoconjugates of ∼200–300 nm. The role of HA was to enable hydrogel formation at pH values of 6.8. Specifically, these nanoconjugates aggregated and grew in size (up to 10 000 nm), leading to network formation as a result of intermolecular hydrogen bonding of HA. The authors showed that by decreasing the pH, carboxyl dimers were formed due to the partial protonation of HA. These intermolecular hydrogen bonds were responsible for the formation of the hydrogel network (figure 2(B)). The transition between nanoconjugate solution and hydrogel was reversible and pH dependent. The anticancer drug doxorubicin was loaded within the porous silica core and in vitro release studies were performed at pH of 6.8 and in the presence of hyaluronidase. A sustained release was observed for several days, with 80% of the drug released at day 7. The authors propose that due to the specific interaction between HA and CD44 receptor, which is over-expressed in many tumor cells, the specificity can be achieved. Slightly acidic environment of tumor tissue triggers the gelation due to the pH-responsive hydrogen bonds. Upon enzymatic degradation of the gel, nanoconjugates are released and subsequently internalized by tumor cells. Eventually, by further degradation of the nanocarrier inside the cells, the loaded drug is released. Furthermore, in vitro cellular experiments showed drug release in human breast cancer cells, whereas in the relative control groups (non-cancerous cells) no drug was released.
Besides drug delivery, hydrogen bond-crosslinked HA hydrogels were also explored for wound dressing applications. A nanocomposite hydrogel composed of PVA (polyvinyl alcohol) and silver (Ag) nanoparticles was reported by Zhang et al, in which HA (1200 kDa) was introduced because of its biocompatibility and swelling properties [36]. The hydrogels were prepared by a freeze-thaw method, known to induce formation of microcrystalline domains in PVA, which act as the crosslinks [68]. In fact, aqueous solutions of HA and PVA in which the content of HA was varied from 0 wt% to 33 wt% were mixed at 60 °C, then frozen at −20 °C and finally allowed to thaw at room temperature. After three freeze-thaw cycles, lyophilization yielded water-free networks that formed supramolecular hydrogels upon rehydration. It was observed that upon increasing HA concentration, crystallinity of PVA in the rehydrated gels decreased, suggesting that hydrogen bonding interactions between PVA and HA were formed. Therefore, the weakening of the hydrogen bonds in the crystalline domains of PVA was counteracted by the formation of additional hydrogen bonds between HA and PVA [36]. HA/PVA hybrid hydrogels were then embedded with silver nanoparticles (size 20–50 nm) by UV-initiated photoreduction of included AgNO3. Antibacterial properties of the resulting hybrid nanocomposite hydrogels were assessed via disk diffusion method against E. coli. The results showed that HA/PVA/Ag hydrogels indeed displayed high antibacterial properties, compared to the HA/PVA and PVA control hydrogels. Therefore, considering good antibacterial properties of these gels, the authors argue their suitability as potential wound-dressing materials.
Other than described above, hydrogen bonding HA-containing supramolecular hydrogels have also been studied for 3D printing of constructs for tissue engineering applications. An example is given by the hydrogel ink based on gallol-modified HA and gelatin synthesized by Shin et al [35]. Here, the authors made use of the hydrogen bonding between gallol units. HA (151–300 kDa) and gelatin were modified with 5-hydroxydopamine via EDC/NHS coupling, resulting in gallol content of around 10%–15% as determined by means of H-NMR and Folin–Ciocalteu phenol assay [69]. Hydrogels were formulated at different concentrations and at different ratios between HA and gelatin. In general, by mixing the gallol-modified polymers, self-assembly takes place, resulting in the formation of a hydrogel within minutes (under physiological conditions) governed by hydrogen bonding formed between gallol units, as well as between gallol and protein backbone of gelatin (figure 2(C)). Such a gel initially exhibits dynamic character, due to hydrogen bonds, which are then replaced by covalent crosslinks when gallols undergo auto-oxidation. Therefore, a short-term supramolecular hydrogel becomes stabilized in time (stabilization by oxidation) (figure 2(C)). Immediately after the gelation, due to reversible hydrogen bonds, the hydrogels display shear-thinning behavior, which makes them suitable for 3D extrusion-based printing. For example, 3D printing was successfully achieved (at 6 wt%) with encapsulated fibroblasts resulting in high cell viability (95%). Therefore, the present HA and gelatin based bioink seems to be a promising approach to fabricate constructs for soft tissue engineering applications, due to the fast and efficient hydrogen bonding self-assembly, with subsequent covalent crosslinking and stabilization.
For ease of discussion, we include in this section a specific type of supramolecular gels, based on short peptides which undergo gelation through efficient β-sheet formation, a synergistic effect between hydrogen bonds, π–π stacking and hydrophobic interactions. Such peptides were not chemically coupled to HA, but rather HA was added as an additional component in the peptide solution, prior to gel formation. It is noted however, that the resulting hydrogels are not based solely on hydrogen bonding. A specific type of a hybrid supramolecular system, given by HA and dipeptide fluorenylmethoxycarbonyl-diphenylalanine (Fmoc-FF), was developed by Nadernezhad et al [37]. Fmoc-FF peptides create a network through β-sheet formation [70]. The authors observed that the peptide fibrous network was reinforced by the presence of HA chains (388 and 765 kDa), most likely due to the stabilizing effect of HA by hydrogen bonding. However, by increasing HA Mw (984 kDa) a weaker network was formed. This is a result of increased solution viscosity, as well as additional physical interactions with the peptide, leading to altered self-assembly process of the fibrous peptide network. Therefore, there is an interplay between the two components, directly affecting the gelation process and the resulting hydrogel viscoelastic properties. Such a system acts as a suitable platform for further development of supramolecular hydrogels with tunable printability and injectability. Although it was not the goal of this study, authors speculate that HA/Fmoc-FF hybrid supramolecular hydrogels could potentially be used as bioinks for 3D biofabrication processes and regenerative medicine applications.
Another example of a hybrid supramolecular hydrogel between HA and a short peptide hydrogelator is given by the study of Rodon Fores et al [38]. Specifically, an enzyme-assisted self-assembly method was employed, where an enzyme produces a hydrogelator in situ, starting from a precursor. A water-soluble tripeptide Fmoc-FFpY (Y = tyrosine; p = phosphate group) was used as a precursor, which upon enzymatic dephosphorylation by alkaline phosphatase (AP), becomes less hydrophilic (Fmoc-FFY) and thus undergoes a self-assembly process and hydrogel formation (β-sheets). Additionally, HA (406 kDa) was incorporated at different concentrations (2–10 mg ml−1) in the peptide solution and hydrogel formation was evaluated. It was seen that hybrid supramolecular hydrogels could be formed upon AP addition. However, the gelation time increased and the stiffness of the resulting gels decreased as HA concentration went up. Upon fibrous network formation, HA formed additional physical interactions with the network, leading to less effective entanglements of the peptide fibers. Moreover, the authors successfully formed micrometer-thick hybrid hydrogel layers based on the localized enzyme-assisted self-assembly method [71, 72]. In this way, by bringing the precursor solution into contact with the surface of a polymer multilayer film with absorbed enzyme, a hydrogel layer was formed. Consequently, this approach holds promise for hydrogel-based coating applications, i.e. to prevent protein adsorption. Finally, it was shown that fibroblasts seeded on top of the hydrogel layers were able to adhere and spread, indicating cytocompatibility of the supramolecular hybrid hydrogel.
3. Hydrophobic interactions
Hydrophobic interactions are a specific type of noncovalent interactions, which as opposed to hydrogen bonding are less geometrically constrained and thus non-directional. They are short-range attractive interactions, typical of nonpolar molecules and responsible for their clustering in aqueous environment. This way, the amount of ordered water molecules on the surface is minimized, thus excluded from the solvation shell [73]. Therefore, the hydrophobic effect is primarily driven by entropy and as such is intrinsically temperature sensitive. The hydrophobic effect is pivotal in different biological systems. For example, it is manifested in many biological processes, including protein folding and formation of lipid membranes [74–77].
Various hydrogels based on solely hydrophobic interactions have been made [78, 79]. An overview of such hydrogels, along with their intended use, is included in table 1 and those will be further discussed below. Generally, the polymers that undergo gelation in aqueous environment by hydrophobic interactions are amphiphilic, possessing both hydrophobic and hydrophilic segments. Gelation is driven by a self-assembly process via hydrophobic interactions between the nonpolar units, which, like other physical interactions, are also reversible and dynamic. Supramolecular hydrogels containing such hydrophobic domains exhibit certain advantages over other types of physical interactions. For example, such gels could be employed for solubilizing and loading apolar molecules, showing promising applications in drug delivery of poorly water-soluble drugs [80–82]. Amphiphilic polymers often display thermo-responsive behavior and the temperature dependent hydration of these polymers can be tuned by adjusting the ratio between the hydrophilic and hydrophobic segments [83, 84]. In case of HA, thermosensitive units have been commonly introduced to produce hydrogels, the majority of them relying on the use of thermo-responsive polymers such as poly(N-isopropylacrylamide) (PNIPAM). PNIPAM is one of the most often used thermo-responsive polymers that undergoes a reversible sol–gel transition at physiologically relevant temperature, considering its lower critical solution temperature (LCST) of around 32 °C. HA is usually coupled to PNIPAM in order to form amphiphilic polymers, where HA imparts hydrophilicity and swelling capacity to the resulting hydrogels and the thermo-responsive properties of PNIPAM are responsible for the self-assembling.
In the study of Chou et al, supramolecular hydrogels based on polymer of PNIPAM, chitosan and HA (1800 kDa) were prepared for post-operative peritendinous adhesion prevention during tendon surgery [39]. In detail, PNIPAM end-capped with a carboxyl group was synthesized by free radical polymerization, subsequently reacted with chitosan via EDC/NHS coupling, and HA was finally grafted on the PNIPAM-chitosan copolymer by means of EDC/NHS chemistry. The final polymer (abbreviated as HACPN) consisted of 81.9 w/w PNIPAM, 12.6 w/w chitosan and 5.5 w/w HA. HA was primarily used because of its water retention properties to increase hydrophilicity of the system and lubrication. In addition, the prepared HACPN-based solution was injectable (below its LCST), and at physiological temperature, a physical hydrogel network was formed. To assess the capacity to prevent post-operative tendon adhesion, the authors showed by in vivo studies using a rabbit animal model that the HACPN hydrogels provided a barrier to prevent peritendinous adhesion while maintaining the intratendinous strength. Compared to a commercial product Seprafilm®, the HACPN thermo-responsive hydrogel displayed better performance, including anti-adhesion and biomechanical properties [39].
The same group of authors also investigated the use of HACPN for prevention of post-operative peritoneal adhesion. In vivo studies in rats (sidewall abrasion model) showed that the hydrogel based on HACPN proved to be an effective barrier, preventing migration of fibroblasts and providing superior anti-adhesion properties, compared to a PNIPAM-based gel and untreated control groups (figure 3(A)) [40]. Therefore, such an injectable hydrogel has potential for intra-abdominal applications. Additionally, the same material was investigated for cartilage and meniscus tissue engineering applications. In particular, HACPN (figure 3(B)) hydrogels proved to be suitable scaffolds for rabbit articular chondrocytes and meniscus cells, showing beneficial effects on cell proliferation and phenotypic morphology, compared to chitosan-g-PNIPAM hydrogel [41].
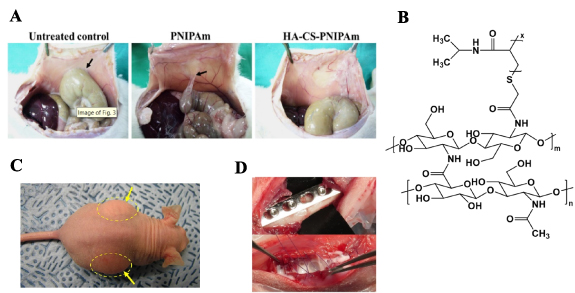
Figure 3. (A) Evaluation of post-operative peritoneal adhesion prevention upon application of thermo-responsive HACPN hydrogel. Control groups (no treatment and only PNIPAM hydrogel) allowed adhesion between peritoneum and cecum to take place. After applying HACPN hydrogel no adhesion was observed. Black arrows indicate the adhesions. Reprinted from [40], © 2017 Elsevier Ltd. All rights reserved. (B) Chemical structure of polymer containing HA, PNIPAM and chitosan (HACPN). PNIPAM segments are responsible for hydrophobic associations, which lead to supramolecular hydrogel network formation [41]. (C) HA-PNIPAM hydrogels injected subcutaneously in athymic nude mice for testing material's biocompatibility. Yellow errors and circles indicate sites where the hydrogel was injected. Reprinted from [44], © 2009 Elsevier Ltd. All rights reserved. (D) Application of gentamicin-loaded HA-PNIPAM hydrogels for protection against bacterial infections. The images show osteotomy surgery procedure in rabbits. The bottom image highlights the presence of the in situ formed hydrogel containing gentamicin. Reprinted from [46], © 2016 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageMortisen et al explored the use of HA-PNIPAM based hydrogels for cell encapsulation and drug delivery applications [42]. PNIPAM was coupled to HA (502 kDa) in order to introduce physical crosslinks via hydrophobic association between PNIPAM segments at physiological temperature. The HA-PNIPAM copolymer was prepared in two steps. First, propargylamide was attached to the carboxylic acid groups on the HA by means of EDC/NHS chemistry and then the copper-catalyzed azide-alkyne cycloaddition reaction was performed between propargyl-HA and azide-PNIPAM that was separately prepared by RAFT polymerization method. The grafting via 'click' chemistry led to a PNIPAM substitution degree of 25%–30%. The same chemistry can be used to 'click' fluorescent or small bioactive molecules in order to further broaden the application of the obtained hydrogel materials. The prepared HA-PNIPAM solutions displayed low viscosity at 20 °C as well as shear thinning behavior, which are suitable properties for suspending cells and injection. The gelling of the polymer solution occurred within minutes at 37 °C (above polymer LCST). The hydrogel degradation products (upon treatment with hyaluronidase) displayed good cytocompatibility towards fibroblasts after incubation at 24 and 48 h. Overall, the HA-PNIPAM hydrogel seems a versatile platform suitable for carrying cells and incorporating small bioactive molecules.
The same research group explored the use of this HA-PNIPAM hydrogel for soft tissue engineering purposes, in particular, for intervertebral disk regenerative therapies [43]. The hydrogel was assessed for its compatibility with nucleus pulposus tissue. Both HA-PNIPAM copolymers and hydrogel degradation products (upon treatment with hyaluronidase) were cytocompatible with nucleus pulposus cells (NPCs). In addition, NPCs were viable after extrusion of cell-laden hydrogels through a 22 G needle, confirming the hydrogel suitability as injectable cell carrier. Finally, NPCs viability was tested in a whole-organ culture model and the results showed that upon introducing the cells in a nucleus pulposus defect site, cell viability of 90% was observed for over a week.
Other hydrophobically assembled HA-PNIPAM hydrogels have been used for tissue engineering applications. Tan et al developed a supramolecular hydrogel based on HA (1600 kDa) and PNIPAM for soft tissue engineering applications [44]. HA was functionalized with adipic acid dihydrazide by means of EDC/HOBt coupling reaction, while PNIPAM-COOH was separately synthesized by free radical polymerization of NIPAM monomers, using 4,4ʹ-azobis(4-cyanovaleric acid) as an initiator. PNIPAM-COOH was subsequently grafted onto hydrazide-HA in the presence of EDC, to reach weight contents of PNIPAM in the copolymers of 28% or 53%. As in the examples reported above, PNIPAM containing polymers were able to form hydrogels when heated above their LCST (30 °C). These hydrogels supported the survival of embedded human adipose-derived stem cells. Besides, in order to determine the material's applicability for tissue engineering purposes, in vivo biocompatibility tests were done by implanting the gels in the dorsal subcutaneous region of mice (figure 3(C)). The results confirmed the in situ formation of physically crosslinked gels upon injection, with no negative effects observed after 5 d. Overall, the authors suggest potential applications of such hydrogels in adipose regeneration and tissue engineering. More recently, the same material was studied by D'Este et al for treating osteochondral defects [45]. A rabbit model was used and the HA-PNIPAM solution was injected in a defect site. The physical hydrogel was formed within seconds and it remained stable in situ, without being displaced or washed out. The results from the animal studies demonstrated that, compared to non-treated control groups, hydrogels displayed high healing response, with no detected local or systemic toxicity. However, these hydrogels exhibit a storage modulus of only a few 1000 Pa, which indicates that the mechanical performance of the gels might not be sufficient for cartilage engineering purposes.
During implant surgeries there is a high risk of infections associated with implants. Therefore, HA-PNIPAM thermo-responsive physical gels have been investigated as reservoirs of antibiotics [46]. In this typical example, amine-terminated PNIPAM was first made by free-radical polymerization, and HA (170.6 kDa) was converted into a less water-soluble HA-tetrabutyl ammonium salt (HA-TBA). Next, the amine-terminated PNIPAM (Mn of 11 kDa) was grafted on the HA-TBA in DMSO using 1,1ʹ-carbonyldiimidazole as a coupling agent. The degree of substitution was determined to be ∼15%. In this study, the gels were used as injectable carriers of gentamicin (an aminoglycoside, broad-spectrum antibiotic). For the in vitro release studies, antibiotic encapsulation was achieved by dissolving the polymer and the drug together in PBS, then injecting the resulting solution in pre-warmed PBS at 37 °C to allow immediate formation of physically crosslinked hydrogel spheres. The results from the release studies showed a burst release of 47% of the initial drug amount after one hour, while complete release was reached after 7 d. This gentamicin-loaded HA-PNIPAM hydrogel was designed for complex trauma procedures, to prevent bacterial infections. Moreover, the authors made use of a rabbit humerus model (mimicking a real trauma case) to investigate the efficacy of antibiotic delivery in vivo (figure 3(D)). The results demonstrated that the HA-PNIPAM hydrogel offered similar prophylactic properties against S.aureus as compared to an existing clinical product (collagen fleece), but with the advantage of being more versatile and possessing better distribution through complex wounds. Such a system seems efficient for local delivery of high concentrations of antibiotics, providing protection against infections in implant surgeries.
The above-discussed hydrogels typically exhibit shear thinning behavior, meaning the viscosity decreases when a shear force is applied, making them injectable [41]. These properties make HA-PNIPAM based hydrogels easy to handle and suitable for biomedical applications. It is evident that several authors showed quite promising data on applicability of the injectable thermo-responsive hydrogels, which potentially could have success in translation to clinics [39, 40]. A possible drawback is related to the non-biodegradability of PNIPAM segments. However, it was shown that if the Mw is low, renal excretion is one of the possible ways to remove PNIPAM fragments from the body [85].
4. Ionic interactions
Ionic (electrostatic) interactions can be an interesting approach for developing supramolecular hydrogels because, they generally have fast kinetics, and are responsive to changes in environment, such as salt concentration, temperature and pH [86]. HA can be used for hydrogels based on these interactions, as it has negatively charged carboxyl groups at physiological pH, which can form complexes with positively charged molecules [47]. An overview of such hydrogels is included in table 1. These interactions have been explored by Tabet et al in combination with chondroitin sulfate (CS) and BSA to model central nervous system tissue [48]. Although the overall charge of BSA is negative at physiological pH, it has cationic binding pockets, which can bind with anionic GAGs like HA and CS. BSA has only one available binding area when it is in its folded form, but when heated (80 °C, 2 h), it can interact with multiple polysaccharide chains (figure 4(A)). The interactions between BSA and CS/HA were first explored separately, with 10 wt% of CS and 50 mg ml−1 BSA being vigorously mixed to form nanoparticles. Gels were formed when 1 wt% HA (1500–1800 kDa) and heated BSA at different concentrations were mixed. When the BSA was not heated, no gels were formed. In order to obtain model CNS tissue and to study diffusion of drugs from a delivery system into this model tissue, dry CS and BSA were mixed together, and then added to HA in solution. To study the diffusion properties, a hydrophobic poly(caprolactone) blend with rhodamine B was placed on top of the HA/CS/BSA gels. The diffusion of rhodamine into the model tissue was observed with UV/vis spectroscopy. The HA/CS/BSA supramolecular gel provided a new and improved way of studying drug diffusion into a hydrophilic environment, compared to the standard diffusion into saline, as it mimics the tissue more accurately.
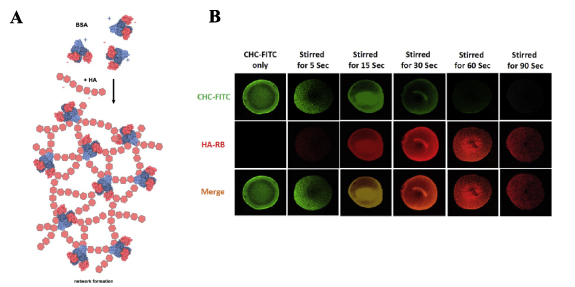
Figure 4. (A) Schematic of the network formation between partially denaturated BSA and HA. Positively charged peptide domains (blue) of BSA show higher affinity towards negatively charged HA chains. Reproduced from [48]. CC BY 4.0. © 2019 Tabet A et al. (B) FRET imaging analysis of the formation of nanoparticles from micelles through stirring of CHC-FITC (green) and HA-RB (red) and the formation of hydrogels. CHC-FITC micelles emit green fluorescence. When interacting with HA-RB, green fluorescence is quenched, as the HA and CHC form colloidal particles. This is represented by the increased red fluorescence when combining the two. The red fluorescence decreases after 60 s, when the two become 'glued' together and form supramolecular gels, indicating that the number of assembled nanoparticles is reduced in the suspension. Reprinted from [47], © 2018 Elsevier Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageA new way to form hydrogels is through micelle aggregation, which can 'glue' polymers, such as HA, into a hydrogel. For example, micelle-forming carboxymethyl hexanyl chitosan (CHC) can form gels in the presence of low Mw HA (LMW HA 15–30 kDa), through electrostatic polymer-nanoparticle interactions [47]. To form such hydrogels, 4 wt% CHC and 4 wt% LMW HA were mixed and stirred in DI water. Shear stresses due to stirring helped the CHC particles to overcome the repulsion barrier keeping them apart, allowing their aggregation and thus gelation due to inter-particle bonds. With increasing shear stress, the storage modulus increased, therefore the rheological behavior of the gels was dependent on the amount of shear stress the mixture was subjected to during mixing. The shear-induced gelation of colloidal CHC particles was improved by HA acting as inter-colloidal crosslinker. Gel formation was confirmed by fluorescent imaging using a mixture of CHC labeled with FTIC, and HA labeled with rhodamine: by the increasing Förster resonance energy transfer over time, the transition from nanoparticles to gels was observed (figure 4(B)). Gels were stable when immersed in pH 5 and 6 but lost weight (i.e. degraded) in pH 7.4 over time, due to less dense packing of the particles and HA, while addition of lysozymes and hyaluronidase further increased degradation rates. To test the use of this hydrogel as a drug delivery vehicle in cartilage, berberine was incorporated into the gel through electrostatic interactions. Berberine decreases sodium nitroprusside (SNP)-induced apoptosis of chondrocytes and protects against cartilage degeneration. Therefore, human chondrocyte primary cells were used to test the protective effect of berberine on chondrocytes. When chondrocytes were exposed to 0.75 mM SNP for 3 d, the apoptotic rate was 54.5%, when 0.75 mM berberine was added, this dropped to 41.5%, and in the case where chondrocytes were exposed to CHC/HA gels with berberine incorporated, apoptotic rate dropped to 32.8%. These experiments showed a significant protective effect of the berberine encapsulated gel, and the potential of incorporating drugs in HA hydrogels through electrostatic interactions for drug delivery.
5. Metal ion coordination
Metal-coordination bonds are a non-covalent type of interaction formed between a ligand and a metal ion. Coordination bonds are highly directional, in contrast to electrostatic interactions, considering their dependence on both the geometry of the ligand and the coordination preferences of the central metal cation [87]. In metal-coordination chemistry, a bond is formed when an electron pair from a ligand is donated to a metal ion. In addition, when the same ligand has two or more binding sites with the same metal ion, multiple coordination bonds are formed, and the resulting interaction is referred to as chelation. The strength of the coordination bonds varies a lot and it depends on the nature of the metal ion, as well as on the type of the ligand [88]. Being non-covalent, these bonds are reversible and thus can be useful to design supramolecular hydrogels. The transient character and the timescales of these bonds are primarily determined by the metal-ligand exchange kinetics [89, 90]. Chelating groups can be attached onto a polymer backbone and physical hydrogels can be formed in the presence of metal ions in solution. Some of the most frequently used chelators are histidine, pyridine (also bipyridine and terpyridine), catechol and bisphosphonate (BP) [91]. In order to fabricate supramolecular hydrogels, a variety of bi- and tri-valent metal ions are commonly employed, such as Cu2+, Ba2+, Ca2+, Mg2+, Fe2+, Ru2+, Co2+, Fe3+, Al3+, Ru2+ [91–93]. It should be noted that the presence of metal ions in such materials might cause toxicity depending on the choice of metal ion. Although the majority of the mentioned metals are essential in humans (e.g. in structural factors and cofactors in many biochemical processes), the amount of used metal ions in hydrogel fabrication should be carefully evaluated, as dose-dependent toxic effects might arise [94].
Coordination chemistry has an important role in biological systems by providing structural integrity of metalloproteins. Coordination bonds are also present in mussel's byssal thread, and their cooperative way of binding is responsible for structural support and adhesion properties [95, 96]. These examples from nature have served as inspiration for scientists to make use of coordination interactions for the design of supramolecular hydrogels, to prepare functional materials with tunable, versatile and dynamic properties. HA is one of the polymers that have been used to design supramolecular hydrogels by means of coordination bonds. A summary of such hydrogels, alongside with their biomedical applications is reported in table 1.
One of the most frequently used polysaccharides for fabricating physical gels is alginate. This biopolymer forms hydrogels upon complexation with Ca2+ ions and has been extensively studied. It has shown to be biocompatible with low toxicity, making it suitable for biomedical applications [97]. However, in the present review, we focus only on HA hydrogels. Therefore, the readers interested in alginate are referred to other published material [97–99]. One of the first examples of a HA-based supramolecular hydrogel with metal coordination bonds was described by Miyazaki et al [49]. The authors developed a hydrogel system starting from a HA polymer (1600 kDa), crosslinked by doxycycline (an antibacterial drug) in the presence of a bivalent metal cation. In more detail, doxycycline contains a positively charged dimethylammonium moiety, which interacts with negatively charged carboxylate groups on HA backbone. In addition to those electrostatic interactions, doxycycline possesses a phenolic diketone functionality that, in the presence of bivalent ions such as Mg2+, allows chelation between two of the doxycycline molecules, creating crosslinks and giving rise to a physical network. The minimal concentration of HA required to form the hydrogel was around 0.05 wt%. The authors observed that in order to form a stable hydrogel, potentially useful as a drug delivery system of doxycycline, an equimolar amount of doxycycline and Mg2+ was necessary. In addition, the release profiles of doxycycline were heavily dependent on the drug to HA ratio. If HA disaccharide units and doxycycline were present in equimolar amounts, doxycycline exhibited a zero-order release profile, with drug release being governed primarily by gel erosion. On the other hand, gels that contained more drug than HA displayed Fickian diffusion release due to the diffusion of the non-bound drug. However, it is noted that in this study the release experiments were not conducted under physiological pH and temperature conditions, therefore the actual drug delivery application might not be fully translatable to an in vivo environment.
HA hydrogels based on metal ion-coordination have also been studied as cell carriers, for neural transplantation therapies. Nakaji-Hirabayashi et al developed a supramolecular hydrogel based on HA and recombinant peptides making use of metal coordination bonds [50]. Specifically, HA was first chemically modified in the presence of EDC/NHS to include N-(5-amino-1-carboxypentyl) iminodiacetic acid (NTA), a known chelating agent for Zn2+ [100]. Subsequently, ZnSO4 was added to the HA-NTA solution to achieve Zn2+ chelation. A peptide containing hexahistidine regions at both termini, as well as brain-derived neurotrophic factor (BDNF) carrying a histidine at C-terminus were prepared by recombinant DNA methods. Thanks to the histidine moieties, both BDNF and the peptide were able to coordinate with Zn2+ present on HA and formed stable chelates. The peptide with hexahistidine was the crosslinker, used to provide network formation with HA, whereas BDNF-histidine was tethered to HA. Thus, a stable supramolecular hydrogel was formed at a HA concentration of 0.6 w/v% and at a peptide concentration of 150 mg ml−1. It was observed that BDNF was stably interacting with Zn2+ and that only 33% of the loaded amount was released in 12 d. The control group, which was Zn2+ deficient released 90% of BDNF within 3 d. To test cell viability in the gels, the authors incorporated neural cells in the gels with and without BDNF and viability was studied for 3 d. In the BDNF containing gels cell survival was above 60% after 3 d, whereas in the BDNF deficient group the viability was only 23%, suggesting that BDNF is indeed crucial for promoting neural cell survival in the hydrogels. The authors speculate that the observed low cell viability could be due to poor cell adhesion and accelerated apoptosis, but further studies are necessary to fully understand cell behavior in these gels. However, in order to further test applicability of the described system, hydrogel should be tested with regards to the enzymatic degradation by hyaluronidase in vivo.
Nanocomposite supramolecular hydrogels based on HA and silica nanoparticles, crosslinked via metal coordination bonds were studied for drug delivery purposes by Shi et al [51]. Such hydrogels were prepared from a BP modified HA (150 kDa) and magnesium silicate nanoparticles (MgSiO3). HA-BP was synthesized in a two-step reaction. First, a thiol containing side chain was grafted via EDC mediated coupling and subsequently an acrylated BP reagent was coupled to HA-thiol via a thiol-ene photo-initiated addition reaction. The degree of substitution of BP was found to be ∼22.5%. Magnesium silica nanoparticles (∼350 nm) were prepared via a hydrothermal treatment of silica nanoparticles with ammonia in the presence of MgCl2. Supramolecular hydrogels were fabricated by mixing HA-BP and MgSiO3 particles in water, due to the formation of coordination bonds between Mg2+ ions present on the particle's surface and BP ligands. In addition, considering that MgSiO3 nanoparticles are very porous, they can be loaded with therapeutic drugs. In this study, the nanoparticles were loaded with doxorubicin as a model drug, with a loading efficiency of 80%. Therefore, the role of MgSiO3 was two-fold, acting both as crosslinker and drug carrier. Interestingly, the release studies showed that the free doxorubicin was not readily released from the hydrogel nor from the free nanoparticles. The authors argue that the observed anticancer effect with cells was due to the cellular uptake of loaded nanoparticles and further release of the drug within cells. However, the aspect of drug release from the described system requires additional investigation. Nonetheless, the system seems to have a promising role as a drug delivery platform. Being based on dynamic bonds, these gels exhibit shear-thinning and self-healing properties, which are beneficial for processing, injectability, ease of fabrication and versatility of such materials.
Hydrogels based on coordination bonds can be useful in 3D printing applications and the development of novel bioinks, considering that such hydrogels are often characterized by dynamic, shear-thinning and self-healing properties, as seen with other non-covalent interactions. In the study conducted by Shi et al, HA-BP (HA 150 kDa) was used in combination with Ca2+ ions to form supramolecular hydrogels based on metal ion-ligand dynamic coordination bonds to be used as a bioink (figure 5(A)) [52]. Such hydrogels displayed dynamic properties, which allowed for their extrusion during so-called free directional printing, i.e. a method that allows for extrusion of a hydrogel within a support hydrogel bath made of HA-BP and Ca2+ in order to create 3D structures. In addition, the extruded HA was functionalized with side acrylamide functionalities, to allow for chemical crosslinking of the 3D constructs by UV light in order to provide better structural and mechanical integrity after printing. Finally, in order to isolate the fixed 3D structure from the hydrogel bath, all coordination bonds between BP and Ca2+ were disrupted by placing the entire construct in acidic PBS (pH 5.0) (figure 5(A)). In this way, the support hydrogel bath was completely dissolved, whereas the printed and fixed hydrogel structure remained intact due to the covalent crosslinks. Osteoblast-like cells were encapsulated in the hydrogel and their viability was determined for the metal coordination crosslinking process and subsequent photochemical fixation step. The results showed that the cells displayed viabilities above 88% under all tested conditions, which suggests that the described system is suitable for hydrogel bioprinting applications.

Figure 5. (A) Schematic representation of the hydrogel network resulting from metal coordination bonding between Ca2+ ions and BP functionalities on HA (left). Free directional printing method: 1st step involves the printing of the physical hydrogel, resulting from Ca2+ and BP coordination bonding, within a support bath hydrogel. The next step comprises UV-induced crosslinking of the acrylamide groups present on the polymer chains of the printed structure to achieve structure fixation. Finally, by disassembling the hydrogel bath in acidic PBS, the 3D printed structure can be isolated (right). Reprinted with permission from [52]. Copyright (2017) American Chemical Society. (B) Schematic representation of the synthesis of the HA-BP derivative via Michael addition reaction between maleimide and thiol groups (left). Schematic depiction of the hydrogel formation upon introduction of AgNO3, by means of metal coordination bonds between Ag+ ions and BP groups present on HA (right). [53] John Wiley & Sons. © 2017, John Wiley and Sons.
Download figure:
Standard image High-resolution imageThe same group of authors also investigated the potential use of metal coordination self-assembled supramolecular hydrogels based on HA for antibacterial and wound healing purposes [53]. In this case, HA (150 kDa) was functionalized with a BP side group using a different approach. First, maleimide was inserted on HA by functionalizing with N-(2-aminoethyl)maleimide by EDC coupling, and BP-acrylamide was functionalized with dithiothreitol to introduce thiol moieties. The final HA-BP polymer was prepared by a chemoselective Michael addition reaction between HA-maleimide and thiol groups present on BP, with a BP degree of substitution of ∼20% (figure 5(B)). In contrast with the previous studies, where the authors used Mg2+ and Ca2+ ions to form supramolecular hydrogels of HA-BP, in this study Ag+ was used. Silver is a well-known antibacterial agent [31], which is an important aspect when considering applications such as wound healing. Supramolecular HA hydrogels were instantaneously formed upon simply mixing HA-BP and Ag(NO3) aqueous solutions, thus forming coordination crosslinking between BP and Ag+ ions (figure 5(B)). The final concentration of HA-BP was 3 w/v%, using a 1:1 molar ratio between BP and Ag+. As seen with the previously described HA-BP hydrogels, in the present system one of the main aspects was the reversible, dynamic nature of the metal coordination bonds, resulting in the injectability, shear-thinning and self-healing properties of the supramolecular hydrogels. These properties make the gels easy to process and apply (i.e. inject or print). For wound dressing applications, the gel could be conveniently introduced in irregular wound sites. A disk diffusion method showed that the gels exhibited antibacterial properties against Gram-positive and Gram-negative bacteria, compared to relative controls, confirming its potential as wound dressings. Therefore, in this system, the Ag+ ions act both as crosslinker and antibacterial agent. Finally, the authors also performed in vivo studies, testing the hydrogels in a rat skin full-thickness defect model. Compared to untreated groups, the healing process was faster, resulting in a thicker regenerated epidermal layer.
As observed from the reported examples, metal coordination based supramolecular hydrogels of HA are mainly characterized by dynamic viscoelastic properties, due to the reversible character of the bonds. As such, most of the above reviewed hydrogels are self-healing, shear-thinning and therefore injectable. These properties are very important for the material processing, handling and non-invasive application, especially when it comes to biomedical and drug delivery applications. However, being solely based on reversible crosslinks, one of the main drawbacks is the lack of mechanical integrity and long-term stability. Therefore, in the system developed by Shi et al, the polymers were crosslinked by UV light after the formation of metal coordination bonds, thus providing improved mechanical integrity of the final hydrogel. Coordination bond dynamics could be optimized to achieve longer timescales and thus better stability, depending on the choice of metal ion. However, for biomedical applications this choice is limited to relative biocompatible metal ions. It should be remarked that specific hydrogel design (physical crosslinking or physical + covalent) and thus resulting material's mechanical properties are governed by the intended application.
6. Inclusion complexes
Among different supramolecular interactions, inclusion complexes, also known as guest–host interactions, seem to produce predictable and tunable interactions, due to a predetermined ratio between host and guest molecules and specific association constants [5]. There are several guest–host inclusion complexes that are commonly used in the design of hydrogels for biomedical applications. Reported HA-based guest–host hydrogels predominantly contain cyclodextrin and adamantane, as shown in table 1. β-cyclodextrin has a hydrophobic cavity, which acts as a 'host' that can interact with hydrophobic 'guest' molecules such as adamantane (figure 6(A)) [55]. Adamantane consists of three cyclohexanes fused together into a spherical group with a diameter of 7 Å, which fits into the cavity of β-cyclodextrin [101]. This guest–host interaction has an association constant of 104 and 105 M−1 in water, which is relatively high in comparison to other supramolecular interactions [55].
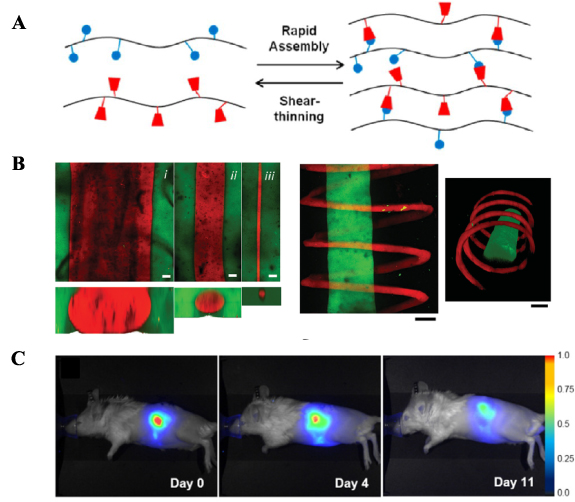
Figure 6. (A) Schematic of the guest–host complexation between β-CD and adamantane, and its dynamics. Reprinted with permission from [55]. Copyright (2013) American Chemical Society. (B) Guest–host writing (GHost-writing) with β-CD:Ad filaments using different percentages of modification (25% and 40%). The 40% modification was labeled with green fluorescence, and 25% modification was labeled with red fluorescence. It was shown that these gels do not mix when printed together. [57] John Wiley & Sons. © 1969, John Wiley and Sons. (C) A labeled β-CD:Ad hydrogel for treatment of chronic kidney disease (CKD) was injected under the renal capsule of BALB/c mice. These mice were imaged, and showed localization of the gel, and material degradation through decrease intensity of the fluorescence. Reprinted from [59], © 2015 Elsevier BV. All rights reserved.
Download figure:
Standard image High-resolution imageRodell et al started to investigate this guest–host interaction as a potential crosslinking strategy for shear-thinning hydrogels, which can be used for minimally invasive drug delivery [55]. In this work, the authors synthesized two versions of modified HA (74 kDa). One part of HA-TBA was modified with adamantane (degree of modification, 20%–50%) through esterification, while other part was modified with β-cyclodextrin (DM = 20%). The degree of modification appeared to have a larger impact on the relaxation time of the hydrogel in comparison to the polymer concentration. However, the polymer concentration did affect the erosion rate of the hydrogel, which is an important parameter to control drug delivery, as it can affect the release kinetics. The authors used fluorescently labeled BSA to model the protein release behavior of multiple compositions of the hydrogel. The hydrogels with 2.5 wt% of polymers took 21 d to release 90% of labeled BSA, while hydrogels with 10 wt% took 53 d to release the same amount. Similar as other supramolecular hydrogels, these HA-based hydrogels also displayed the desirable ability of shear-thinning, and the ability to recover nearly immediately after injection (figure 6(A)). This has resulted in multiple other research papers by Rodell and Burdick, where they further explored the adamantane-β-cyclodextrin interactions in HA-based hydrogels.
In one of the follow-up works, Rodell et al explored the tunability of proteolytic degradation of HA hydrogels, through the addition of peptide tethers to adamantane before coupling it to HA (90 kDa) [56]. These peptides with adamantane were prepared using solid phase peptide synthesis and coupled to maleimide-modified HA. The tethers were named positive Ad (PAd) or negative Ad (NAd), designating if they are degradable by metallopeptidase or not. The presence of peptide tethers did not affect the mechanical properties of the hydrogels. Although the hydrogels containing NAd had a reduced rate of proteolytic degradation compared to hydrogels containing PAd, the surface erosion rate was equal in both versions of the HA-hydrogels.
Highley et al also investigated adamantane–cyclodextrin crosslinked hydrogels to be used for 3D bioprinting, which they called GHost-writing [57]. Two formulations of the Ad:CD HA-hydrogels were used, either to act as a support gel or as a bioink that can be extruded. The support gel was synthesized to contain 40% modified HA repeat units, and the gel was mixed in a 1:1 Ad:CD ratio at a total polymer concentration of 4 wt/v%. The bioink contained 25% modified HA repeat units, which were also mixed in a 1:1 Ad:CD ratio, at a total polymer concentration of 5 wt/v%. Using fluorescent labels, no mixing was seen between the bioink and the support gel after printing (figure 6(B)). Mesenchymal stem cells and fibroblasts were incorporated into the support gel and in the bioink, showing more than 90% viability after the printing process. In addition to those gels, gels were synthesized that included methacrylate moieties conjugated to HA (90 kDa), which allowed for secondary covalent crosslinking. This resulted in a mechanically stronger hydrogel compared to the pure supramolecular hydrogels. The use of guest–host hydrogels for 3D printing could provide interesting opportunities, including mechanically stable structures, and self-healing gels which were also suitable for carrying cells. The support and bioink gels did not interact with each other (1 week), which allowed the printing of high resolution and complex hydrogel structures.
Mealy et al looked at the possibility of delivering small molecules, such as tryptophan (a model compound), using β-cyclodextrin-adamantane HA (90 kDa) hydrogels [58]. The hydrogels were all formed through mixing 25 wt% CD-HA and 25 wt% Ad-HA. Because small molecules and peptides can also bind within the pocket of CD in CD-HA, the effect of drug loading on the mechanics of Ad:CD hydrogels was investigated, by loading the 5 wt% hydrogels with tryptophan. No significant effect on the mechanical properties was observed, most likely due to the higher association constant of the Ad-CD complex (105 M−1) compared to the association constant between CD and tryptophan. The diffusivity and mobility of tryptophan and a peptide with a lower affinity for CD-HA were investigated in gels with an excess of CD units (i.e. CD content was increased from 1:1 to 1:2 or 1:3 Ad:CD). This increase in CD content resulted in reduced peptide mobility, i.e. 1:1 Ad:CD released 70%, 1:2 released 35%, and 1:3 released only 20% of the peptide within 24 h, while covalently crosslinked methacrylated HA without CD moieties released the peptides for 90% within 24 h. At the same time, tryptophan showed reduced diffusion compared to the lower affinity peptide, showing that tryptophan is indeed complexed more tightly by CD compared to the lower affinity peptide within hydrogels. Drug release properties were further studied by encapsulating doxycyclin or doxorubicin in 1:1 and 1:2 hydrogel formulations. Doxycyclin had a slower release profile than doxorubicin due to its higher affinity to CD. Nevertheless, both drugs were still present within the 1:2 hydrogel after 14 d. These results showed the potential of these hydrogels to be used for the controlled and sustained release of small molecules.
The ability to add therapeutic molecules in hydrogel networks can be an interesting approach to treat diseases such as CKD [59]. Rodell et al looked into the application of HA guest–host hydrogels for local immunotherapy with interleukin-10 (IL-10) and anti-TGFβ for CKD. Hydrogels (3.5 wt%) were formed by mixing HA (74 kDa) functionalized with 25% Ad and 25% CD in the presence of IL-10 or/and anti-TGFβ at concentrations of 0.3 µg µl−1 and 6.7 µg µl−1, respectively. The release of the drugs was studied, and 90% of either/both biotherapeutic agents were released by day 13. The gels were injected under the renal capsule of the left kidney in nude mice with unilateral ureteral obstruction (UUO) (figure 6(C)). One group of mice was injected with the hydrogel, one was injected with the hydrogel containing IL-10, another with the hydrogel containing anti-TGFβ, and the last with the hydrogel containing both drugs. The hydrogels were degraded for 60% within 4 d and were cleared from the kidney within 18 d. Histological analysis of the kidneys was performed to see the effect of the different treatments on UUO on macrophage infiltration, apoptosis, and fibrosis after 7, 21, and 35 d. After 21 d, the treatment groups had decreased macrophage infiltration and showed decreased apoptosis. Interestingly, dual treatment with IL-10 and anti-TGFβ caused increased fibrosis after 21 d. After 35 d, only the hydrogels with IL-10 or anti-TGFβ had a decreased fibrotic response. The study showed the possibility for these hydrogels to be used as an injectable drug delivery vehicle for CKD.
Gold nanorods (AuNRs) are being incorporated in supramolecular hydrogel systems to create thermo-responsive systems, that release drugs when the AuNRs are heated by a photothermal reaction upon near-infrared radiation (NIR) [60]. Highley et al prepared a 1:1 Ad-HA and CD-HA (HA 90 kDa) guest–host hydrogel (4 wt% of the polymers with DM = 40% for both components), in which AuNRs of 30 nm long and 9 nm wide were incorporated through a seed growth method. In addition to bulk hydrogels, microgels were formed using a microfluidic device, where the polymer solutions were supplied in different microfluidic channels that came together to form microgels of 80 µm. When the AuNRs within the gel were illuminated and consequently heated up, the supramolecular forces between Ad-HA and CD-HA were disrupted, allowing for the release of therapeutics. Molecules with varying Mw were encapsulated in the hydrogels, to see the effect of NIR radiation (60 s and with an intensity of 3.4 W cm−2) on the release profile. The morphology of the AuNR hydrogels showed clear changes after NIR exposure, i.e. the gels swelled, morphology became more irregular and voids were observed. Release of 5(6)-carboxyfluorescein (376 Da) and 20 kDA FITC-dextran increased two-fold from hydrogels upon NIR radiation, as compared to hydrogels that were not irradiated or did not contain AuNRs. For 500 kDa FITC-dextran, the release rate increased even three-fold. The released amount also increased with increasing light exposure time and/or intensity. In microgels similar results were found for the release profiles. To summarize, the release of different payloads from bulk or microgels all benefited from the addition of AuNRs NIR exposure, resulting in the enabling of externally triggered release properties.
Another commonly used family of host molecules is the cucurbituril (CB) family. It has a higher binding affinity and higher selectivity to its guests (i.e. alkylammonium ions) than the complexes formed by adamantane and cyclodextrin. The most popular CB host molecules are CB[8] and CB[6], which can bind selectively to alkylammonium ions and polyamines like 1,6-diaminohexane (DAH) and spermine (SPM), resulting in stable guest–host complexes [61]. Park et al used these properties to develop a supramolecular hydrogel for cellular engineering by conjugating CB[6], DAH, and SPM with HA (100 kDa). Thiol-modified HA was reacted with (allyloxy)12CB[6] through a thiol-ene click reaction to produce CB[6]-HA (DM = 6%). Modification of HA with DAH and SPM resulted in DM = 50% and 52%, respectively. The hydrogels were formed by mixing equal volumes of 2 wt% CB[6]-HA and 2 wt% SPM-HA or DAH-HA. To check cytocompatibility of the hydrogel, cells from a mouse embryonic fibroblast cell line (NIH3T3) were encapsulated within the hydrogel. The CB[6]/DAH-HA hydrogel displayed 93% cell viability, whereas the CB[6]/SPM-HA hydrogel displayed 62% cell viability after 3 d of culture. DAH-HA was present in excess, leaving DAH moieties open for complexation with other CB[6] 'host' molecules. In this way, fibronectin motifs were introduced, which promoted adhesion and proliferation of the NIH3T3 cells, while the hydrogels without this motif had lower proliferation rates and poor cell adhesion. Also, FITC-labeled CB[6] tags were introduced into the matrix at 0.1 equivalence, which barely affected the storage modulus of the hydrogels. These fluorescent tags were used to test the hydrogel in vivo where it was injected subcutaneously in nude mice. The hydrogel emitted fluorescence for 11 d post injection locally. Histological analysis was done to further ensure the safety of the hydrogel, and negligible inflammation was found. This hydrogel combination therefore represents an interesting option for 3D cellular engineering.
Hydrogels with CB[6]-HA and DAH-HA polymers were further explored for bioengineered stem cell therapy [62]. The cells introduced into these hydrogels were engineered MSCs (eMSCs), which were transduced to express EGFP and mutant IL-12 (IL-12 M). To stimulate the expression of IL-12 M and EGFP, dexamethasone (Dexa) and all-trans-retinoic acid (RA) were incorporated into the hydrogels: Dexa was coupled with CB[6], and all-trans-RA was coupled to DAH-HA to form RA-DAH-HA (HA 100 and 210 kDa). The performance of the hydrogels was compared with commercially available Matrigel and Matrixen. When eMSCs were encapsulated within the gels, the cells did not spread in the HA-based gels, while they did in Matrigel and Matrixen. However, Matrigel degraded completely within 3 weeks, and cell viability within Matrixen fell below 50% after 3 weeks. On the other hand, the HA-based hydrogels took 3 months to degrade, which is much longer than Matrixen and Matrigel. As a consequence, HA-based hydrogels, with and without RA and Dexa, displayed much longer cell viability of more than 75% at the 3 weeks time point and transgene expression was detected for up to 5 weeks. The hydrogels were then tested in vivo using fluorescence as the result of EGFP expression. Mice were injected in the ventral dermis with the hydrogel precursor mixtures (Matrigel, Matrixen, and HA ± RA and Dexa) and EGFP-expressing eMSCs. With Matrigel and Matrixen, cells stopped expressing EGFP within 20 d, while cells within the HA hydrogel continued expressing EGFP for 60 d. The hydrogel loaded with eMSCs was further studied as a potential cancer therapy in vivo. Melanoma cells were injected in mice, which developed into tumors. The hydrogel mixtures with eMSCs were then injected into the tumors. The authors found that for the mice injected with the cells encapsulated in HA hydrogels showed a reduced tumor growth, and increased survival rate, in comparison to mice treated with free MSCs and eMSCs (IL-12 M expressing) and cells encapsulated in Matrigel and Matrixen. IL-12 M produced by the eMSCS has a therapeutic effect, which indicated the potential to act as a combination therapy with RA that releases as the hydrogel degrades. Multiple injections of the hydrogel over the course of 10 d showed a further decrease in tumor growth. The HA-based hydrogels and IL-12 M also showed negligible immunological reactions, cementing the potential of these hydrogels for cell therapy.
The capacity to encapsulate cells within these hydrogels also opens up the possibility to use them as scaffolds for tissue engineering. A 2 wt% CB[6]/DAH-HA hydrogel, as previously mentioned [62], was used to encapsulate hMSCs to study chondrogenesis [63]. This system showed 95% cell viability after 10 d and cells were actively proliferating. FITC-CB[6] was added to the hydrogel mixture as a fluorescent label, and was maintained in the hydrogel for a significant amount of time, i.e. more than 10 d. Dexa was attached to CB[6], and added into the hydrogel to act as a potential modulator for chondrogenesis. Dexa-CB[6] was released for 50% in the 1st week, and continued to release for 3 weeks, while free Dexa was released completely within 2 h. This also affected the hMSCs, as more proliferation was seen in the hydrogel with Dexa-CB[6] present, compared to free drug. The gels were tested subcutaneously in nude mice, with either Dexa-CB[6] or free Dexa. The gels formed in vivo and were detected for up to 4 weeks post-injection. In hydrogels containing Dexa-CB[6], the levels of GAGs, type 2 collagen (COL II), cartilage oligomeric matrix protein (COMP), transcription factor SOX9 and aggrecan expression were all significantly higher than in hydrogels with free Dexa. Besides, the addition of physically loaded TGF-β showed a large increase in chondrogenic markers compared to the hydrogels only containing Dexa-CB[6], indicating a synergistic effect of TGF-β and Dexa on chondrogenesis (figure 7(A)). As a result, the combination of Dexa-CB[6] and TGF-β in the hydrogel allowed for a more even distribution of the cells within the constructs, compared to gels with neither Dexa-CB[6] or TGF-β, as the cells could more actively proliferate into the gel. In conclusion, Jung et al showed that CB[6]/DAH-HA gels could stimulate chondrogenesis in hMSCs, and that these gels were injectable and stable in vivo.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
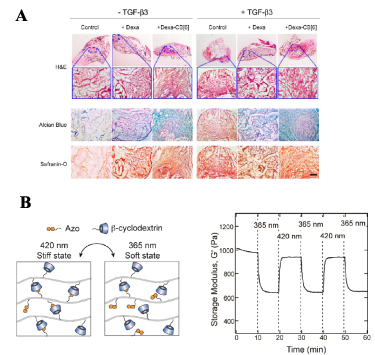
Figure 7. (A) Histological analysis of the neocartilage formed in vivo by hMSCs in monoCB[6]/DAH-HA hydrogels under different conditions. The staining showed highest neocartilage formation in the presence of TGF-β3 and Dexa-CB[6]. Reprinted with permission from [63]. Copyright (2014) American Chemical Society. (B) Schematic diagram showing the cis-trans switch in the β-CD:Azo hydrogel (left), and the effect of this change on the storage modulus of the resulting gel (right). Reprinted with permission from [66]. Copyright (2018) American Chemical Society.
Download figure:
Standard image High-resolution image{kind=link}
Rowland et al also focused on cucurbit[n]uril guest–host interactions, but they used one with a larger cavity, CB[8], which can bind two guest molecules via π–π stacking [64], for example phenylalanine or tryptophan. Cysteine-phenylalanine (CF) was conjugated to HA-methacrylate (DM = 10%) (HA 1500 kDa) and hydrogels were formed through mixing of HA-CF with CB[8]. These hydrogels showed to be mechanically stable, and displayed shear-thinning behavior similar as other supramolecular hydrogels based on guest–host interactions.
The above-mentioned hydrogel from 2 wt% HA-CF and 0.5eq of CB[8] (HA 1500 kDa) was studied as a potential delivery device for chemotherapeutics after glioblastoma resection [65]. The hydrogel could be injected locally and mold to the area of injection. The injected gel material was softer than the brain tissue, which allowed easy diffusion of chemotherapeutics. The cytotoxicity of the gel without chemotherapeutics was tested on primary cell lines derived from patient glioblastoma material, and no significant cell death was observed. Proliferative markers were detected, whereas inflammatory markers were barely present. When doxorubicin or cisplatin were added at 1 mg ml−1, cell viability dropped to 40%–60%. For the hydrogel with doxorubicin, immunocytochemistry showed a decrease in proliferative markers and an increase in inflammatory markers as compared to the empty gel. After 10 h, 8% of the loaded therapeutics were released, and 11% after 50 h. This release profile can be affected by the addition of hyaluronidase, which can help to break down the hydrogel. The release of the chemotherapeutics was further explored by injecting FITC-labeled hydrogels containing rhodamine B or doxorubicin, into ex vivo tumor samples. Through imaging it was confirmed that the hydrogels did not diffuse into the tissue, but the cargo did.
In addition to the above-mentioned interactions, there are several other guest–host interactions that have been explored. For example, β-cyclodextrin can also complex with 2-napthylacetic acid (2-NAA) to form supramolecular crosslinks [54]. Chen et al synthesized mixed hydrogels from β-CD-HA and 2-NAA-dextran for use as cell scaffolds, with the aim to improve the hydrogel's integrity by the more stable dextran. HA-CD (500 kDa) was produced by coupling ethylene diamine β-CD to HA through EDC/NHS coupling, while 2-NAA-dex was synthesized through esterification of 2-NAA and dextran. The two polymers were mixed, with 10 wt% HA-CD and 5 wt%–30 wt% 2-NAA-dex. Increasing amounts of 2-NAA-dex increased the amount of inclusion complexes within the hydrogel, and improved the mechanical properties. However, the higher weight fraction of 2-NAA-dex did decrease the water storage capability of the hydrogel. The precursors and the formed hydrogels were tested for cytotoxicity using fibroblasts. Neither the precursors nor the hydrogels had an adverse effect on the cells, and in both cases the cell viability was more than 90% after 48 h. The gels were found to protect and sustain encapsulated cells sufficiently and can therefore be used as a potential scaffold for tissue engineering. As a last example, a photo-responsive hydrogel was formed by complex formation between azobenzene in the trans conformation and β-cyclodextrin [66]. When these complexes are irradiated with UV light, the conformation of azobenzene changes to cis, lowering the binding affinity 2.5 times (figure 7(B)). The hydrogels were formed by 8%, 20%, or 28% modified 4-phenylazobenzoic acid HA (Azo), and β-CD-HA (DM = 28%) (HA 75 kDa). Hydrogels were formed at a 1:1 ratio of β-CD:Azo using a combination of manual mixing and centrifugation. Upon irradiation at λ = 365 nm, absorption spectra showed a rapid trans-cis transition and the gels became weaker (figure 7(B)). The gels only require a short burst of irradiation to undergo conformational changes, reducing the possibility of free radical generation. The decrease in crosslinks could be used as a potential drug release mechanism, which was tested using FITC-BSA as a model drug. Release was studied for 4 d, then the gels were irradiated for 10 min every 4 h, and it was found that after 1 d post-irradiation, the irradiated gels initially released twice as much BSA compared to the non-irradiated state. After this initial burst, the release profile returned to similar levels as seen with non-irradiated gels, due to crosslinks reverting to its trans state within 1 d. Finally, cytocompatibility was tested using fibroblasts in a 6 wt% hydrogel, showing that a high cell viability was maintained for more than 3 d.
A similar hydrogel system was developed by Zhao et al [67]. A photo-responsive supramolecular hydrogel based on HA was formed between β-CD-HA (DM = 42%) and Azo-HA (DM = 11%), whereas HA-Ad (DM = 11%) was used to make a non-photo-responsive supramolecular hydrogel (control) (HA 200–400 kDa). The hydrogels were formed at 10 wt% (1:1 v/v starting polymer solutions), and were evaluated for UV-irradiation-induced controlled release of epidermal growth factor (EGF). Both formulations contained 7.25 × 103 IU of EGF-FITC. Under ambient conditions, both gels exhibited a similar release profile of EGF, but following UV irradiation (365 nm, 50 mW cm−2), the photo-responsive formulation (with Azo-HA) displayed two to three fold increase in release, whereas illumination did not affect the release from the gel without Azo-HA component. The UV induced conformational change of azobenzene, resulted in lower affinity towards CD, thus led to a less crosslinked and softer hydrogel, from which more EGF could be released. Cytocompatibility was tested with fibroblasts cultured in the presence of hydrogels, resulting in cell viabilities above 80%. In addition, the gels were incubated with saline containing red blood cells and hemolysis was assessed. It was shown that UV irradiation had a very limited hemolysis effect (<5%) when compared to a positive control. During the healing process of wounds, EGF plays an important role, thus controlled EGF release from supramolecular gels could potentially be used for accelerating wound healing process. With this in mind, the authors also investigated EGF-loaded gels for in vivo wound healing efficacy. A rat model with full-thickness wounds on the back was used and the photo-responsive hydrogel formulation (upon UV irradiation for 15 min) exhibited 96% wound closure ratio after 7 d, which was significantly higher when compared to the relative control groups. The acceleration of wound healing was due to a higher accumulative release of EGF at the wound bed following UV irradiation. Therefore, the described photo-responsive EGF-loaded gel can be considered as a smart dressing material with potential application in a clinical setting.
7. Conclusions and perspectives
This review provides an overview of the hydrogels fabricated by means of physical crosslinking mechanisms and containing HA as one of the main components. Being a naturally occurring polymer, present in many tissues and ECM, HA makes such supramolecular materials attractive for biomedical applications. The fact that the gels are supramolecular makes them also interesting in terms of processing technologies. Many of the discussed examples exhibit shear-thinning behavior, which allows for the gels to be extruded through a needle and to recover their original stiffness following extrusion. This translates to the ease of manageability in filling defect sites with gels for example. Moreover, this also implicates the possibility for the gels to adopt different shapes without breaking their structure or disrupting the crosslinked network, as is usually the case with covalently crosslinked gels. The large toolbox of physical interactions allows for diversity in the design process of the supramolecular gels. Tuning the material's viscoelastic response, stress relaxation, response to stimuli (pH, temperature) are just some of the features easily achieved by incorporating reversible, temporary crosslinks. Specifically, these aspects are very important and come in handy when the gel should support delivery of drugs. Their reversible nature also makes them efficient in dissipating stress during mechanical deformations, however supramolecular gels still fall behind traditional covalent and double network gels when it comes to mechanical properties and supporting stresses at the MPa range. In order to achieve better mechanical stability, covalent crosslinks could be incorporated within supramolecular gels allowing for fixation of the initially reversible and processable material [35, 57].
Supramolecular hydrogels are also very suitable for 3D printing (due to their reversible nature) and therefore 3D printing constitutes an important processing technology for producing novel biomaterials [52, 57]. Further developments are necessary in order to achieve the full potential of 3D printed hydrogels and biomaterials to be used in clinics.
In general, supramolecular hydrogels have important implications in the biomedical field [8, 9]. However, in the present review we narrowly focus solely on HA containing gels, because there is a huge potential in using this biopolymer to produce clinically relevant materials. Although there is a significant progress in the development of such materials in the recent years, there is still a lot of room for improvement, optimization and design of novel multifunctional materials.
Considering translation to clinics, several of the reported examples showed quite promising results. For example, NIPAM-based HA gels for antiadhesion applications in surgery seem to be very attractive and safe materials for clinical translation [39, 40]. However, more effort has to be made in order for supramolecular gels to get to the market. Development of reproducible and validated methods for materials production and characterization have to be in place, as well as suitable upscaling protocols. Rigorous GMP processes have to be established and followed. Overall, considering the great potential and interesting properties of the examined materials, it is worth putting more efforts in the design of new supramolecular HA-based biomaterials, allowing for quicker translation into clinics.
Acknowledgments
This work was performed under the framework of Chemelot InSciTe, supported by the partners of Regenerative Medicine Crossing Borders (www.regmedxb.com) and powered by Health∼Holland, Top Sector Life Sciences and Health.
Data availability statement
No new data were created or analyzed in this study.