
Abstract
Functional nanomaterials (NM) of different size, shape, chemical composition, and surface chemistry are of increasing relevance for many key technologies of the twenty-first century. This includes polymer and silica or silica-coated nanoparticles (NP) with covalently bound surface groups, semiconductor quantum dots (QD), metal and metal oxide NP, and lanthanide-based NP with coordinatively or electrostatically bound ligands, as well as surface-coated nanostructures like micellar encapsulated NP. The surface chemistry can significantly affect the physicochemical properties of NM, their charge, their processability and performance, as well as their impact on human health and the environment. Thus, analytical methods for the characterization of NM surface chemistry regarding chemical identification, quantification, and accessibility of functional groups (FG) and surface ligands bearing such FG are of increasing importance for quality control of NM synthesis up to nanosafety. Here, we provide an overview of analytical methods for FG analysis and quantification with special emphasis on bioanalytically relevant FG broadly utilized for the covalent attachment of biomolecules like proteins, peptides, and oligonucleotides and address method- and material-related challenges and limitations. Analytical techniques reviewed include electrochemical titration methods, optical assays, nuclear magnetic resonance and vibrational spectroscopy, as well as X-ray based and thermal analysis methods, covering the last 5–10 years. Criteria for method classification and evaluation include the need for a signal-generating label, provision of either the total or derivatizable number of FG, need for expensive instrumentation, and suitability for process and production control during NM synthesis and functionalization.
Graphical abstract

Similar content being viewed by others

Introduction
Need for and importance of functional group quantification
Functionalized nanomaterials (NM) are of increasing industrial and economic importance in the life sciences and the health sector as well as for applications in nano(bio)technology, optical and sensor technologies, solid state lighting and photovoltaics, as well as opto-electronic and electronic devices and security applications. Nowadays, NM are used as catalysts, hydrogen storage and energy conversion materials, contrast agents and drug carriers for imaging and therapy in medicine, signal-generating reporters in bioanalysis, molecular diagnostics and sensing, as additives for food and cosmetics, in textile industry, and as phosphors for lighting and display technologies [1,2,3,4,5,6,7,8,9,10,11,12,13]. This comprises all types of core and core/shell NM such as organic polymer and inorganic silica or silica-coated nanoparticles (NP) with covalently bound surface groups as well as other inorganic NP like metal and metal oxide NP, semiconductor quantum dots (QD), and lanthanide-based NP with coordinatively or electrostatically bound ligands [14,15,16]. It also includes different types of encapsulated nanostructures like inorganic NP with hydrophobic surface ligands wrapped with amphiphilic (co)polymers or lipid coatings that can also be crosslinked, yielding micellar-type systems, or coated with alternating layers of differently charged polyelectrolytes by the so-called layer-by-layer (LbL) approach [17,18,19].
Decisive for most applications of NM are their specific surface properties, which are largely controlled by the chemical nature and number of ligands and functional group (FG) on the NM surface. The surface chemistry and surface FG determine the charge, dispersibility, and colloidal stability of NM, as well as their hydrophilicity/hydrophobicity, processability, and interaction with their environment [15, 18, 20,21,22,23]. In addition, FG enable the controlled modification and functionalization of NM by covalent binding of functional molecules such as hydrophilic ligands, anti-fouling agents, sensor dyes, and biomolecules like proteins, peptides, or oligonucleotides, e.g., for the preparation of nanosensors and targeted nanoprobes [5, 10, 24,25,26]. Control of the surface chemistry is also relevant for the minimization of unspecific adsorption, increase of colloidal and/or dissolution stability, and the design of drug carriers and triggered release systems [15, 22, 27,28,29,30]. For example, the reactivity and stability of NM can be altered intentionally and rationally by surface passivation strategies utilizing special coatings such as silica or polymeric shells, or via tailored modifications of the surface charge via the density of FG and ligands. This underlines the crucial importance of surface chemistry and surface functionalities for many NM applications in the life and material sciences and nano(bio)technology and their relevance for the rational design and tuning of the properties of functional NM. Knowledge of NM surface chemistry presents not only a key issue to understand the nano-bio interface largely controlling NM functionality and performance in (bio)applications, but is also relevant to assess the fate, exposure, dissolution, transformation, and accumulation of NM, and thus, NM toxicity and potential risks for human health and the environment [31,32,33,34,35,36]. This also includes the evaluation of risks associated with the application of engineered NM in consumer, food, and biomedical products [37,38,39,40,41]. Here, also unintentional changes and modifications in NM surface by time- and environment-dependent aging effects and transformations during the material’s life-cycle must be considered, that can affect NM safety aspects [42]. This is addressed by the increasingly pursued safe-by-design (SbD) concept of NM, which integrates considerations of material safety and performance as early as possible into the innovation process [43,44,45], thereby balancing safety, functionality, and costs for the development of better nanotechnology-enabled products throughout their life-cycle [46].
The increasing importance of NM in fundamental research and technological applications makes the sustainable development of functional and safe(r) NM as well as a comprehensive understanding of the structure-function and structure-safety relationships mandatory [43, 47]. Reliable, robust, and simple methods for the adequate characterization of such materials are key requirements to overcome challenges associated with the rapidly diversifying development of NM and to address still existing uncertainties and knowledge gaps [48, 49]. This is also essential for quality assurance and production control of engineered NM in support of the SbD concept [9, 18, 23, 50, 51]. In this context, the development of harmonized and standardized characterization methods not only simplifies to rank, prioritize, and choose safer alternatives during the innovation process of engineering NM, but is similarly beneficial for regulatory frameworks and the confidence in NM [41, 46, 52, 53]. Nevertheless, there is still a lack of normative measurement and characterization regulations, validated and standardized measurement protocols, reference materials of known surface chemistry for FG quantification, and reference data on application-relevant NM. Together with the often contradictorily literature in this field, this presents growing technological and economic challenges for manufacturers and users of NM. This has been increasingly recognized not only by scientists from different disciplines all over the world, but also by European legislation as well as national and international standardization organizations like ISO, IEC, and OECD [50, 54]. This makes the assessment of analytical methods for FG analysis on NM, including the determination of method-inherent limitations and NM-specific requirements and limitations as well as achievable method uncertainties, an increasingly important topic for NM-based technologies and NM risk assessment and regulation.
Nanomaterial surface functionalization and bioanalytically relevant FG
The reactive FG and surface ligands relevant for NM-based (bio)analytical applications typically correspond with the complementary FG on the functional molecules utilized in typical (bio)conjugation reactions, e.g., biomolecules used as target-specific recognition moieties [24, 26, 55]. For biomolecules like peptides and proteins including antibodies and enzymes, this includes amino groups (-NH2) at the N-terminus of the polyamide backbone and at the side chains of the amino acids arginine, histidine, lysine, and tryptophan, carboxy groups (-COOH) at the C-terminus of the polyamide backbone and at the side chains of aspartic acid and glutamic acid, the hydroxyl and phenol groups (-OH) of serine, threonine, and tyrosine, the thiol (-SH) and thioether groups of cysteine and methionine, as well as chemically introduced thiol groups, e.g., via reductive cleavage of disulfide bridges [56, 57]. For carbohydrates like mono- and disaccharides, oligo- and polysaccharides, and as part of glycolipids and glycoproteins, the most abundant native FG are hydroxyl groups. Some monosaccharide derivatives such as non-acetylated amino sugars provide additional reactive FG for bioconjugation. Also, vicinal diols can be oxidized to aldehyde groups (-CHO). Oligonucleotides like DNA and RNA consist of a sugar-phosphate polymer backbone with a reactive phosphate group at the 5′-terminus and a OH group in the case of DNA or a vicinal diol for RNA at the 3′-terminus, respectively. The native nucleic acids cannot be chemically modified as easily as the amino acids in proteins. However, synthetic oligonucleotides can be prepared via solid-phase synthesis with aminoalkyl- or thioalkyl-containing linkers attached to the nucleobases, the phosphate backbone, or the 3′- or 5′-terminus, which allow for further modifications and labeling [58]. Besides native FG, there are several FG which can also be chemically introduced into biomolecules that are suitable for chemoselective labeling and bio-orthogonal chemistry such as azide (-N3), alkyne (-C≡CH), or maleimide groups for cycloaddition reactions [26, 57,58,59]. Other interesting FG for (bio)analytical applications are silanol (≡Si-OH, =Si(OH)2) and siloxane (≡Si-O-Si≡) groups present on silica particles and silica-based surface modifications and coatings used for all types of NM to improve or tune their stability and dispersibility in aqueous media, for SbD concepts and the supply of surface FG for further functionalization reactions [60,61,62].
Quantifying total vs. derivatizable FG on nanomaterials
For the quantification of FG on NM, it needs to be distinguished between methods that provide the total number of FG present on the NM surface, and methods that determine the number of derivatizable FG [63, 64]. The total FG number largely determines NM charge (zeta potential), and thus, colloidal stability, dispersibility, and hydrophobicity/ hydrophilicity, and is therefore an important and application-relevant parameter for all types of NM in addition to size, size distribution, and shape/morphology. The number of derivatizable FG, in turn, controls the number of groups available for covalent attachment of functional molecules such as hydrophilic ligands, anti-fouling agents, or biomolecules. Hence, the number of derivatizable FG is important for all (bio)labeling reactions as well as the functionality and performance of the resulting surface-modified NP and NP (bio)conjugates.
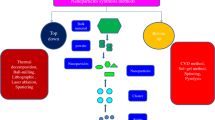
Relevant for the selection of suitable analytical methods for FG and surface ligand quantification is the signal generation principle, i.e., whether these methods can quantify FG directly without a signal generating reporter (label-free methods), or whether they require a reporter for readout (label-based methods) that is covalently bound or interacts with the FG via electrostatic or adsorptive interactions [51]. In Fig. 1a, the analytical methods for FG quantification covered by this review are displayed and highlighted according to the principle of signal generation including electrochemical methods, dye-based optical methods, and other instrumental analytical techniques. For label-based methods, reporter properties such as molecule size, shape, and charge as well as the coupling efficiency and yield of the chemical reaction used for reporter conjugation can influence the analytical result. Especially the size and shape of the reporter utilized to determine the number of derivatizable FG can play an important role, as the obtained labeling density, and hence, number of derivatizable FG can be affected by the bulkiness of the label and steric effects [63], as shown in Fig. 1b. This, however, is also the case for any other covalently bound (bio)molecule of interest, so that the results can be correlated if a suitable reporter is applied, or can at least be estimated from the size and shape of the reporter and the (bio)molecule of interest. Moreover, it must be distinguished between absolute, i.e., calibration-free analytical methods and methods that require a calibration for analyte quantification. The latter is by far more common but can introduce additional challenges and uncertainties due to the need of a suitable reference material or standards, particularly for optical methods like absorption and fluorescence that yield signals which are affected by reporter environment. Depending on the method used, this can also determine whether the whole nanoobject can be analyzed as prepared, or whether the NM has to be dissolved prior to FG quantification.

Brief overview of the bioanalytically relevant FG and the analytical methods covered by this review including typical reporters. a Method classification according to the principle of signal generation, i.e., electrochemical methods (blue), dye-based optical methods (yellow), and other instrumental analytical techniques (green). b Schematic presentation of the influence of the reporter size used to determine the number of FG depending on FG density or ligand bulkiness (steric hindrance) on the NP surface. The sizes of the labels can range from very small reporters like protons (H+) and metal (Mn+) ions, to small and medium-sized reporters like organic dyes, that are still smaller than large biomolecules
Also, the type of bond between the NM and the FG-bearing ligand must be considered. For FG tightly bound to NP via covalently attached ligands, only steric effects affect the accessibility of the FG. For silica NP, it must be kept in mind that not all hydroxy groups are quantitatively involved in the grafting procedure and free ethoxy or methoxy groups remain on the silica NM. For electrostatically or coordinatively bound ligands, in turn, excess ligands must be removed and potential influences of NP concentration and dilution steps that can shift the ligand adsorption/desorption equilibrium must be excluded or properly considered for FG analysis [65]. For NP encapsulated in micellar structures, as often utilized for inorganic NP like iron oxide, semiconductor QD, and lanthanide-based upconversion NP [17, 18, 66, 67], even the orientation of the FG can play a role for FG analysis, depending on the chemical nature of the respective organic coating. Such systems can consist of buried FG pointing inwards that interact with the surface atoms of the encapsulated NP, and FG pointing outwards to the NP microenvironment. Only the latter ones are relevant for the covalent attachment of functional molecules. For such nanostructures, the information provided by the applied analytical method must be carefully evaluated regarding its information content to decide whether the whole nanoobject should be analyzed as prepared or whether the encapsulated NM should be dissolved prior to FG quantification.
Besides FG/ligand concentration, also the NM number concentration and surface area of the NM are of importance, as they determine the number of FG or ligands per particle as well as the FG or ligand density on the NM. Typically, the measured number of FG/ligands is divided by the number of particles or their total surface area. The NP number concentration can be determined via counting methods such as resistive pulse sensing (Coulter counter), nanoparticle tracking analysis (NTA), flow cytometry (FCM), as well as by absorption spectroscopy (if the molar absorption coefficient or the molar absorption cross section is known). The mean particle concentration can also be calculated indirectly from the NM mass (dry weight) or the concentration of certain NM-specific elements that can be quantified, e.g., with inductively coupled plasma mass spectrometry (ICP-MS) or optical emission spectroscopy (ICP-OES). The surface area, in turn, can be directly measured using gas sorption methods, or can be calculated from the NP concentration and the particle dimensions, obtained by sizing techniques such as transmission or scanning electron microscopy (TEM/SEM), small-angle X-ray scattering (SAXS), NTA, or dynamic light scattering (DLS). All these NM characterization techniques regarding NP size and concentration are well-known and have been reviewed in the literature [68,69,70,71,72], and we will focus here solely on methods for FG and ligand quantification.
For the determination of the total and derivatizable number of FG or ligands per particle, always another parameter needs to be considered, namely the distribution of NM size (and shape) and the corresponding variations of the surface-to-volume ratios within one particle batch, which directly influence the total surface area of the NM, and thus, the determined FG/ligand density (number of FG/ligands per particle). A perfect FG quantification method should be able to count the groups/ligands of interest per particle for a large number of individual particles to yield a histogram of FG or ligands per NM independent of particle size/size distribution and shape. A few sophisticated analytical techniques such as single-particle ICP-MS (sp-ICP-MS) or flow methods like FCM are in principle capable of measuring NM properties like elemental composition (for suited elements) and scattering and fluorescence (intensity) features on a particle-by-particle basis. These methods, however, still face limitations in NM surface characterization, e.g., related to the lack of sensitivity or influences of labeling chemistries (vide infra). Even if FG/ligand counting on single particles was possible, different morphological features (i.e., exposed crystal facets, local curvature radii of the surface) that can be present even at various surface areas of a single particle will still impact the ligand density on the NM surface. Thus, most analytical techniques applied for FG/ligand quantification and described here are ensemble techniques that yield only a mean value of FG/ligands for a given (mean) particle size and shape. As smaller particles have a smaller surface area, but a larger surface-to-volume ratio compared with larger particles, the distribution of FG/ligands per (individual) particles can strongly differ for particles with the same (mean) size but different size distributions. However, as the particle size has to be determined to calculate the surface area (vide supra), the obtained number-based size distribution can also be used to calculate the distribution of FG/ligands (assuming a similar surface morphology) and even to consider the size- and shape-dependent curvature of the NM surface.
In this review, typical methods for FG quantification on NM are presented (cf. Fig. 1a), and their working principles, advantages, and limitations are described, focusing on publications from the last 5–10 years and selected, representative examples to underline the versatility of the respective methods. Analytical techniques covered include electrochemical titration methods, optical assays, nuclear magnetic resonance (NMR), ICP-MS and ICP-OES, infrared (IR) and Raman spectroscopy, X-ray photoelectron spectroscopy (XPS) and X-ray fluorescence spectroscopy (XRF), as well as thermal analysis methods and elemental analysis. Other mass spectrometry techniques like laser ablation ICP-MS (LA-ICP-MS) or time-of-flight secondary ion mass spectrometry (ToF-SIMS) are not considered here, as these techniques are commonly used for the surface analysis of 2D-supports. Parameters addressed and used for method classification and evaluation include whether the respective analytical method (i) provides the total or derivatizable number of FG and (ii) is label-free or requires a signal-generating reporter; (iii) whether the reporter is covalently bound to the FG, giving rise to a possible influence of the efficiency of the conjugation reaction; (iv) the influence of reporter size; and (v) the need for method calibration. Special emphasis is dedicated to simple FG quantification methods with inexpensive instrumentation that are broadly accessible and can be used for routine analysis and process control during NM production and surface functionalization, like electrochemical and optical methods. We do not intend to cover NM bioconjugation strategies utilized for preparing nano-bioconjugates nor methods to quantify NM-bound biomolecules or to assess biomolecule functionality, which have already been excellently described in other review articles [56, 57].
Electrochemical titrations for the quantification of (de)protonable FG on dispersed nanomaterials
Electrochemical titrations methods like potentiometric titrations (measurement of the electrochemical potential(s) of the sample), conductometric titrations (measurement of the sample conductivity), and the so-called Boehm titration that is specifically employed for carbon materials are commonly used as inexpensive and precise methods for the quantification of (de)protonable surface FG such as carboxylic acids, amines, or thiols. Closely related Zeta potential measurements that provide NM surface charge, which presents a measure for colloidal stability and can be used for the monitoring of reactions on nano- and microparticles [73], are not further detailed here.
During the course of an electrochemical titration, defined amounts of a titrant (typically acids or bases) are added to the sample, and the resulting changes in the electrochemical properties of the sample are monitored. As electrochemical acid-base titrations are typically carried out over a broad pH range, often including the isoelectric point of the sample (i.e., the pH value at which the net surface charge is zero), they can only be applied for NM that are stable under the given pH conditions. For example, the determination of amino groups (pKa about 9–11) on silica-based NM can be challenging as silica dissolves at basic pH values, and some metal oxide and other chalcogenide-based NM can dissolve at acidic pH values where carboxylic groups (pKa about 5) are typically detected. Nevertheless, for many other NM like carbon-based and polymeric particles, electrochemical titrations are well suited for FG and ligand quantification. Another limitation of electrochemical methods is their lack of specificity and selectivity, as all (de)protonable species with comparable pKa values are detected which can distort the obtained results. This can include surfactants, initiators, or stabilizers from the NM synthesis, excess ligands with the (de)protonable group of interest, or the presence of other (de)protonable FG having a similar pKa value. In addition, electrochemical titration methods require a relatively large amount of sample (typically about 10–20 mg/mL of NM sample).
Potentiometric titration
In a potentiometric titration, the electrochemical potential(s) of the analyte solution is measured with two electrodes, normally in the form of pH measurements, upon addition of defined amounts of acid or base as titrant which yields a pH titration curve. The equivalence points of the titration curves provide the amount and the pKa values of the (de)protonable FG of the analyzed sample. Also, the use of other ion-selective electrodes is possible [74, 75]. Potentiometric titrations have been used for characterizing the surface chemistry of different types of organic and inorganic NM with various FG, and have been applied to determine the number and nature of acidic sites (carboxy, lactone, phenol, and ester groups) on carbon-based materials like carbon dots (CD), nanocellulose/nanobentonite composites, biochar particles, multi-walled carbon nanotubes, or cellulose nanocrystals [76,77,78,79,80], or to quantify hydroxy (silanol) and thiol groups on hybrid silica particles [81]. Potentiometric titrations have also been used to determine the total number of acidic sites on different catalyst materials like phosphotungstic acid-functionalized Sn-TiO2 and organic-inorganic polyoxometalate NP, SrTiO3 particles used to catalyze condensation, hydrogenation, and amination reactions, functionalized silica particles employed as catalysts for the esterification of linoleic acid, and photocatalytic TiO2/S-doped carbon hybrids [82,83,84,85,86]. For example, Wang et al. potentiometrically quantified carboxy and amino groups on fluorescent CD prepared with different amounts of l-arginine or l-glycine (see Fig. 2a) [87]. By addition of Fe(III) ions before the titration and comparison of the results with measurements done without metal ions, the authors could also derive information on metal ion-CD interactions. Renner et al. compared potentiometric pH titrations with Zeta potential and conductivity measurements to quantify the number of hydroxy groups on silica and iron oxide NP [88]. The results obtained for silica particles, shown in Fig. 2b, demonstrate that Zeta potential values are closely linked to pH and conductivity of a sample, which is reflected by the respective curves changing at the same titrant volumes added. The titration with HCl used to protonate surface hydroxy groups was reversed by back titration with NaOH to ensure the reversibility of the process. The slightly negative zeta potential at full protonation was attributed to non-accessible hydroxy groups.

Representative examples for the FG quantification on NM using potentiometric titrations. a Results for the potentiometric FG quantification for carbon dots functionalized with different concentrations of either l-arginine (left) or l-glycine (right) using NaOH as titrant. Adapted with permission from ref. [87]. Copyright 2019, American Chemical Society. b Reversible deprotonation of a colloidal silica dispersion using HCl/NaOH titrants as detected by zeta potential (black) and pH (red, left) or conductivity (red, right) measurements. Adapted with author permission from ref. [88] (CC BY-NC 4.0)
Conductometric titration
In a conductometric titration, the conductivity of a sample is measured as a function of the added amount of acid or base. Typical examples present the quantification of the total amount of amino and carboxy groups on polystyrene (PS) particles [64, 89, 90]. The suitability of conductometry for carboxy group quantification on polymeric particles has been validated for polymethylmethacrylate (PMMA) particles grafted with polyacrylic acid (PAA) by comparison with quantitative NMR spectroscopy (qNMR) [63] and for PS particles by comparison with Zeta potential measurements [90]. Conductometric titrations are also commonly used for quantifying sulfate half-esters as well as carboxy and amino groups on cellulose nanocrystals (CNC), e.g., to achieve a tailored surface charge and to control CNC surface modification, or to study the effect of surface treatment on the dispersion rheology of CNC [91,92,93,94,95]. Other groups applied conductometric titrations to characterize CNC regarding their applicability for acid-base organo-catalysis [96, 97], or to determine the hydroxy group content on the surface of hydrogels consisting of modified cellulose nanofibrils suitable for controlled and pH-responsive release of a chemotherapeutic agent [98]. A general procedure for the determination of the sulfate half-ester content on CNC via conductometric titration, consisting of dialysis followed by treatment with a strong acid to ensure full protonation, was developed by Beck et al. [99], and a protocol to for the conductometric quantification of the sulfur and sulfate half-ester content on CNC was validated in an interlaboratory comparison [100]. The difference in the sulfur content determined by conductometry and by ICP-OES was attributed to sulfur in the CNC interior that is not conductometrically accessible.
Boehm titration
Boehm titration is a method developed by Boehm et al. in 1964 [101] suitable for the quantification of acidic, oxygen-containing surface groups on various carbon-based materials such as graphene, carbon nanotubes (CNT), CD, and carbon-coated particles. This method allows not only to quantify common FG relevant for biolabeling and bioanalytical applications, but also other FG such as lactone or phenol groups [101,102,103,104]. The Boehm method is based on the treatment of a dispersed carbon sample with titration bases of different pKa values like NaOH, Na2CO3, and NaHCO3 [101, 105], followed by the back titration of the unconsumed amount of the titrant. It is assumed that each base only neutralizes FG that are more acidic than the respective base, and the ratios of the amounts of the different oxygen-containing FG can then be calculated directly from the base consumption. Boehm titration has been utilized to characterize the FG on different kinds of CNT [106,107,108] as well as on other carbon-based materials like ozone-treated nanodiamonds, carbon NP derived from organic resin, graphite-decorated MnFe2O4 nanocomposites, and natural char nano- and microparticles [109,110,111,112]. To evaluate and standardize Boehm titration regarding accuracy, robustness, repeatability, and precision, Schönherr et al. investigated the FG on oxidized multi-walled CNT using different reaction bases, treatment times, and amounts of carbon material [105, 113]. A major concern of these studies was the dissolution of CO2 from air which leads to the formation of HCO3− and CO32−, that can considerably influence the titration results. To quantify this effect, a direct and an indirect approach to the Boehm titration procedure were compared. The results, shown in Fig. 3, underline the influence of CO2 particularly for the direct titration curve with NaOH. To circumvent such distortions, a medium-strong base like Na2CO3 was proposed and a protocol for an indirect titration approach with this base using an autotitrator was developed. Schönherr et al. also compared Boehm titration to other analytical techniques suitable for the quantification of oxygen-containing FG like XPS or temperature-programmed desorption mass spectrometry (TPD-MS), underlining its superior precision [105, 113].

Boehm titration curves (potentiometric detection) obtained for the direct titration (left) and indirect titration (right) with HCl as analyte solution and NaHCO3, Na2CO3, or NaOH as titrant solutions, underlining the strong impact of CO2 from air on the results of the direct approach. Adapted from ref. [105] (CC BY 4.0)
Boehm titration is presently the only electrochemical titration method considered by international standardization organizations like IEC TC 113: Nanotechnology for Electrotechnical Products and Systems for surface FG analysis and quantification in the currently evaluated standardization document 62607-6-13: Nanomanufacturing – Key control characteristics – Part 6-13: Determination of Oxygen Functional Groups Content of Graphene Materials with Boehm titration method. The main purpose of this document is to provide a standardized method for the determination of surface oxygen FG on graphene materials prepared by, e.g., oxidation-reduction method, solution-phase exfoliation, micro mechanical exfoliation, and organic synthesis using the Boehm titration method and to obtain quantitative information about the acidic oxides at the surface of graphene materials, including carboxy groups (also in the form of their cyclic anhydrides), lactone groups, hydroxyl groups and reactive carbonyl groups.
FG quantification with photometric and fluorometric assays and different optical reporters
FG quantification with optical spectroscopy relies on the measurement of the absorption (spectrophotometry; photometric or colorimetric assay) or emission (fluorometry; fluorometric assay) of a dye label (also called reporter or probe). Typically, fluorometric measurements are considered more sensitive than photometric measurements, as emission can in principle be detected down to the single molecule level, while absorption measurements utilizing the Beer-Lambert law for quantification commonly require a higher reporter concentration, depending on the reporter’s molar absorption coefficient. The dye label is either covalently bound to the FG on the NM surface requiring a reporter with a complementary reactive group, or interacts with the FG electrostatically in the case of adsorption/desorption assays [63]. In all cases, only the number of derivatizable FG is obtained (see also Fig. 1), which can considerably differ from the total amount of FG particularly for higher FG densities or concentrations, as most dye labels are much larger than the FG to be quantified. To correlate the measured optical properties with label concentration, optical quantification always requires a calibration with a dye closely matching the optical reporter used for FG quantification since the signal relevant optical properties of most reporters are influenced by reporter environment. Calibration can be carried out either with the free (unbound) dye itself, the reacted (bound) dye, or with a model system consisting of the optical label bound to a molecule mimicking the NM surface chemistry, given that the absorption and emission features are closely matching those of the sample.
Optical assays for FG quantification on NM can be distorted by interferences originating from light scattering by the NM, which in turn depends on NM size, excitation wavelength, and the difference in refractive index between the NM and its environment, as well as from NM absorption and/or emission. Only for very small NM (< 25 nm), light scattering is negligible and optical reporters bound to the NM surface can be quantified directly, if the NM does not absorb/emit at the same wavelengths as the dye reporter (spectral discrimination) and if dye-dye interactions at the NM surface can be excluded. For larger particles, light scattering can hamper a reliable and accurate quantification in the presence of the NM. In these cases, the NM has to be removed prior to optical dye quantification by either filtration or centrifugation. Alternatively, the particles must be dissolved, so that only the reporter dyes present in the transparent solution are detected. Also, FG determination via optical reporters can be done by an indirect quantification of unbound labels, or with the aid of cleavable probes and catch-and-release assays where the optical reporter is readout in a transparent solution as detailed in the following sections.
A broad variety of optical assays for different FG on NM and 2D-supports has been developed which utilize different types of absorbing or fluorescent labels. As summarized in Fig. 4, this includes (i) conventional (“always ON”) dyes, (ii) chromogenic/fluorogenic (“chameleon”-type) dyes and activatable (“turn-ON”) dye reporters that change either the spectral position of their absorption and/or emission bands upon reaction with the respective FG or become absorptive (colored) or emissive upon the binding event [64, 114, 115], and (iii) cleavable probes that can be quantitatively cleaved off from the NM surface and subsequently quantified in solution [64, 89]. In addition, (iv) adsorption/desorption assays relying on negatively or positively charged reporters and electrostatic interactions with oppositely charged FG are utilized [116]. These different types of optical assays are subsequently described and compared including representative examples.

Schematic presentation of the working principles of different photometric and/or fluorometric assays for FG quantification on NM using different optical reporters including typical examples for respective dye-based reporters and their absorption and/or emission spectra
FG quantification with conventional “always ON” dyes
Conventional dyes used for FG quantification on NM are fluorophores with a reactive group that allow for the covalent coupling of the label to the FG on the NM surface. Due to the large toolbox of commercial dyes available from different fluorophore classes bearing different reactive groups, that were developed for bioconjugation reactions ranging from simple NHS chemistry to biorthogonal reactions and Click chemistry, this approach can be utilized for all types of bioanalytically relevant FG. The optical properties of conventional dye labels such as the spectral position of their absorption and emission bands as well as their absorption and emission intensities, determined by their molar absorption coefficients and photoluminescence quantum yields (QYPL) commonly change only slightly upon NM conjugation. The size of such changes, particularly in QYPL, depend on dye class, the optical transitions involved, and on the length of the linker between the reactive group of the reporter binding to the NM surface and the dye’s chromophore system. This is advantageous and disadvantageous at the same time. As NM-bound dyes and unbound (free) dyes cannot be spectroscopically distinguished, a separation of bound and unbound dye molecules is necessary prior to optical quantification [64].
Conventional dyes have been applied for the quantification of derivatizable FG on various kinds of inorganic, organic, and hybrid NM, e.g., amino groups on a silane surface using a self-made BODIPY dye and a commercial Rhodamine B dye [117], or aldehyde and azide-containing ligands on the surface of CdSe-ZnS QD using 2-hydrozinopyridine (forming a stable hydrazone chromophore with aldehydes) or an NHS-activated Cy3 dye in conjunction with amino-dibenzocyclooctyne crosslinkers, respectively [118]. Felbeck et al. utilized various NHS-activated conventional dyes to quantify amino groups on the surface of laponite nanoclays modified with 3-(aminopropyl)triethoxysilane (APTES), as shown in Fig. 5 [114]. The authors compared dyes of different charge like the negatively charged hemi-cyanine DY681, the zwitterionic BODIPY 581/591, a neutral dansyl derivative, and the positively charged pyrylium dye Chromeo P503. While charged dyes were prone to aggregation or did not react with the FG on the laponite surface due to electrostatic repulsion, the neutral dansyl dye enabled efficient labeling of the amino groups of APTES.

Quantification of the derivatizable amino groups on APTES-modified laponite disks using differently charged NHS-activated conventional dyes. Reprinted with permission from ref. [114]. Copyright 2015, American Chemical Society
For conventional dye labels, an indirect quantification, i.e., the quantification of the amount of unbound dye molecules, is recommended as the most effective and reliable way to determine the number of accessible FG [64]. Depending on the NM, alternatively, the dye-functionalized sample can be dissolved after removal of unbound label, followed by optical quantification of the residual reporter molecules. Only if light scattering is negligible (e.g., due to a small NP size) and dye-dye interactions can be excluded (e.g., due to a low FG density on the NM surface), a direct quantification of the particle-bound reporters leads to reliable and accurate results [119]. A strategy to circumvent dye-dye interactions for higher FG densities presents NM labeling with a mixture of dye molecules and non-functional molecules bearing the same reactive group, thereby diluting the dye reporters at the NM surface [63]. In this case, FG quantification relies on the assumption of identical coupling efficiencies of both reactants, which needs to be validated individually.
Activatable (“turn-ON”) and chromogenic (“chameleon”) reporter dyes
Activatable reporters are dye precursors that become strongly absorbing (“colored”) or emissive (so-called turn-ON dyes) after covalent coupling to the respective FG on the NM surface, while chromogenic dyes display significant spectral shifts in their absorption and/or emission bands upon the covalent attachment to FG. The latter dyes are sometimes also referred to as “chameleon dyes.” Well-known examples for activatable dyes utilized as reporters in photometric and fluorometric assays are Ninhydrin (2,2-dihydroxyindane-1,3-dione) and Fluram (4′-phenylspiro[2-benzofuran-3,2′-furan]-1,3′-dione) that both form optically detectable products upon reaction with primary amino groups.
Ninhydrin, that has been initially used in protein assays, forms the dye Ruhemann’s Purple with primary amino groups, absorbing at about 570 nm [120]. The photometrically detectable colored species is released and the absorption measurement is done in the supernatant, thereby circumventing interferences from possible scattering of the excitation light by the NM. As the Ninhydrin reaction is an equilibrium reaction, Ruhemann’s Purple is continuously generated in the presence of primary amino groups reacting to aldehyde functionalities, which reduces the impact of steric crowding of multiple Ninhydrin molecules occupying neighboring FG. Ninhydrin was, e.g., used to monitor the reproducibility of silica particle synthesis and their modification with APTES in comparison to NMR measurements [121], and to study the FG density and colloidal stability of surface functionalized silica NP over a period of time of 30 days [122]. Sun et al. compared the quantification potential of two optical assays (Ninhydrin and 4-nitrobenzaldehyde) with 19F solid state NMR measurements to quantify FG on amino-modified silica NP of different sizes (see Fig. 6) [123].

Comparison of two optical assays utilizing dye reporters (Ninhydrin, 4-nitrobenzaldehyde) and quantitative 19F NMR using the F-containing label trifluoromethyl benzaldehyde for FG quantification on amino-modified silica NP of different sizes. Reprinted from ref. [123] with permission from the authors (CC BY-NC 3.0)
Fluram, that is also referred to as Fluorescamine, is a colorless dye precursor that forms a yellow product with primary amines with a strong emission between 400 and 600 nm. As the amino group is integrated into the fluorophore in a ring-formation mechanism, the emissive Fluram product needs to be measured directly bound to the NM surface. As unreacted Fluram itself is not emissive, no washing or purification steps are required to remove the unreacted precursor dye prior to assay readout. As the emissive dye product is of limited stability, the assay should be read out at a constant time point after the reaction with Fluram. Moreover, particle light scattering and dye-dye interactions can interfere with the Fluram assay, as has been demonstrated in comparison to the results obtained with the cleavable fluorenylmethyloxycarbonyl protecting group (Fmoc, vide infra) [124]. Fluram was applied to study the influence of the FG density on the biocompatibility of aminated silica NP [125], and to quantify primary amino groups on nanoclays (see Fig. 5) as well as on PS nano- and microparticles [64, 114]. In the latter case, a reliable and accurate FG quantification with the Fluram assay involved the dissolution of the polymer particles in an organic solvent and a correlation of the subsequently detected dye signal with a calibration curve obtained with a suitable model system consisting of the dye bound to a small molecule such as propylamine bearing a primary amino group.
A related method, here for the quantification of thiol groups, is the Ellman’s assay which exploits the reaction of 5,5′-dithiobis-2-nitrobenzoic acid (DTNB, also called Ellman’s reagent) with thiolate anions to a mixed disulfide and 2-nitro-5-thiobenzoic acid, which can be detected photometrically at about 410 nm. The Ellman’s assay, which has been initially developed for the quantification of thiol groups on proteins, has been used to quantify thiol groups (directly) or maleimide groups (indirectly after reaction with l-cysteine) on functionalized PS particles [126, 127], and to determine the number of thiol ligands like mercaptopropionic acid (MPA) or dithiol dihydrolipoic acid (DHLA) on semiconductor QD and noble metal particles [65, 127].
Chameleon dyes possess an electron-withdrawing group conjugated with the chromophore π-system (e.g., a halogen atom such as -Cl) that is transformed upon the reaction with a FG, e.g., a primary amino group, into an electron-donating group, resulting in strong blue shifts in absorption and emission. The spectral shifts as well as the changes in the molar absorption coefficients and QYPL values are considerably influenced by the exact chemical structure of the analyte or FG (i.e., the electron donating amino group-containing ligand) substituting the electron-withdrawing group. Cyanine-based chameleon dyes have been utilized for the labeling and subsequent detection of biomolecules containing primary amino groups [128] and to confirm the amino modification of silica and PS NP both photometrically and fluorometrically [129]. The chameleon dye IR797 was used by us to quantify the amount of accessible amino groups on PS nano- and microbeads [64]. As for activatable reporters, a reliable and accurate FG quantification required the dissolution of the dye-bound particles and a thorough calibration with a suitable model system. Another class of chameleon dyes also suited for FG quantification are pyrylium reporters that react with primary amino groups to form pyridinium dyes with strongly blue shifted absorption and emission bands [115].
The advantage of activatable and chromogenic dye reporters compared to conventional dye labels are the different optical properties of the NM-bound and free dyes, allowing for a straightforward spectroscopic discrimination between these species. For activatable dyes, only a quantification of the NM-bound reporters is possible, which can be hampered by particle light scattering, while for chromogenic labels a spectroscopic quantification of both the NM-bound dyes and the unreacted free dyes is feasible. Moreover, some NM such as metal particles or semiconductor QD exhibit strong absorption and/or emission bands that can interfere with the dye spectra. As for conventional labels, FG quantification requires a calibration curve from a model system with absorption and/or emission properties that closely match those of the NM-bound activatable or chromogenic dyes to consider the environment dependence of the absorption and emission features of the reporter, particularly its QYPL.
Labeling with cleavable probes, catch-and-release assays, and indicator displacement assays
Modularly built cleavable probes consist of a reactive group that can be coupled to the FG of interest, a cleavable linker that can be cleaved fast and quantitatively after the conjugation reaction, and a reporter unit subsequently released which can be quantified photometrically or fluorometrically. Suitable cleavable linkers can be taken from established drug release concepts like pH-cleavable hydrazone bonds or reductively cleavable disulfide bridges. Another type of cleavable probes are optically detectable protection groups like Fmoc, which is frequently used to determine resin substitution in solid-phase peptide synthesis. Fmoc can be used for the quantification of amino groups on NM by cleaving off the NM-bound Fmoc protecting groups with piperidine in DMF followed by photometric or fluorometric detection of the released dibenzofulvene-piperidine adduct [130,131,132,133,134,135,136]. Meanwhile, variations of the reaction solvent [137] and other suitable detection wavelengths [138] have been reported. To increase the sensitivity of the assays, that was initially read out photometrically, a Fmoc-Cl fluorescence assay was developed that can be performed in aqueous solution. This assay is approximately 50–200-fold more sensitive than the photometric method, but the separation of excess Fmoc-Cl and its strongly fluorescent reaction products is still challenging [124, 139].
We rationally designed the cleavable probes N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP) and N-(aminoethyl)-3-(pyridin-2-yldisulfanyl)-propane amide (N-APPA) by combining a reactive NHS- or amino group, a reductively cleavable disulfide linker, and a simple 2-thiopyridone reporter unit. [64] To demonstrate the advantages of these cleavable probes, we compared their performance with that of conventional dye labels and activatable/chromogenic reporter dyes (vide supra) for the quantification of amino and carboxy groups on PS nano- and microparticles, as shown in Fig. 7. This comparison confirmed the advantages of SPDP and N-APPA for FG quantification, i.e., the possibility for determining mass balances and a straightforward method validation with other analytical methods like 32S ICP-OES. These cleavable probes are also suited for quantifying amino and carboxy groups at various FG densities and even on absorbing and fluorescent NM like dye-stained fluorescent particles.[89]. Moreover, this design principle can be easily adapted to other FG. For example, we developed the SPDP derivative 3-(2-pyridyldithio)propionyl hydrazide (PDPH) bearing a reactive hydrazide group for the quantification of aldehyde groups on a set of PMMA microparticles [140]. Validation was done by comparison with another catch-and-release assay utilizing a hydrazide-functionalized fluorescent BODIPY dye (BDP-hzd) as reporter that proved to be even more sensitive due to the fluorometric readout.

Comparison of optical FG quantification using cleavable probes, conventional dyes, and activatable/chromogenic reporters. Validation of the former approach was done with ICP-OES and the Ellman’s assay. Reprinted with permission from ref. [64]. Copyright 2018, American Chemical Society. Further permissions related to the material excerpted should be directed to the American Chemical Society
Other examples for optical assays utilizing cleavable probes present the use of 7-methoxycoumarin-3-carboxylic acid conjugated to an alkyne group for the fluorometric quantification of azide groups on solid substrates, silica particles, and biomolecules after cleaving off the dye under basic conditions [141], and the use of 4-nitrobenzaldehyde (4-NBA) for the photometric quantification of amino groups on silica particles of different sizes (see Fig. 6) after dye hydrolysis [123]. A similar type of assay that also relies on reporter detection in solution after particle removal is the so-called indicator displacement assay that exploits supramolecular host-guest chemistry, e.g., the competitive binding of the reporter dye acridine orange (AO) and the high affinity guest aminomethyladamantane (AMADA) to the macrocycle host cucurbit[7]uril (CB7) for the optical quantification of azide groups on PMMA particles [142].
Cleavable probes and catch-and-release assays are ideally suited for FG quantification on NM, since quantification of both the unbound (unreacted) dye and the initially particle-bound reporter (after cleavage) can be performed in solution after removal of the NM. Hence, these methods allow for FG quantification without interferences from light scattering, absorbing and/or emitting NM, or dye-dye interactions of surface-bound reporters. The cleavable reporter approach enables to generate a mass balance from the known amount of applied label and the measured amount of unbound and bound reporters, which increases the accuracy and reliability of this FG quantification method and simplifies method validation. In addition, the modular design of the cleavable probes offers the opportunity to specifically choose the reactive group, the cleavable linker, and the reporter unit according to the desired application and sample-specific requirements (specific type of target FG, limitations due to particle material-related properties, etc.).
Optical adsorption/desorption assays
Alternatives to optical assays involving covalent labeling are adsorption/desorption assays with photometric or fluorometric readout. In an adsorption/desorption assay, optically detectable reporter dyes or, less common, small metal ions with charges complementary to that of the FG of interest, are allowed to adsorb at the charged NM surface. The reporters must not bear a reactive group and should not penetrate the particle matrix. Subsequently, non-adsorbed probe molecules are removed by several washing steps, followed by quantitative desorption of the adsorbed reporter by addition of a surfactant. Then, the desorbed reporter is quantified photometrically or fluorometrically in the supernatant after removal of the NM by centrifugation or filtration. In the case of metal ions, with few exceptions [143], optical detection is achieved by addition of an indicator dye that forms a colored or fluorescent product of defined stoichiometry with the metal ion. Alternatively, the non-adsorbed amount of the reporter can be quantified after removal of the NM containing the fraction of the adsorbed reporter. In conjunction with a fluorescent dye and readout with fluorescence microscopy or flow cytometry, also the amount of particle-adsorbed fluorophore can be measured directly [144]. The use of metal ions as reporters in adsorption/desorption assays for charged FG like carboxy and amino groups exploits the much smaller size of metal ions compared to organic dyes which is expected to provide a reporter-to-FG stoichiometry close to 1, and thus, a number of (accessible) FG approaching the total number of FG.
A popular dye-based adsorption/desorption assay for the photometric quantification of carboxy groups is based on the cationic dye toluidine blue (TBO) that displays an intense blue color with an absorption maximum at around 630 nm [63]. An example for a dye-based adsorption/desorption assay with fluorometric detection utilizes the red emissive cyanine-type nucleic acid stain SYTO-62 [144]. Metal ion-based adsorption/desorption assays have been reported by several research groups, e.g., using Mg2+ ions to quantify tryptophan ligands on gold NP [143], or Ni2+ ions to quantify carboxy groups on polymeric microparticles [116, 145].
Advantages of adsorption/desorption assays are their simplicity, as they are in principle suitable for all types of NM bearing charged FG independent of their chemical composition, if dye penetration into the NM matrix can be excluded. Thus, such assays are not suited for porous materials like mesoporous silica NP. They are very versatile and require only one calibration for different NM samples. A drawback presents the time-consuming washing steps needed for quantitative dye desorption. Moreover, as typically more than one FG interacts with one reporter molecule, FG quantification requires the determination of a stoichiometry factor by comparison with the results obtained by another method yielding the total number of FG [63]. This stoichiometry factor is most likely NM-specific, which can affect the reliability of FG quantification with this type of assay without a thorough validation. Adsorption/desorption assays are well suited for quality assurance and process control (including control of product reproducibility) of NM bearing charged FG as well as the monitoring of aging effects affecting surface FG, as such conclusions can be drawn based upon relative comparisons.
Other methods for FG quantification on nanomaterials
Other analytical techniques used for FG analysis and quantification on NM surfaces include NMR spectroscopy, ICP-MS and ICP-OES, IR and Raman spectroscopy, X-ray-based methods such as XPS and XRF, as well as thermal analysis methods and elemental analysis [18, 23, 51]. Depending on the chemical nature of the NM and the FG of interest, these analytical techniques can either utilize intrinsically present moieties (label-free methods) or specific reporters (label-based methods) for signal generation and quantification, and thereby, provide the total or derivatizable number of FG. These analytical methods are very valuable tools for FG quantification and for validation of the results obtained with simpler methods (method validation). A straightforward approach to simplify method comparisons for method validation and calibration is the utilization of multimodal labels and reporters that are designed for the readout by different analytical techniques relying on different signal generation principles. Multimodal reporters can be realized, e.g., by including heteroatoms like sulfur, nitrogen, fluorine, or certain metal ions into molecular labels like organic dyes used for chemical derivatization reactions or reporters utilized for the design of cleavable probes. In the following, also examples for this strategy including its use to provide a traceability chain of FG quantification to the SI unit mole are highlighted.
Nuclear magnetic resonance spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy measures the intrinsic magnetic moments of certain nuclei such as hydrogen (1H), carbon (13C), fluorine (19F), or phosphorus (31P) in the presence of a strong magnetic field. NMR spectroscopy can provide chemical, physical, and structural information about the NM, its organic ligand shell and surface FG, as well as information on dynamic interactions with the environment [146]. Moreover, it can distinguish between surface bound and free (excess) ligands which has been exploited to gain a deeper understanding of the NM-ligand interface including ligand binding sites and dynamics, particularly for semiconductor QD with their surface-dependent luminescence properties [29, 147,148,149,150].
The quantification of FG and ligands on NM surfaces can be carried out by solution phase NMR techniques and by solid-state NMR. It typically involves the addition of an internal standard of known concentration and known, high purity, that is chosen to reveal NMR signals (chemical shift) well separated from the NMR signals originating from the FG or ligands of interest and the matrix [51, 89]. Particularly for solution NMR, it must also be considered that the NMR signals of the FG or ligands bound to a NM surface can significantly change compared to the signals of the free molecule or FG in solution. The NMR signals obtained typically show differences in both chemical shift and linewidth due to homogeneous and/or inhomogeneous line broadening, which can hamper peak assignment and integration [151]. The size of such effects depends on NM size. Since the percent weight of the bound surface ligands decreases with increasing particle size, quantification of larger size NM can require a relatively large amount of sample compared to other analytical methods. To overcome these limitations, dissolution methods for FG and ligand quantification prior to NMR analysis have been developed [121, 152]. Particularly for organic polymer particles, the distinction of NMR signals originating from surface FG and the NM matrix can present a challenge, which can be met by using isotope-enriched reagents for surface functionalization [63], or by applying a multi-Lorentzian-splitting algorithm [153]. Also, combinations of multinuclear and multidimensional NMR techniques are a promising approach to identify and quantify FG/ligands, and to study their interaction with and binding to the NM surface [154,155,156].
Quantitative NMR (qNMR) is particularly attractive for FG quantification, due to its potential for an absolute quantification. Moreover, it can provide traceability to the SI unit mole if suitable calibration materials of very high and known purity are available [157, 158]. For instance, qNMR was used to study the FG or ligand density on gold NP [159, 160], semiconductor QDs [161], and silica NPs [123, 162]. However, qNMR requires special measurement conditions. Prior to collecting NMR spectra, the T1 relaxation times of the components must be determined with a series of inversion-recovery experiments. These T1 times then have to be considered for the recording of the NMR spectra used for signal quantification which commonly requires a relatively high number of scans, and thus, elongated measurement times. For data evaluation, the integral values of the evaluated signals must be baseline-corrected and the purity of the internal standard added in a precisely known amount must be determined, for example by comparing its NMR signals to that of a reference material of certified purity. Despite the need for expensive equipment operated by well-trained scientists, qNMR has become increasingly popular for the quantification of FG on NM due to its inherent chemical selectivity and the provision of the total number of FG without the need for a calibration curve. Also, this technique is increasingly used for the calibration and validation of other more simple analytical methods, particularly for NM where electrochemical titrations cannot be utilized due to interferences from the NM matrix, or for the quantification of FG that are electrochemically not accessible. For example, the quantification of carboxylate and amino functionalities on silica NM is not feasible by electrochemical titrations as the pKa values of the inherently present silanol groups and carboxylate groups cannot be well separated and as silica NM dissolve at alkaline pH. Here, qNMR is the method of choice for the quantification of the total amount of carboxylate and amino groups [89, 123, 156, 163, 164].
An example for a possible traceability chain for FG analysis with different analytical methods including NMR is shown in Fig. 8, using multimodal reporters that can be read out by different analytical techniques, here solid-state 19F NMR, emission spectroscopy, and XPS, and a certified NMR reference standard containing both 19F and 1H [157, 158].

Example for a traceability chain for FG quantification, linking measurements (blue arrows) of XPS (a) and fluorometry (b) to quantitative solid-state 19F NMR (solid red arrows). The use of a certified NMR reference standard containing both 19F and 1H provides the link to the SI unit mole (dotted red arrows). Reprinted from Ref. [158] (CC BY 3.0 unported)
Mass spectrometry and atomic spectroscopy
Mass spectrometry (MS) and optical emission spectrometry (OES; also referred to as atomic emission spectroscopy, AES) can quantitatively measure the total number of atoms of certain elements within a sample. This can be utilized for the quantification of surface FG and ligands on NM as well as for the determination of NP concentration. Typically, both techniques use inductively coupled plasma (ICP) to produce excited atoms and ions, which are then identified based upon their characteristic mass-to-charge ratios (ICP-MS) or atomic spectral emission lines (ICP-OES). ICP-MS and ICP-OES directly measure the element concentration with unparalleled sensitivity over a wide linear dynamic range regardless of NM size or surface chemistry down to the parts-per-trillion level. However, the achievable detection limits (LOD) depend on the element(s) of interest, instrument, experimental conditions, and possible spectroscopic interferences between the analyte elements. For NM analysis, ICP-MS and ICP-OES are often used in connection with an upstream particle separation method like high performance liquid chromatography (HPLC) or asymmetric flow field-flow fractionation (AF4) [165,166,167,168,169,170].
ICP-MS and ICP-OES are often applied to determine the NM concentration from the measured element concentration of the sample in combination with the known NM dimensions, typically determined with sizing techniques such as TEM, SAXS, DLS, or NTA [166, 171, 172]. However, both methods can also be applied to detect elements that directly correspond to ligands and FG native on the NM surface, and thus, the total amount of FG, or to detect specific labels conjugated or associated to the FG, and hence, the number of derivatizable FG. Examples for the latter case present the quantification of elements such as sulfur present in certain dyes or cleavable probes [64, 89, 127], or the use of metal ion containing reporters [173]. ICP-OES has been used to quantify FG on various NM such as carbon-based NM, noble metal NP, polymer beads, and lanthanide-based NP [127, 174,175,176,177]. ICP-MS has been frequently used to quantify FG-bearing ligands on gold NP [169, 173, 178,179,180], but has also been applied to quantify FG and ligands on other NM such as silica NP, polymeric beads, and semiconductor QD [126, 166,167,168]. Particularly single-particle ICP-MS (sp-ICP-MS) is a promising technique as in principle it allows to simultaneously measure the particle number concentration and the number of FG/ligands on the surface of individual particles. However, especially for light elements often present in commonly used organic FG and ligands, that are difficult to ionize (large ionization potential) and are prone to a high background, quantification is very challenging and detections limits are higher compared to other heavier elements. Further instrument improvements and methodological advances can make sp-ICP-MS a very well-suited method for NM characterization [181,182,183].
In principle, atomic absorption spectroscopy (AAS) with either flame-based or electrothermal (graphite tube) atomizers can also be used to quantify FG and ligands, but as AAS is mostly limited to metallic and semi-metallic elements that are typically not present in organic ligands and surface groups, this technique is of very limited use for FG analysis [184]. However, there exists a special AAS technique that is very sensitive to fluorine [185], which can be utilized for the quantification of FG derivatized with fluorine-containing labels, such as BODIPY dyes for the validation of optical assays or other elemental tags bearing CF3 groups for the comparison with XPS measurements or 19F-NMR (see Fig. 8).
Other MS methods that can also be used for NM surface characterization, but are not further detailed here, are time-of-flight secondary ion mass spectrometry (ToF-SIMS) and matrix-assisted laser desorption/ionization (MALDI) in combination with time-of-flight (ToF) analysis of the released ions by mass spectrometry (MALDI-ToF-MS). These methods can provide molecular information of FG and surface bound molecules [186,187,188], but up to now, have been rarely utilized for NP analysis.
Vibrational spectroscopy
Vibrational spectroscopy measures the absorption or (inelastic) scattering of incident light due to vibrational stretching and/or bending modes of molecules within a sample. Infrared (IR) spectroscopy relies on the absorption of IR radiation, typically in a wavelength region between 2500 nm and 25 μm (4000–400 cm1 on the wavenumber scale). The fundamental vibrations of most chemical bonds occur within this spectral region. IR spectroscopy is frequently being used for qualitative and quantitative analysis of organic compounds in agriculture, food products, polymers, pharmaceuticals, cosmetics and the petroleum industry as well as for the monitoring of chemical reactions in process analysis [189,190,191]. Nowadays, commonly Fourier-transform infrared (FTIR) spectroscopy is used, allowing for the simultaneous detection of all vibrational frequencies [192, 193]. Raman spectroscopy is a complimentary vibrational spectroscopic technique, which detects inelastically scattered photons (Raman scattering) from a monochromatic light source (usually a laser) in the near-UV to near-IR range. Due to the different selection rules valid for both methods, Raman spectroscopy yields similar—yet complementary—information compared to IR spectroscopy [194]. Both methods are relatively fast and non-destructive, and can be coupled with other analysis methods. For FG and ligand analysis on NM, surface-sensitive variants of these techniques are of particular importance like attenuated total reflection (ATR-) FTIR [195, 196], diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) [197, 198], and surface-enhanced Raman spectroscopy (SERS) [199,200,201]. While SERS is limited to electrically conducting materials such as graphene or noble metals, FTIR spectroscopy is frequently employed to study a wide range of NM. Both DRIFTS and ATR-FTIR overcome the shortcomings of sample preparation complexity in classical FTIR spectroscopy, and ATR-FTIR further allows in situ characterization of particle surfaces in biologically and environmentally relevant media [196]. Although vibrational spectroscopy is widely accepted for qualitative analysis of NM surfaces [202,203,204,205], there are only few literature reports on the application of these methods for quantitative analysis. Nevertheless, with modern instrumentation and combined approaches, the quantitative determination of FG and ligands at the nano- to picogram level should be feasible [206, 207]. Examples for FG determination with ATR-FTIR spectroscopy include the measurement of the density of thiol and bromoalkyl FG on silica particles [208] and the amount of APTES on silica-coated iron oxide NP [209]. Furthermore, ATR-FTIR can be applied to characterize and (semi-)quantify the chemical composition of mixed ligand layers at NM surfaces [210, 211]. To quantify FG and surface ligands with FTIR spectroscopy, a calibration curve is generally required and samples that obey the Beer-Lambert law. A combined use of chemometric tools such as convolutional neural networks and ensemble learning, with different vibrational spectroscopy techniques, might contribute to an increased accuracy of quantitative FG analysis in the future [212].
X-ray-based methods
X-ray photoelectron spectroscopy (XPS) measures the number and kinetic energy of electrons that escape from the near-surface region of a sample, like a planar substrate or particles deposited on a substrate, up to an information depth of 5–10 nm upon irradiation with an X-ray beam in vacuum. In laboratories, typically Mg Kα or Al Kα sources with photon energies of 1253.6 eV or 1486.6 eV are used. The information depth of XPS is determined by the inelastic mean free path (IMFP) of the photo-excited electrons in solid matter. Using higher photon energies between 3 and 15 keV as in hard X-ray photoelectron spectroscopy (HAXPES) can extend the information depth up to about 100 nm [213]. XPS measurements provide information on surface composition, the enrichment or depletion of elements at the surface, the presence and/or thickness of coatings, and the chemical states of the elements [23, 214, 215]. Therefore, information on the chemical composition and amount of FG or certain chemical species at the particle surface can be obtained [216]. Due to the sensitivity of XPS for all elements except H and He, not only inorganic surface coatings, passivation shells, and surface modifications by chemical processes like oxidation or etching can be determined [217,218,219], but also organic coatings, ligands, and surface-bound biomolecules [220, 221]. Depending on the chemical composition of the sample and the surface ligands or FG, XPS can be done without the need for a label. A reliable quantification requires calibration with suitable reference material or standards [222]. Alternatively, theoretically derived values for the cross sections (Scofield factors), the IMFP and the transmission function of the spectrometer can be used [214, 223,224,225]. It must be noted that all these quantification procedures are only valid to homogeneous planar surfaces; however, there are different approaches which can be used for NP [214]. To enhance the sensitivity and selectivity of XPS and/or to enable the comparison with other more quantitative methods like NMR for method calibration and validation, for example fluorine-containing reactive labels like 3,5-bis(trifluoromethyl)phenyl isothiocyanate [226], trifluoroacetic anhydride (TFAA) [227], and 2,2,2-trifluoroethylamine (TFEA) [158] can be employed for the chemical derivatization of specific surface FG like amino, hydroxyl, and carboxy FG, respectively. The use of TFEA to quantify the amount of carboxy groups on PMMA particles with a grafted shell of PAA also provides a link to the SI unit mole, as shown in Fig. 8 [157, 158].
In the last years, an increasing number of examples for the applicability of XPS to determine the elemental distribution of small inorganic NM and the chemical surface modification of various NP by light-induced chemical changes, ligand exchange, or the presence of certain species like chloride ions has been reported [23, 28, 219, 228]. XPS was also used to derive the thickness of inorganic passivation shells, e.g., on semiconductor QD like CdSe/CdS by simulation of the spectra [214]. As the size of many NM is larger than the XPS information depth, and as NP size, shape, and morphology determine the fraction of surface elements accessible within this information depth, the use of XPS for the quantification of NM ligand shells and FG is challenging and requires mathematical modeling of the measured data, e.g., with a software like SESSA (Simulation of Electron Spectra for Surface Analysis) [229], thereby also considering the size- and shape-dependent curvature of the NM surface. New approaches for the quantification of XPS at nanostructured materials take these effects into account, but further developments are necessary for more complex materials [214, 230,231,232,233]. Another challenge of XPS measurements is the need of an appropriate sample preparation and handling to prevent changes of the NM due to undesired influences from the surrounding of the particles [23]. Also, NM agglomeration and aggregation as well as decomposition or changes induced by the X-ray beam can influence the reliability of the obtained results.
Another X-ray based analytical technique that is in principle capable of quantitatively measuring the elemental composition of a sample presents X-ray fluorescence (XRF) spectroscopy, which detects the secondary (“fluorescent”) X-rays emitted from a material after excitation with high-energy X-rays (or sometimes gamma rays) using either energy-dispersive (EDXRF) or wavelength-dispersive (WDXRF) spectroscopy [234]. As the secondary radiation from lighter elements (with Z < 12) is of relatively low energy (< 3 keV) and has a low penetration depth, it is often reabsorbed by the sample and severely attenuated by any matter between the sample and the detector. Hence, XRF is usually only applied to characterize the chemical composition or impurities on inorganic materials such as metal, glass, ceramic, and semiconductor surfaces, films, or layers. However, using vacuum technique, synchrotron radiation, and/or special detector windows also lighter elements such as carbon, nitrogen, oxygen, and fluorine can be detected [234,235,236]. Particularly the surface-sensitive methods total reflection X-ray fluorescence (TXRF) and grazing-incidence X-ray fluorescence (GIXRF) spectroscopy using X-ray standing waves (XSW) are principally well suited for a reference-free quantification of FG on NM [237,238,239], but for particle samples additional corrections are necessary to account for absorption and shadowing effects occurring on the nanostructured surfaces [240].
Other techniques to determine the total number of FG
Other techniques that can be applied for FG and ligand characterization and are more or less well suited for a quantitative analysis include elemental analysis (EA), thermal analysis methods such as thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), and separation techniques like asymmetrical flow field-flow fractionation (AF4).
EA is an inexpensive method to determine the elemental composition of a material by combusting the sample under controlled conditions and analyzing the combustion products quantitatively. Particularly the so-called CHNS analysis, that provides the mass fractions of carbon, hydrogen, nitrogen, and sulfur via their combustion gases under high temperature and high oxygen conditions (converting these elements to their oxidized form, i.e., to CO2, H2O, NO or NO2, and SO2, or reducing, e.g., NO or NO2 to N2), is in principle well suited for FG or ligand quantification on NM, as long as the NM matrix does not contain the FG-specific element(s). These gases are then detected by a thermal conductivity detector that typically is calibrated daily with suitable standards or reference materials. However, CHNS analysis has only rarely been used for FG quantification, e.g., for carboxy groups on alumina particles, [241] or amino groups on graphene oxide [242] as well as silica particles [243, 244], due to relatively large amounts of sample needed and the relatively low detection sensitivity compared to other methods like ICP-MS [126]. For NM characterization, CHNS analysis is often used only in conjunction with other analytical methods such as FTIR, TGA, or XPS [126, 242, 243, 245].
TGA and DSC are typically used to obtain information on the thermal stability, chemical composition, and purity of a sample as well as on kinetic parameters. DSC can also be employed to determine phase transitions, e.g., of liquid crystals. TGA detects the resulting mass changes as a function of temperature under defined conditions, while DSC measures the difference in the amount of heat required to increase the temperature of the sample [246]. Recent results of the EU funded project ProSafe identified TGA as a very useful, simple, and reliable method to study the surface chemistry particularly of inorganic NM like silica, metal, and lanthanide-based NP as well as QD [247]. For TGA measurements, no complex sample preparation is needed. However, due to the underlying measurement principle, all contaminants present in the sample such as remaining dispersion media/solvents, impurities originating from NM synthesis, and free ligands/molecules can contribute to the mass changes, and thus, influence the results. Therefore, a careful work-up and clean-up procedure of the NM sample to be analyzed is mandatory. Also control samples and precise heating steps at lower temperatures are often included to overcome the inherent challenge of undesired mass contributions from matters other than the organic surface ligand/FG [164, 248]. Another drawback is the amount of NM needed for a single TGA analysis, i.e., commonly several milligrams, which can make TGA less suitable for small-scale samples of functionalized NM used in biomedical applications. Modern TGA methods address these limitations by a higher sensitivity. Mansfield et al. [246] developed a microscale TGA method (μ-TGA) using a quartz crystal microbalance, that needs a 1000-fold reduced amount of sample. With this method, the authors could obtain results for commercial CNT, polymer-coated gold NP as well as polymer-modified gold/silica NP which were comparable with those determined by conventional TGA [246, 249, 250]. They also used μ-TGA to quantify the amount of surface-bound poly-l-lysine and DNA on gold NP intended for potential use in biomedical applications [250]. Another advanced TGA approach presents the coupling with mass spectrometry (TGA-MS) or FTIR spectroscopy (TGA-FTIR) to enable the identification of the species responsible for the observed mass loss upon heating. For example, TGA-MS has been used to determine the amount of aromatic molecules adsorbed on CNT [251] and to identify the organic compounds extractable from various NM [252]. Moreover, TGA has been used in multi-method approaches for quantitative analysis to enhance the reliability of the results by comparing them with information from FTIR, NMR, ICP-MS/OES, and XPS measurements [164, 175, 186, 253,254,255,256]. And just recently, TGA has been applied to quantify adsorbed citrate molecules on the surface of gold NP, which provided new insights into mechanistic details of the well-known Turkevich gold NP synthesis method [257].
As the density of FG/ligands on the NM surface determines its charge and hydrophilicity/hydrophobicity, also NP separation techniques like AF4, a fractionation method that is used for the characterization of polymers, proteins, and NP, coupled with capillary electrophoresis (CE) can be used for FG quantification. Combining AF4 and CE provides separation by size and surface charge. The correlation of these parameters requires a calibration curve with similarly sized particles of known FG density to correlate the CE retention times with FG density [258]. This method is, however, only suited for surface FG that control particle charge, e.g., for (de)protonable functionalities such as carboxylic acids and amines. Moreover, in addition to the method-inherent limitation of AF4, this method combination faces the same limitations as electrochemical titration methods, except for signal contributions from the presence of other (de)protonable molecules due to the coupling with a NP separation technique.
Conclusion and future challenges
All analytical techniques presented here for the analysis and quantification of functional groups (FG) and ligands on nanomaterial (NM) surfaces possess specific method- and material-related requirements and limitations. For a straightforward, efficient, and reliable quantification of FG and ligands on NM, these parameters need to be considered. Also, the distribution of NM size, shape, and surface morphology must be taken into account for a thorough determination of the FG/ligand density (number of FG/ligands per particle or surface area), which can vary from particle to particle in a NM population. In addition, it must be kept in mind whether the total number or the derivatizable number of FG is desired for which purpose/application and with which uncertainty. For example, quality control during NM fabrication and surface modification or the bioconjugation of NM reporters to bioligands for fluorescence assays do not necessarily require the knowledge of the total number of FG. For the production and characterization of nanoscale reference materials, e.g. as negative and/or positive controls in toxicity tests or for the application in the health sector or as food additives can impose more stringent requirements on NM characterization. To provide guidelines for the choice of the optimum method(s), an overview of the most relevant criteria for the choice of suitable methods for FG and ligand quantification on NM is summarized in Table 1.
Electrochemical titration methods are inexpensive, rapid, require only simple and widely available laboratory instrumentation, and can be performed by technical staff under ambient conditions at constant temperature. Also, data interpretation is comparatively easy and straightforward. This makes these methods well suited for routine analysis as well as production and quality control of synthesized or functionalized NM. Sometimes the removal of CO2 from air is mandatory by purging sample solutions and dispersions with nitrogen or argon. Moreover, it must be assured that complete (de)protonation has been reached after each titration step. All electrochemical methods provide the total amount of the FG of interest present in the sample, i.e., on the NM surface, as the reporters used for signal generation, namely protons or hydroxide ions, are very small [64, 89]. This has been verified, e.g., by comparing the results from conductometry and quantitative solid state NMR measurements [63]. In principle, also the number of derivatizable FG can be obtained via electrochemical measurements by comparing the results prior to and after FG derivatization, combining electrochemical titrations with other quantification methods such as optical spectroscopy utilizing dye reporters. A general drawback of electrochemical methods is their lack of specificity and selectivity, as they measure all (de)protonable species with comparable pKa values present in the sample, and hence, not necessarily only the number of FG on the NM surface. As previously mentioned, other species remaining from NM synthesis like surfactants, initiators, stabilizers, or excess ligands with (de)protonable groups can also contribute to the measured signals, and thus, can distort the obtained results if not removed prior to analysis. Also, other (de)protonable FG on NM can interfere with the signals originating from the target. For example, for NM bearing a mixture of different types of FG like carboxy and amino groups, the simultaneous quantification of the total amount of both types of FG is very challenging if not impossible [64]. This implies also, that certain particle matrices can interfere with electrochemical titrations like mesoporous silica where the close match of the pKa values of the silanol groups (pKa 4.5–5.5) and carboxy groups pKa (about 4.8) prevents a discrimination between these FG. Moreover, the NM needs to be colloidally stable during the course of the acid-base titration, i.e., at the pH conditions necessary for (de)protonation of the FG/ligand of interest. For example, the dissolution of the silica matrix at alkaline pH values required for the determination of amino groups (pKa about 9–11) renders the electrochemical quantification of amino FG on silica not feasible [89]. In addition, electrochemical titration methods like conductometry require a relatively large amount of sample (about 10–20 mg/mL of the surface functionalized NM), which renders these methods suitable only for relatively simple and self-made particles that are either not expensive or can be easily prepared in larger quantities. For expensive NM that are difficult to obtain on a large scale or for surface ligands which are either costly or difficult to synthesize, other methods that require less amount of sample present a better choice like optical assays.
Optical assays, particularly fluorometric assays, are very sensitive, require only small amounts of sample, and can be performed by technical staff with standard bench-top instrumentation available in most laboratories. This makes them ideal for routine process monitoring during particle manufacturing and for quality control of surface functionalization. Moreover, optical measurements are simple and fast, and the chemical derivatization step necessary for labeling-based assays can provide an additional selectivity. Meanwhile, there is a large toolbox of differently sized optical reporters with various reactive groups commercially available including the different types of reporters introduced in the section on optical assays. However, for all optical assays as well as for other analytical methods relying on labeling reactions, only the number of derivatizable FG is obtained and the result can be affected by a combination of the reactivity of the label or reporter, the underlying reaction mechanism, the reaction yield, and particularly for crowded surfaces, also by reporter size, shape, and charge. Depending on the NM application, the influence of reporter size and shape must not be a disadvantage but can provide a more realistic estimate of the number of FG that can be derivatized with the molecule of interest. For bioconjugation reactions involving large biomolecules, commonly, the number of derivatizable FG determined with a reporter such as an organic dye provides an upper limit of the FG on the NM surface that can be coupled to the respective biomolecule. A general drawback of optical methods is the need for a suitable calibration to correlate the intensity of the measured optical signal to the analyte concentration, i.e., the amount of FG or ligands. The calibration needs to consider the sensitivity of the reporter’s optical properties to its microenvironment, which can change the spectroscopic properties relevant for quantification, i.e., the reporter’s molar absorption coefficient and especially its QYPL values. This can be accomplished by choosing a standard for assay calibration that closely matches the reporter and its environment in the optical assay. This can make an accurate calibration (i.e., measurement uncertainties < 5%) tedious and can render the calibration sample specific. However, if larger uncertainties exceeding 20% are acceptable, a universal calibration could be sufficient. Also, optical signals can be distorted by interferences from the sample material like size- and wavelength-dependent light scattering as well as NM absorption and/or emission. For fluorescent labels that are detected when bound to the NM surface, quantification can also be hampered by reporter-specific and labeling density-dependent dye-dye interactions resulting in fluorescence quenching. Such sources of uncertainty can be elegantly circumvented with the aid of cleavable probes and catch-and-release assays.
Analytical techniques like NMR spectroscopy, ICP-MS, and XPS are very valuable tools for FG quantification and can yield the number of total and/or derivatizable FG depending on the respective surface-modified NM and the reporter used for signal generation. As detailed before, utilizing intrinsically present moieties provides the total amount of a certain FG, while the use of specific reporters in conjunction with chemical derivatization/labeling reactions yields the number of derivatizable FG. Moreover, particularly NMR and XPS can simultaneously provide information on mixed ligand shells and different FG. Drawbacks of these methods are, however, the need for expensive and sophisticated instrumentation, well-trained scientific staff, and elaborated data analysis. Therefore, such methods are often not the optimum choice for routine analysis and quality control; here, simple electrochemical methods and optical assays are better suited. However, these analytical methods are essential for validating the results obtained with simpler methods. Elegant tools for method validation present multimodal reporters that can be read out with different analytical techniques varying in the principle of signal generation, as summarized in the previous sections. Also, methods like NMR are mandatory to establish a traceability chain to the SI unit mole.
In the future, the increasingly recognized importance of FG and ligand quantification on NM and its direct correlation with the safe(r) use of NM will require more interlaboratory comparisons using different analytical methods to determine accomplishable uncertainties for broadly used NM and typical surface modifications. This is particularly relevant for applications of NM in the life sciences, health sector, and as food additives, and the corresponding quality control of NM production and long-term stability. Due to the application-specific importance of information on the total and derivatizable number of FG/ligands, analytical methods for both types of surface functionalities are needed. For the latter, the information obtained is influenced by the size, shape, and charge of the applied reporter, also in comparison to the respective properties of the molecule(s) of interest that are to be attached to the FG/ligands on the NM surface. Thus, models are desired that enable to consider these features. A relatively simple approach would be to calculate or estimate the steric demand (footprint) of the reporter label and the molecule of interest, which, however, needs to be verified at least for a set of commonly used reporters and application-relevant target (bio)molecules such as typical peptides, oligonucleotides, and proteins including antibodies and enzymes. In any case, the overall aim should be to establish protocols for surface FG/ligand analysis and quantification (with known uncertainties) for commonly employed methods, and to eventually standardize these methods. Such activities are currently being pushed forward, e.g., by ISO/TC 229 Nanotechnologies, the Nanomaterials Study Group of ISO TC201 SG1, and ISO TC 201 Surface Chemical Analysis as well as different working groups of IEC. Other activities are being pursued by national standardization organizations, e.g., by the XPS community. Particularly attractive and efficient tools for surface group analysis are multimodal reporters, which enable to correlate the results obtained with different analytical methods, thereby simplifying method comparison, validation, and traceability, as exemplarily shown in Fig. 8.
Protocols and recommended methods for surface analysis are also needed for establishing nanoscale reference materials for method calibration and/or validation, as reliable control for toxicity tests, and the supply of reference data of NM. Other properties of NM, that are closely linked to surface chemistry and will be of increasing importance for nanotechnology and nanobiotechnology in the future, are NM hydrophobicity/hydrophilicity, NM stability (including the possible release of potentially toxic constituents), and environment-induced changes in NM surface chemistry (including adsorption of (bio)molecules, (bio)corona formation, etc.) for representative and application-relevant test scenarios. Here, also overall accepted methods are needed. Although these needs have been meanwhile recognized by metrological institutes, standardization organizations, and regulatory agencies worldwide, the constantly increasing number of new and more advanced NM developed make it difficult to keep track. A categorization or classification of NM could present an appropriate tool that has been addressed by different EU consortia, and stronger requirements on the quality of the analytical data to be provided for scientific publications involving NM could be beneficial to improve the overall confidence in “nano” data [31, 32, 41, 247, 259].
Data availability
Not applicable.
Code availability
Not applicable.
References
Shin SJ, Beech JR, Kelly KA (2013) Targeted nanoparticles in imaging: paving the way for personalized medicine in the battle against cancer. Integr Biol 5(1):29–42
Pelaz B, Alexiou CH, Alvarez-Puebla RA et al (2017) Diverse applications of nanomedicine. ACS Nano 11(3):2313–2381
Farka Z, Juriik T, Kovaar D, Trnkova L, Sklaadal P (2017) Nanoparticle-based immunochemical biosensors and assays: recent advances and challenges. Chem Rev 117(15):9973–10042
Giner-Casares JJ, Henriksen-Lacey M, Coronado-Puchau M, Liz-Marzan LM (2016) Inorganic nanoparticles for biomedicine: where materials scientists meet medical research. Mater Today 19(1):19–28
Howes PD, Chandrawati R, Stevens MM (2014) Colloidal nanoparticles as advanced biological sensors. Science 346(6205):1247390
Kim D, Kim J, Park YI, Lee N, Hyeon T (2018) Recent development of inorganic nanoparticles for biomedical imaging. ACS Cent Sci 4(3):324–336
Peng HS, Chiu DT (2015) Soft fluorescent nanomaterials for biological and biomedical imaging. Chem Soc Rev 44(14):4699–4722
Petryayeva E, Algar WR (2015) Toward point-of-care diagnostics with consumer electronic devices: the expanding role of nanoparticles. RSC Adv 5(28):22256–22282
Stark WJ, Stoessel PR, Wohlleben W, Hafner A (2015) Industrial applications of nanoparticles. Chem Soc Rev 44(16):5793–5805
Wolfbeis OS (2015) An overview of nanoparticles commonly used in fluorescent bioimaging. Chem Soc Rev 44(14):4743–4768
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR (2016) Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res 33(10):2373–2387
Pietryga JM, Park YS, Lim JH, Fidler AF, Bae WK, Brovelli S, Klimov VI (2016) Spectroscopic and device aspects of nanocrystal quantum dots. Chem Rev 116(18):10513–10622
Geißler D, Hildebrandt N (2016) Recent developments in Forster resonance energy transfer (FRET) diagnostics using quantum dots. Anal Bioanal Chem 408(17):4475–4483
Andresen E, Resch-Genger U, Schaferling M (2019) Surface modifications for photon-upconversion-based energy-transfer nanoprobes. Langmuir 35(15):5093–5113
Heuer-Jungemann A, Feliu N, Bakaimi I, Hamaly M, Alkilany A, Chakraborty I, Masood A, Casula MF, Kostopoulou A, Oh E, Susumu K, Stewart MH, Medintz IL, Stratakis E, Parak WJ, Kanaras AG (2019) The role of ligands in the chemical synthesis and applications of inorganic nanoparticles. Chem Rev 119(8):4819–4880
Ghosh Chaudhuri R, Paria S (2011) Core/shell nanoparticles: classes, properties, synthesis mechanisms, characterization, and applications. Chem Rev 112(4):2373–2433
Wilhelm S, Kaiser M, Würth C, Heiland J, Carrillo-Carrion C, Muhr V, Wolfbeis OS, Parak WJ, Resch-Genger U, Hirsch T (2015) Water dispersible upconverting nanoparticles: effects of surface modification on their luminescence and colloidal stability. Nanoscale 7(4):1403–1410
Hühn J, Carrillo-Carrion C, Soliman MG, Pfeiffer C, Valdeperez D, Masood A, Chakraborty I, Zhu L, Gallego M, Yue Z, Carril M, Feliu N, Escudero A, Alkilany AM, Pelaz B, del Pino P, Parak WJ (2017) Selected standard protocols for the synthesis, phase transfer, and characterization of inorganic colloidal nanoparticles. Chem Mater 29(1):399–461
Zhao S, Caruso F, Dahne L et al (2019) The future of layer-by-layer assembly: a tribute to ACS Nano associate editor Helmuth Mohwald. ACS Nano 13(6):6151–6169
Hildebrandt N, Spillmann CM, Algar WR, Pons T, Stewart MH, Oh E, Susumu K, Díaz SA, Delehanty JB, Medintz IL (2017) Energy transfer with semiconductor quantum dot bioconjugates: a versatile platform for biosensing, energy harvesting, and other developing applications. Chem Rev 117(2):536–711
Soenen SJ, Parak WJ, Rejman J, Manshian B (2015) (intra)cellular stability of inorganic nanoparticles: effects on cytotoxicity, particle functionality, and biomedical applications. Chem Rev 115(5):2109–2135
Gagner JE, Shrivastava S, Qian X, Dordick JS, Siegel RW (2012) Engineering nanomaterials for biomedical applications requires understanding the Nano-bio Interface: a perspective. J Phys Chem Lett 3(21):3149–3158
Baer DR, Engelhard MH, Johnson GE, Laskin J, Lai J, Mueller K, Munusamy P, Thevuthasan S, Wang H, Washton N, Elder A, Baisch BL, Karakoti A, Kuchibhatla SVNT, Moon DW (2013) Surface characterization of nanomaterials and nanoparticles: important needs and challenging opportunities. J Vac Sci Technol A 31(5):050820
Silvi S, Baroncini M, La Rosa M, Credi A (2016) Interfacing luminescent quantum dots with functional molecules for optical sensing applications. Top Curr Chem 374(5):65
Steichen SD, Caldorera-Moore M, Peppas NA (2013) A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci 48(3):416–427
Algar WR, Prasuhn DE, Stewart MH, Jennings TL, Blanco-Canosa JB, Dawson PE, Medintz IL (2011) The controlled display of biomolecules on nanoparticles: a challenge suited to bioorthogonal chemistry. Bioconjug Chem 22(5):825–858
Zhou J, Liu Y, Tang J, Tang WH (2017) Surface ligands engineering of semiconductor quantum dots for chemosensory and biological applications. Mater Today 20(7):360–376
Lim SJ, Ma L, Schleife A, Smith AM (2016) Quantum dot surface engineering: toward inert fluorophores with compact size and bright, stable emission. Coord Chem Rev 320:216–237
Boles MA, Ling D, Hyeon T, Talapin DV (2016) The surface science of nanocrystals. Nat Mater 15(2):141–153
Alkilany AM, Zhu L, Weller H, Mews A, Parak WJ, Barz M, Feliu N (2019) Ligand density on nanoparticles: a parameter with critical impact on nanomedicine. Adv Drug Deliv Rev 143:22–36
Dekkers S, Oomen AG, Bleeker EA et al (2016) Towards a nanospecific approach for risk assessment. Regul Toxicol Pharmacol 80:46–59
Oomen AG, Bleeker EA, Bos PM, van Broekhuizen F, Gottardo S, Groenewold M, Hristozov D, Hund-Rinke K, Irfan MA, Marcomini A, Peijnenburg W, Rasmussen K, Jiménez A, Scott-Fordsmand J, van Tongeren M, Wiench K, Wohlleben W, Landsiedel R (2015) Grouping and read-across approaches for risk assessment of nanomaterials. Int J Environ Res Public Health 12(10):13415–13434
Zielinska A, Costa B, Ferreira MV et al (2020) Nanotoxicology and nanosafety: safety-by-design and testing at a glance. Int J Environ Res Public Health 17(13):4657
Lead JR, Batley GE, Alvarez PJJ, Croteau MN, Handy RD, McLaughlin MJ, Judy JD, Schirmer K (2018) Nanomaterials in the environment: behavior, fate, bioavailability, and effects-an updated review. Environ Toxicol Chem 37(8):2029–2063
Geißler D, Wegmann M, Jochum T, Somma V, Sowa M, Scholz J, Fröhlich E, Hoffmann K, Niehaus J, Roggenbuck D, Resch-Genger U (2019) An automatable platform for genotoxicity testing of nanomaterials based on the fluorometric gamma-H2AX assay reveals no genotoxicity of properly surface-shielded cadmium-based quantum dots. Nanoscale 11(28):13458–13468
Fubini B (1997) Surface reactivity in the pathogenic response to particulates. Environ Health Perspect 105:1013–1020
del Pino P, Yang F, Pelaz B et al (2016) Basic physicochemical properties of polyethylene glycol coated gold nanoparticles that determine their interaction with cells. Angew Chem Int Edit 55(18):5483–5487
Henriksen-Lacey M, Carregal-Romero S, Liz-Marzan LM (2017) Current challenges toward in vitro cellular validation of inorganic nanoparticles. Bioconjug Chem 28(1):212–221
Lewinski N, Colvin V, Drezek R (2008) Cytotoxicity of nanoparticles. Small 4(1):26–49
Rivera-Gil P, De Aberasturi DJ, Wulf V et al (2013) The challenge to relate the physicochemical properties of colloidal nanoparticles to their cytotoxicity. Acc Chem Res 46(3):743–749
Isigonis P, Afantitis A, Antunes D, Bartonova A, Beitollahi A, Bohmer N, Bouman E, Chaudhry Q, Cimpan MR, Cimpan E, Doak S, Dupin D, Fedrigo D, Fessard V, Gromelski M, Gutleb AC, Halappanavar S, Hoet P, Jeliazkova N et al (2020) Risk governance of emerging technologies demonstrated in terms of its applicability to nanomaterials. Small 16(36):2003303
Lin S, Yu T, Yu Z, Hu X, Yin D (2018) Nanomaterials safer-by-design: an environmental safety perspective. Adv Mater 30(17):1705691
Tavernaro I, Dekkers S, Soeteman-Hernandez LG et al. (2021) Safe-by-design part II: a strategy for balancing safety and functionality in the different stages of the innovation process. NanoImpact (accepted for publication)
Dekkers S, Wijnhoven SWP, Braakhuis HM, Soeteman-Hernandez LG, Sips AJAM, Tavernaro I, Kraegeloh A, Noorlander CW (2020) Safe-by-design part I: proposal for nanospecific human health safety aspects needed along the innovation process. NanoImpact 18:100227
Sánchez Jiménez A, Puelles R, Pérez-Fernández M, Gómez-Fernández P, Barruetabeña L, Jacobsen NR, Suarez-Merino B, Micheletti C, Manier N, Trouiller B, Navas JM, Kalman J, Salieri B, Hischier R, Handzhiyski Y, Apostolova MD, Hadrup N, Bouillard J, Oudart Y et al (2020) Safe(r) by design implementation in the nanotechnology industry. NanoImpact 20:100267
Kraegeloh A, Suarez-Merino B, Sluijters T, Micheletti C (2018) Implementation of safe-by-design for nanomaterial development and safe innovation: why we need a comprehensive approach. Nanomaterials 8(4):239
Gilbertson LM, Zimmerman JB, Plata DL, Hutchison JE, Anastas PT (2015) Designing nanomaterials to maximize performance and minimize undesirable implications guided by the principles of green chemistry. Chem Soc Rev 44(16):5758–5777
Johnston LJ, Gonzalez-Rojano N, Wilkinson KJ, Xing B (2020) Key challenges for evaluation of the safety of engineered nanomaterials. NanoImpact 18:100219
Comandella D, Gottardo S, Rio-Echevarria IM, Rauscher H (2020) Quality of physicochemical data on nanomaterials: an assessment of data completeness and variability. Nanoscale 12(7):4695–4708
Lopez-Serrano A, Olivas RM, Landaluze JS, Camara C (2014) Nanoparticles: a global vision. Characterization, separation, and quantification methods. Potential environmental and health impact. Anal Methods 6(1):38–56
Smith AM, Johnston KA, Crawford SE, Marbella LE, Millstone JE (2017) Ligand density quantification on colloidal inorganic nanoparticles. Analyst 142(1):11–29
Soeteman-Hernandez LG, Apostolova MD, Bekker C, Dekkers S, Grafström RC, Groenewold M, Handzhiyski Y, Herbeck-Engel P, Hoehener K, Karagkiozaki V, Kelly S, Kraegeloh A, Logothetidis S, Micheletti C, Nymark P, Oomen A, Oosterwijk T, Rodríguez-LLopis I, Sabella S et al (2019) Safe innovation approach: towards an agile system for dealing with innovations. Mater Today Commun 20:100548
Miernicki M, Hofmann T, Eisenberger I, von der Kammer F, Praetorius A (2019) Legal and practical challenges in classifying nanomaterials according to regulatory definitions. Nat Nanotechnol 14(3):208–216
Modena MM, Rühle B, Burg TP, Wuttke S (2019) Nanoparticle characterization: what to measure? Adv Mater 31(32):1901556
Palui G, Aldeek F, Wang WT, Mattoussi H (2015) Strategies for interfacing inorganic nanocrystals with biological systems based on polymer-coating. Chem Soc Rev 44(1):193–227
Sapsford KE, Tyner KM, Dair BJ, Deschamps JR, Medintz IL (2011) Analyzing nanomaterial bioconjugates: a review of current and emerging purification and characterization techniques. Anal Chem 83(12):4453–4488
Sapsford KE, Algar WR, Berti L, Gemmill KB, Casey BJ, Oh E, Stewart MH, Medintz IL (2013) Functionalizing nanoparticles with biological molecules: developing chemistries that facilitate nanotechnology. Chem Rev 113(3):1904–2074
Algar WR (2017) A brief introduction to traditional bioconjugate chemistry. In: Algar WR, Dawson P, Medintz IL (eds) Chemoselective and bioorthogonal ligation reactions - concepts and applications, 1st edn. Wiley-VCH, Weinheim. https://doi.org/10.1002/9783527683451
Devaraj NK (2018) The future of bioorthogonal chemistry. ACS Cent Sci 4(8):952–959
Rubio L, Pyrgiotakis G, Beltran-Huarac J, Zhang Y, Gaurav J, Deloid G, Spyrogianni A, Sarosiek KA, Bello D, Demokritou P (2019) Safer-by-design flame-sprayed silicon dioxide nanoparticles: the role of silanol content on ROS generation, surface activity and cytotoxicity. Part Fibre Toxicol 16(1):40
Pavan C, Delle Piane M, Gullo M, Filippi F, Fubini B, Hoet P, Horwell CJ, Huaux F, Lison D, Lo Giudice C, Martra G, Montfort E, Schins R, Sulpizi M, Wegner K, Wyart-Remy M, Ziemann C, Turci F (2019) The puzzling issue of silica toxicity: are silanols bridging the gaps between surface states and pathogenicity? Part Fibre Toxicol 16(1):32
Pavan C, Santalucia R, Leinardi R, Fabbiani M, Yakoub Y, Uwambayinema F, Ugliengo P, Tomatis M, Martra G, Turci F, Lison D, Fubini B (2020) Nearly free surface silanols are the critical molecular moieties that initiate the toxicity of silica particles. Proc Natl Acad Sci U S A 117(45):27836–27846
Hennig A, Borcherding H, Jaeger C, Hatami S, Würth C, Hoffmann A, Hoffmann K, Thiele T, Schedler U, Resch-Genger U (2012) Scope and limitations of surface functional group quantification methods: exploratory study with poly(acrylic acid)-grafted Micro- and nanoparticles. J Am Chem Soc 134(19):8268–8276
Moser M, Nirmalananthan N, Behnke T, Geißler D, Resch-Genger U (2018) Multimodal cleavable reporters versus conventional labels for optical quantification of accessible amino and Carboxy groups on Nano- and Microparticles. Anal Chem 90(9):5887–5895
Leubner S, Hatami S, Esendemir N, Lorenz T, Joswig JO, Lesnyak V, Recknagel S, Gaponik N, Resch-Genger U, Eychmüller A (2013) Experimental and theoretical investigations of the ligand structure of water-soluble CdTe nanocrystals. Dalton Trans 42(35):12733–12740
Poselt E, Schmidtke C, Fischer S et al (2012) Tailor-made quantum dot and Iron oxide based contrast agents for in vitro and in vivo tumor imaging. ACS Nano 6(4):3346–3355
Schmidtke C, Poselt E, Ostermann J et al (2013) Amphiphilic, cross-linkable diblock copolymers for multifunctionalized nanoparticles as biological probes. Nanoscale 5(16):7433–7444
Mahmood S, Mandal UK, Chatterjee B, Taher M (2017) Advanced characterizations of nanoparticles for drug delivery: investigating their properties through the techniques used in their evaluations. Nanotechnol Rev 6(4):355–372
Mourdikoudis S, Pallares RM, Thanh NTK (2018) Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties. Nanoscale 10(27):12871–12934
Maguire CM, Rosslein M, Wick P, Prina-Mello A (2018) Characterisation of particles in solution—a perspective on light scattering and comparative technologies. Sci Technol Adv Mater 19(1):732–745
Shang J, Gao XH (2014) Nanoparticle counting: towards accurate determination of the molar concentration. Chem Soc Rev 43(21):7267–7278
Varga Z, Yuana Y, Grootemaat AE, van der Pol E, Gollwitzer C, Krumrey M, Nieuwland R (2014) Towards traceable size determination of extracellular vesicles. J Extracell Vesicles 3(1):23298
Thielbeer F, Donaldson K, Bradley M (2011) Zeta potential mediated reaction monitoring on Nano and Microparticles. Bioconjug Chem 22(2):144–150
Charron G, Huhn D, Perrier A et al (2012) On the use of pH titration to quantitatively characterize colloidal nanoparticles. Langmuir 28(43):15141–15149
Fedin I, Talapin DV (2014) Probing the surface of colloidal nanomaterials with Potentiometry in situ. J Am Chem Soc 136(32):11228–11231
Alves LA, de Castro AH, de Mendonca FG, de Mesquita JP (2016) Characterization of acid functional groups of carbon dots by nonlinear regression data fitting of potentiometric titration curves. Appl Surf Sci 370:486–495
Anirudhan TS, Deepa JR, Christa J (2016) Nanocellulose/nanobentonite composite anchored with multi-carboxyl functional groups as an adsorbent for the effective removal of cobalt(II) from nuclear industry wastewater samples. J Colloid Interface Sci 467:307–320
Chen ZM, Xiao X, Chen BL, Zhu LZ (2015) Quantification of chemical states, dissociation constants and contents of oxygen-containing groups on the surface of biochars produced at different temperatures. Environ Sci Technol 49(1):309–317
Castro VG, Costa IB, Medeiros FS et al (2019) Improved functionalization of multiwalled carbon nanotubes in ultra-low acid volume: effect of solid/liquid Interface. J Braz Chem Soc 30(11):2477–2487
Torlopov MA, Martakov IS, Mikhaylov VI, Legki PV, Golubev YA, Krivoshapkina EF, Tracey C, Sitnikov PA, Udoratina EV (2019) Manipulating the colloidal properties of (non-)sulfated cellulose nanocrystals via stepwise surface cyanoethylation/carboxylation. Eur Polym J 115:225–233
Nedeljko P, Turel M, Kosak A, Lobnik A (2016) Synthesis of hybrid thiol-functionalized SiO2 particles used for agmatine determination. J Sol-Gel Sci Technol 79(3):487–496
Hassan SM, Ahmed AI, Mannaa MA (2019) Surface acidity, catalytic and photocatalytic activities of new type H3PW12O40/Sn-TiO2 nanoparticles. Colloid Surf A 577:147–157
Khoshnavazi R, Bahrami L, Havasi F, Naseri E (2017) H3PW12O40 supported on functionalized polyoxometalate organic-inorganic hybrid nanoparticles as efficient catalysts for three-component Mannich-type reactions in water. RSC Adv 7(19):11510–11521
Srilakshmi C, Saraf R, Shivakumara C (2018) Structural studies of multifunctional SrTiO3 nanocatalyst synthesized by microwave and oxalate methods: its catalytic application for condensation, hydrogenation, and amination reactions. ACS Omega 3(9):10503–10512
Aboelhassan MM, Peixoto AF, Freire C (2017) Sulfonic acid functionalized silica nanoparticles as catalysts for the esterification of linoleic acid. New J Chem 41(9):3595–3605
Bandosz TJ, Policicchio A, Florent M, Poon PS, Matos J (2019) TiO2/S-doped carbons hybrids: analysis of their interfacial and surface features. Molecules 24(19):3585
Wang Z, Xie Y, Lei Z, Lu Y, Wei G, Liu S, Xu C, Zhang Z, Wang X, Rao L, Chen J (2019) Quantitative analysis of surface sites on carbon dots and their interaction with metal ions by a potentiometric titration method. Anal Chem 91(15):9690–9697
Renner AM, Schutz MB, Moog D, Fischer T, Mathur S (2019) Electroacoustic quantification of surface bound ligands in functionalized silica and Iron oxide nanoparticles. Chemistryselect 4(40):11959–11964
Nirmalananthan-Budau N, Rühle B, Geißler D, Moser M, Kläber C, Schäfer A, Resch-Genger U (2019) Multimodal cleavable reporters for quantifying carboxy and amino groups on organic and inorganic nanoparticles. Sci Rep 9(1):17577
Zhu SC, Panne U, Rurack K (2013) A rapid method for the assessment of the surface group density of carboxylic acid-functionalized polystyrene microparticles. Analyst 138(10):2924–2930
Spinella S, Maiorana A, Qian Q, Dawson NJ, Hepworth V, McCallum SA, Ganesh M, Singer KD, Gross RA (2016) Concurrent cellulose hydrolysis and esterification to prepare a surface-modified cellulose nanocrystal decorated with carboxylic acid moieties. ACS Sustain Chem Eng 4(3):1538–1550
Zhao FP, Repo E, Song Y, Yin D, Hammouda SB, Chen L, Kalliola S, Tang J, Tam KC, Sillanpää M (2017) Polyethylenimine-cross-linked cellulose nanocrystals for highly efficient recovery of rare earth elements from water and a mechanism study. Green Chem 19(20):4816–4828
Sahlin K, Forsgren L, Moberg T, Bernin D, Rigdahl M, Westman G (2018) Surface treatment of cellulose nanocrystals (CNC): effects on dispersion rheology. Cellulose 25(1):331–345
Jordan JH, Easson MW, Condon BD (2019) Alkali hydrolysis of sulfated cellulose nanocrystals: optimization of reaction conditions and tailored surface charge. Nanomaterials 9(9):1232
Trache D, Tarchoun AF, Derradji M, Hamidon TS, Masruchin N, Brosse N, Hussin MH (2020) Nanocellulose: from fundamentals to advanced applications. Front Chem 8:392
Ellebracht NC, Jones CW (2018) Amine functionalization of cellulose nanocrystals for acid-base organocatalysis: surface chemistry, cross-linking, and solvent effects. Cellulose 25(11):6495–6512
Ellebracht NC, Jones CW (2019) Optimized cellulose nanocrystal organocatalysts outperform silica-supported analogues: cooperativity, selectivity, and bifunctionality in acid-base aldol condensation reactions. ACS Catal 9(4):3266–3277
Hujaya SD, Lorite GS, Vainio SJ, Liimatainen H (2018) Polyion complex hydrogels from chemically modified cellulose nanofibrils: structure-function relationship and potential for controlled and pH-responsive release of doxorubicin. Acta Biomater 75:346–357
Beck S, Methot M, Bouchard J (2015) General procedure for determining cellulose nanocrystal sulfate half-ester content by conductometric titration (vol 22, pg 101, 2015). Cellulose 22(1):117
Johnston LJ, Jakubek ZJ, Beck S, Araki J, Cranston ED, Danumah C, Fox D, Li H, Wang J, Mester Z, Moores A, Murphy K, Rabb SA, Rudie A, Stephan C (2018) Determination of sulfur and sulfate half-ester content in cellulose nanocrystals: an interlaboratory comparison. Metrologia 55(6):872–882
Boehm HP, Heck W, Sappok R, Diehl E (1964) Surface oxides of carbon. Angew Chem Int Edit 3(10):669
Boehm HP (1966) Chemical identification of surface groups. Adv Catal 16:179–274
Boehm HP, Voll M (1968) Studies on basic surface oxides of carbon. Carbon 6(2):226
Voll M, Boehm HP (1971) Basic surface oxides on carbon .4. Chemical Reactions for Identification of Surface Groups. Carbon 9(4):481
Schönherr J, Buchheim JR, Scholz P, Adelhelm P (2018) Boehm titration revisited (part I): practical aspects for achieving a high precision in quantifying oxygen-containing surface groups on carbon materials. Carbon 4(2):21
Hou JF, Xu LX, Han YX, Tang Y, Wan H, Xu Z, Zheng S (2019) Deactivation and regeneration of carbon nanotubes and nitrogen-doped carbon nanotubes in catalytic peroxymonosulfate activation for phenol degradation: variation of surface functionalities. RSC Adv 9(2):974–983
Tang ZC, Boer DG, Syariati A, Enache M, Rudolf P, Heeres HJ, Pescarmona PP (2019) Base-free conversion of glycerol to methyl lactate using a multifunctional catalytic system consisting of Au-Pd nanoparticles on carbon nanotubes and Sn-MCM-41-XS. Green Chem 21(15):4115–4126
Kolanowska A, Wasik P, Zieba W, Terzyk AP, Boncel S (2019) Selective carboxylation versus layer-by-layer unsheathing of multi-walled carbon nanotubes: new insights from the reaction with boiling nitrating mixture. RSC Adv 9(64):37608–37613
Ackermann J, Krueger A (2019) Efficient surface functionalization of detonation nanodiamond using ozone under ambient conditions. Nanoscale 11(16):8012–8019
Bergaoui M, Aguir C, Khalfaoui M, Enciso E, Duclaux L, Reinert L, Fierro JLG (2017) New insights in the adsorption of bovine serum albumin onto carbon nanoparticles derived from organic resin: experimental and theoretical studies. Microporous Mesoporous Mater 241:418–428
Pham V, Nguyen HTT, Nguyen DTC et al (2019) Process optimization by a response surface methodology for adsorption of Congo red dye onto exfoliated graphite-decorated MnFe2O4 nanocomposite: the pivotal role of surface chemistry. Processes 7(5):305
Motamedi E, Motesharezedeh B, Shirinfekr A, Samar SM (2020) Synthesis and swelling behavior of environmentally friendly starch-based superabsorbent hydrogels reinforced with natural char nano/micro particles. J Environ Chem Eng 8(1):103583
Schönherr J, Buchheim JR, Scholz P, Adelhelm P (2018) Boehm titration revisited (part II): a comparison of Boehm titration with other analytical techniques on the quantification of oxygen-containing surface groups for a variety of carbon materials. Carbon 4(2):22
Felbeck T, Hoffmann K, Lezhnina MM, Kynast UH, Resch-Genger U (2015) Fluorescent Nanoclays: covalent functionalization with amine reactive dyes from different fluorophore classes and surface group quantification. J Phys Chem C 119(23):12978–12987
Hoffmann K, Mix R, Resch-Genger U, Friedrich JF (2007) Monitoring of amino functionalities on plasma-chemically modified polypropylene supports with a chromogenic and Fluorogenic Pyrylium reporter. Langmuir 23(16):8411–8416
Hennig A, Hoffmann A, Borcherding H, Thiele T, Schedler U, Resch-Genger U (2011) Simple colorimetric method for quantification of surface Carboxy groups on polymer particles. Anal Chem 83(12):4970–4974
Fischer T, Dietrich PM, Unger WES, Rurack K (2016) Multimode surface functional group determination: combining steady-state and time-resolved fluorescence with X-ray photoelectron spectroscopy and absorption measurements for absolute quantification. Anal Chem 88(2):1210–1217
Zhan NQ, Palui G, Merkl JP, Mattoussi H (2016) Bio-orthogonal coupling as a means of quantifying the ligand density on hydrophilic quantum dots. J Am Chem Soc 138(9):3190–3201
Laux EM, Behnke T, Hoffmann K, Resch-Genger U (2012) Keeping particles brilliant—simple methods for the determination of the dye content of fluorophore-loaded polymeric particles. Anal Methods 4(6):1759–1768
Cisneros-Covarrubias CA, Palestino G, Gomez-Duran CFA, Rosales-Mendoza S, Betancourt-Mendiola ML (2021) Optimized microwave-assisted functionalization and quantification of superficial amino groups on porous silicon nanostructured microparticles. Anal Methods 13(4):516–525
Hristov DR, Rocks L, Kelly PM, Thomas SS, Pitek AS, Verderio P, Mahon E, Dawson KA (2015) Tuning of nanoparticle biological functionality through controlled surface chemistry and characterisation at the bioconjugated nanoparticle surface. Sci Rep 5:17040
Miller PJ, Shantz DF (2020) Covalently functionalized uniform amino-silica nanoparticles. Synthesis and validation of amine group accessibility and stability. Nanoscale Adv 2(2):860–868
Sun Y, Kunc F, Balhara V, Coleman B, Kodra O, Raza M, Chen M, Brinkmann A, Lopinski GP, Johnston LJ (2019) Quantification of amine functional groups on silica nanoparticles: a multi-method approach. Nanoscale Adv 1(4):1598–1607
Zhang Y, Chen Y (2012) Fmoc-Cl fluorescent determination for amino groups of nanomaterial science. IET Nanobiotechnol 6(2):76–80
Hsiao IL, Fritsch-Decker S, Leidner A et al (2019) Biocompatibility of amine-functionalized silica nanoparticles: the role of surface coverage. Small 15(10):1805400
Moser M, Behnke T, Hamers-Allin C, Klein-Hartwig K, Falkenhagen J, Resch-Genger U (2015) Quantification of PEG-Maleimide ligands and coupling efficiencies on nanoparticles with Ellman's reagent. Anal Chem 87(18):9376–9383
Moser M, Schneider R, Behnke T, Schneider T, Falkenhagen J, Resch-Genger U (2016) Ellman's and Aldrithiol assay as versatile and complementary tools for the quantification of thiol groups and ligands on nanomaterials. Anal Chem 88(17):8624–8631
Gorris HH, Saleh SM, Groegel DBM, Ernst S, Reiner K, Mustroph H, Wolfbeis OS (2011) Long-wavelength absorbing and fluorescent chameleon labels for proteins, peptides, and amines. Bioconjug Chem 22(7):1433–1437
Saleh SM, Ali R, Wolfbeis OS (2011) New silica and polystyrene nanoparticles labeled with longwave absorbing and fluorescent chameleon dyes. Microchim Acta 174(3–4):429–434
Behrendt R, White P, Offer J (2016) Advances in Fmoc solid-phase peptide synthesis. J Pept Sci 22(1):4–27
Eissler S, Kley M, Bachle D et al (2017) Substitution determination of Fmoc-substituted resins at different wavelengths. J Pept Sci 23(10):757–762
Yoon TJ, Yu KN, Kim E et al (2006) Specific targeting, cell sorting, and bioimaging with smart magnetic silica core-shell nanomaterials. Small 2(2):209–215
Yang H, Zhuang Y, Hu H, du X, Zhang C, Shi X, Wu H, Yang S (2010) Silica-coated manganese oxide nanoparticles as a platform for targeted magnetic resonance and fluorescence imaging of Cancer cells. Adv Funct Mater 20(11):1733–1741
Cao T, Yang Y, Gao Y, Zhou J, Li Z, Li F (2011) High-quality water-soluble and surface-functionalized upconversion nanocrystals as luminescent probes for bioimaging. Biomaterials 32(11):2959–2968
Liu Y, Zhou S, Tu D, Chen Z, Huang M, Zhu H, Ma E, Chen X (2012) Amine-functionalized lanthanide-doped zirconia nanoparticles: optical spectroscopy, time-resolved fluorescence resonance energy transfer biodetection, and targeted imaging. J Am Chem Soc 134(36):15083–15090
Paris JL, Manzano M, Cabanas MV, Vallet-Regi M (2018) Mesoporous silica nanoparticles engineered for ultrasound-induced uptake by cancer cells. Nanoscale 10(14):6402–6408
Fidecka K, Giacoboni J, Picconi P, Vago R, Licandro E (2020) Quantification of amino groups on halloysite surfaces using the Fmoc-method. RSC Adv 10(24):13944–13948
Szczepanska E, Grobelna B, Ryl J et al (2020) Efficient method for the concentration determination of Fmoc groups incorporated in the core-shell materials by Fmoc-Glycine. Molecules 25(17):3983
Chen Y, Zhang Y (2011) Fluorescent quantification of amino groups on silica nanoparticle surfaces. Anal Bioanal Chem 399(7):2503–2509
Roloff A, Nirmalananthan-Budau N, Rühle B, Borcherding H, Thiele T, Schedler U, Resch-Genger U (2019) Quantification of aldehydes on polymeric microbead surfaces via catch and release of reporter chromophores. Anal Chem 91(14):8827–8834
Sakai R, Iguchi H, Maruyama T (2019) Quantification of azide groups on a material surface and a biomolecule using a clickable and cleavable fluorescent compound. RSC Adv 9(8):4621–4625
Zhang S, Dominguez Z, Assaf KI et al (2018) Precise supramolecular control of surface coverage densities on polymer micro- and nanoparticles. Chem Sci 9(45):8575–8581
Kim DY, Shinde S, Ghodake G (2017) Colorimetric detection of magnesium (II) ions using tryptophan functionalized gold nanoparticles. Sci Rep 7:3966
Rodiger S, Ruhland M, Schmidt C et al (2011) Fluorescence dye adsorption assay to quantify carboxyl groups on the surface of poly(methyl methacrylate) microbeads. Anal Chem 83(9):3379–3385
Stelmach E, Maksymiuk K, Michalska A (2016) Copolymeric hexyl acrylate-methacrylic acid microspheres—surface vs. bulk reactive carboxyl groups. Coulometric and colorimetric determination and analytical applications for heterogeneous microtitration. Talanta 159:248–254
Marbella LE, Millstone JE (2015) NMR techniques for noble metal nanoparticles. Chem Mater 27(8):2721–2739
Hines DA, Kamat PV (2014) Recent advances in quantum dot surface chemistry. ACS Appl Mater Interfaces 6(5):3041–3057
Kopping JT, Patten TE (2008) Identification of acidic phosphorus-containing ligands involved in the surface chemistry of CdSe nanoparticles prepared in tri-n-octylphosphine oxide solvents. J Am Chem Soc 130(17):5689–5698
Ji XH, Copenhaver D, Sichmeller C, Peng XG (2008) Ligand bonding and dynamics on colloidal nanocrystals at room temperature: the case of alkylamines on CdSe nanocrystals. J Am Chem Soc 130(17):5726–5735
Morris-Cohen AJ, Malicki M, Peterson MD, Slavin JWJ, Weiss EA (2013) Chemical, structural, and quantitative analysis of the ligand shells of colloidal quantum dots. Chem Mater 25(8):1155–1165
De Roo J, Yazdani N, Drijvers E et al (2018) Probing solvent-ligand interactions in colloidal nanocrystals by the NMR line broadening. Chem Mater 30(15):5485–5492
Crucho CIC, Baleizao C, Farinha JPS (2017) Functional group coverage and conversion quantification in nanostructured silica by H-1 NMR. Anal Chem 89(1):681–687
Lu J, Xue Y, Shi R, Kang J, Zhao CY, Zhang NN, Wang CY, Lu ZY, Liu K (2019) A non-sacrificial method for the quantification of poly(ethylene glycol) grafting density on gold nanoparticles for applications in nanomedicine. Chem Sci 10(7):2067–2074
Davidowski SK, Holland GP (2016) Solid-state NMR characterization of mixed phosphonic acid ligand binding and organization on silica nanoparticles. Langmuir 32(13):3253–3261
Hens Z, Martins JC (2013) A solution NMR toolbox for characterizing the surface chemistry of colloidal nanocrystals. Chem Mater 25(8):1211–1221
Lehman SE, Tataurova Y, Mueller PS, Mariappan SVS, Larsen SC (2014) Ligand characterization of covalently functionalized mesoporous silica nanoparticles: an NMR toolbox approach. J Phys Chem C 118(51):29943–29951
Huber A, Behnke T, Würth C, Jaeger C, Resch-Genger U (2012) Spectroscopic characterization of coumarin-stained beads: quantification of the number of fluorophores per particle with solid-state 19F-NMR and measurement of absolute fluorescence quantum yields. Anal Chem 84(8):3654–3661
Hennig A, Dietrich PM, Hemmann F, Thiele T, Borcherding H, Hoffmann A, Schedler U, Jäger C, Resch-Genger U, Unger WES (2015) En route to traceable reference standards for surface group quantifications by XPS, NMR and fluorescence spectroscopy. Analyst 140(6):1804–1808
Guo C, Yarger JL (2018) Characterizing gold nanoparticles by NMR spectroscopy. Magn Reson Chem 56(11):1074–1082
Wu M, Vartanian AM, Chong G, Pandiakumar AK, Hamers RJ, Hernandez R, Murphy CJ (2019) Solution NMR analysis of ligand environment in quaternary ammonium-terminated self-assembled monolayers on gold nanoparticles: the effect of surface curvature and ligand structure. J Am Chem Soc 141(10):4316–4327
Chen Y, Ripka EG, Franck JM, Maye MM (2019) Ligand surface density decreases with quantum rod aspect ratio. J Phys Chem C 123(38):23682–23690
Kong N, Zhou J, Park J, Xie S, Ramström O, Yan M (2015) Quantitative fluorine NMR to determine carbohydrate density on glyconanomaterials synthesized from perfluorophenyl azide-functionalized silica nanoparticles by click reaction. Anal Chem 87(18):9451–9458
Kunc F, Balhara V, Brinkmann A, Sun Y, Leek DM, Johnston LJ (2018) Quantification and stability determination of surface amine groups on silica nanoparticles using solution NMR. Anal Chem 90(22):13322–13330
Kunc F, Balhara V, Sun Y, Daroszewska M, Jakubek ZJ, Hill M, Brinkmann A, Johnston LJ (2019) Quantification of surface functional groups on silica nanoparticles: comparison of thermogravimetric analysis and quantitative NMR. Analyst 144(18):5589–5599
Galazzi RM, Chacon-Madrid K, Freitas DC, da Costa LF, Arruda MAZ (2020) Inductively coupled plasma mass spectrometry based platforms for studies involving nanoparticle effects in biological samples. Rapid Commun Mass Spectrom 34:8726
Bartczak D, Vincent P, Goenaga-Infante H (2015) Determination of size- and number-based concentration of silica nanoparticles in a complex biological matrix by online techniques. Anal Chem 87(11):5482–5485
Bouzas-Ramos D, Menendez-Miranda M, Costa-Fernandez JM, Encinar JR, Sanz-Medel A (2016) Precise determination of the nanoparticle concentration and ligand density of engineered water-soluble HgSe fluorescent nanoparticles. RSC Adv 6(24):19964–19972
Garcia-Cortes M, Gonzalez ES, Fernandez-Arguelles MT et al (2017) Capping of Mn-doped ZnS quantum dots with DHLA for their stabilization in aqueous media: determination of the nanoparticle number concentration and surface ligand density. Langmuir 33(25):6333–6341
Zhou HY, Li X, Lemoff A, Zhang B, Yan B (2010) Structural confirmation and quantification of individual ligands from the surface of multi-functionalized gold nanoparticles. Analyst 135(6):1210–1213
Wilschefski SC, Baxter MR (2019) Inductively coupled plasma mass spectrometry: introduction to analytical aspects. Clin Biochem Rev 40(3):115–133
Fabricius AL, Duester L, Meermann B, Ternes TA (2014) ICP-MS-based characterization of inorganic nanoparticles-sample preparation and off- line fractionation strategies. Anal Bioanal Chem 406(2):467–479
Costo R, Heinke D, Gruttner C et al (2019) Improving the reliability of the iron concentration quantification for iron oxide nanoparticle suspensions: a two-institutions study. Anal Bioanal Chem 411(9):1895–1903
Xia XH, Yang MX, Wang YC, Zheng Y, Li Q, Chen J, Xia Y (2012) Quantifying the coverage density of poly(ethylene glycol) chains on the surface of gold nanostructures. ACS Nano 6(1):512–522
Elzey S, Tsai DH, Rabb SA, Yu LL, Winchester MR, Hackley VA (2012) Quantification of ligand packing density on gold nanoparticles using ICP-OES. Anal Bioanal Chem 403(1):145–149
Tong L, Lu E, Pichaandi J, Cao P, Nitz M, Winnik MA (2015) Quantification of surface ligands on NaYF4 nanoparticles by three independent analytical techniques. Chem Mater 27(13):4899–4910
Vargas KM, San KA, Shon YS (2019) Isolated effects of surface ligand density on the catalytic activity and selectivity of palladium nanoparticles. ACS Appl Nano Mater 2(11):7188–7196
Yang R, Lin Y, Liu BY, Su Y, Tian Y, Hou X, Zheng C (2020) Simple universal strategy for quantification of carboxyl groups on carbon nanomaterials: carbon dioxide vapor generation coupled to microplasma for optical emission spectrometric detection. Anal Chem 92(5):3528–3534
Hinterwirth H, Kappel S, Waitz T, Prohaska T, Lindner W, Lämmerhofer M (2013) Quantifying thiol ligand density of self-assembled monolayers on gold nanoparticles by inductively coupled plasma-mass spectrometry. ACS Nano 7(2):1129–1136
Nicolardi S, van der Burgt YEM, Codee JDC et al (2017) Structural characterization of biofunctionalized gold nanoparticles by ultrahigh-resolution mass spectrometry. ACS Nano 11(8):8257–8264
Tsai DH, Shelton MP, DelRio FW et al (2012) Quantifying dithiothreitol displacement of functional ligands from gold nanoparticles. Anal Bioanal Chem 404(10):3015–3023
Goenaga-Infante H, Bartczak D (2020) Single particle inductively coupled plasma mass spectrometry (spICP-MS). In: characterization of nanoparticles: measurement processes for nanoparticles. Pp 65-77. https://doi.org/10.1016/B978-0-12-814182-3.00003-1
Mozhayeva D, Engelhard C (2020) A critical review of single particle inductively coupled plasma mass spectrometry—a step towards an ideal method for nanomaterial characterization. J Anal At Spectrom 35(9):1740–1783
Peters R, Herrera-Rivera Z, Undas A, van der Lee M, Marvin H, Bouwmeester H, Weigel S (2015) Single particle ICP-MS combined with a data evaluation tool as a routine technique for the analysis of nanoparticles in complex matrices. J Anal At Spectrom 30(6):1274–1285
Bings NH, Bogaerts A, Broekaert JAC (2010) Atomic spectroscopy: a review. Anal Chem 82(12):4653–4681
Abad C, Florek S, Becker-Ross H, Huang MD, Buzanich AG, Radtke M, Lippitz A, Hodoroaba VD, Schmid T, Heinrich HJ, Recknagel S, Jakubowski N, Panne U (2018) Zirconium permanent modifiers for graphite furnaces used in absorption spectrometry: understanding their structure and mechanism of action. J Anal At Spectrom 33(12):2034–2042
Aslund AKO, Sulheim E, Snipstad S et al (2017) Quantification and qualitative effects of different PEGylations on poly(butyl cyanoacrylate) nanoparticles. Mol Pharm 14(8):2560–2569
Ju S, Yeo WS (2012) Quantification of proteins on gold nanoparticles by combining MALDI-TOF MS and proteolysis. Nanotechnology 23(13):135701
Kim YP, Shon HK, Shin SK, Lee TG (2015) Probing nanoparticles and nanoparticle-conjugated biomolecules using time-of-flight secondary ion mass spectrometry. Mass Spectrom Rev 34(2):237–247
Cozzolino D (2015) Infrared spectroscopy as a versatile analytical tool for the quantitative determination of antioxidants in agricultural products, foods and plants. Antioxidants 4(3):482–497
Deidda R, Sacre P-Y, Clavaud M, Coïc L, Avohou H, Hubert P, Ziemons E (2019) Vibrational spectroscopy in analysis of pharmaceuticals: critical review of innovative portable and handheld NIR and Raman spectrophotometers. Trac-Trends Anal Chem 114:251–259
Kiefer J (2015) Recent advances in the characterization of gaseous and liquid fuels by vibrational spectroscopy. Energies 8(4):3165–3197
Berthomieu C, Hienerwadel R (2009) Fourier transform infrared (FTIR) spectroscopy. Photosynth Res 101(2–3):157–170
Lopez-Lorente AI, Mizaikoff B (2016) Recent advances on the characterization of nanoparticles using infrared spectroscopy. Trac-Trends Anal Chem 84:97–106
Gouadec G, Colomban P (2007) Raman spectroscopy of nanomaterials: how spectra relate to disorder, particle size and mechanical properties. Prog Cryst Growth Ch 53(1):1–56
Rytwo G, Zakai R, Wicklein B (2015) The use of ATR-FTIR spectroscopy for quantification of adsorbed compounds. J Spectrosc 2015:1–8
Mudunkotuwa IA, Al Minshid A, Grassian VH (2014) ATR-FTIR spectroscopy as a tool to probe surface adsorption on nanoparticles at the liquid-solid interface in environmentally and biologically relevant media. Analyst 139(5):870–881
Dengo N, Vittadini A, Natile MM, Gross S (2020) In-depth study of ZnS nanoparticle surface properties with a combined experimental and theoretical approach. J Phys Chem C 124(14):7777–7789
Altmann L, Kunz S, Bäumer M (2014) Influence of organic amino and thiol ligands on the geometric and electronic surface properties of Colloidally prepared platinum nanoparticles. J Phys Chem C 118(17):8925–8932
Garcia-Rico E, Alvarez-Puebla RA, Guerrini L (2018) Direct surface-enhanced Raman scattering (SERS) spectroscopy of nucleic acids: from fundamental studies to real-life applications. Chem Soc Rev 47(13):4909–4923
Feliu N, Hassan M, Garcia Rico E, Cui D, Parak W, Alvarez-Puebla R (2017) SERS quantification and characterization of proteins and other biomolecules. Langmuir 33(38):9711–9730
Zou S, Ma L, Li J, Xie Z, Zhao D, Ling Y, Zhang Z (2018) Quantification of trace chemicals in unknown complex systems by SERS. Talanta 186:452–458
Wang Y, Li P, Kong L (2013) Chitosan-modified PLGA nanoparticles with versatile surface for improved drug delivery. AAPS PharmSciTech 14(2):585–592
Palo E, Lahtinen S, Pakkila H et al (2018) Effective shielding of NaYF4:Yb(3+),Er(3+) upconverting nanoparticles in aqueous environments using layer-by-layer assembly. Langmuir 34(26):7759–7766
Podila R, Chacón-Torres J, Spear JT, Pichler T, Ayala P, Rao AM (2012) Spectroscopic investigation of nitrogen doped graphene. Appl Phys Lett 101(12):123108
Jaramillo AF, Baez-Cruz R, Montoya LF, Medinam C, Pérez-Tijerina E, Salazar F, Rojas D, Melendrez MF (2017) Estimation of the surface interaction mechanism of ZnO nanoparticles modified with organosilane groups by Raman spectroscopy. Ceram Int 43(15):11838–11847
Joshi AS, Gahane A, Thakur AK (2016) Deciphering the mechanism and structural features of polysorbate 80 during adsorption on PLGA nanoparticles by attenuated total reflectance—Fourier transform infrared spectroscopy. RSC Adv 6(110):108545–108557
Andersen AJ, Yamada S, Pramodkumar EK, Andresen TL, Boisen A, Schmid S (2016) Nanomechanical IR spectroscopy for fast analysis of liquid-dispersed engineered nanomaterials. Sensors Actuators B Chem 233:667–673
Mangos DN, Nakanishi T, Lewis DA (2014) A simple method for the quantification of molecular decorations on silica particles. Sci Technol Adv Mater 15(1):015002
Demin AM, Koryakova OV, Krasnov VP (2014) Quantitative determination of 3-aminopropylsilane on the surface of Fe3O4 nanoparticles by attenuated total reflection infrared spectroscopy. J Appl Spectrosc 81(4):565–569
Valkenier H, Malytskyi V, Blond P, Retout M, Mattiuzzi A, Goole J, Raussens V, Jabin I, Bruylants G (2017) Controlled functionalization of gold nanoparticles with mixtures of calix[4]arenes revealed by infrared spectroscopy. Langmuir 33(33):8253–8259
Tsai DH, Davila-Morris M, DelRio FW et al (2011) Quantitative determination of competitive molecular adsorption on gold nanoparticles using attenuated total reflectance-Fourier transform infrared spectroscopy. Langmuir 27(15):9302–9313
Yuanyuan C, Zhibin W (2018) Quantitative analysis modeling of infrared spectroscopy based on ensemble convolutional neural networks. Chemom Intell Lab Syst 181:1–10
Hard X-ray photoelectron spectroscopy (HAXPES) (2016). In: Woicik JC (ed) Springer series in surface sciences. vol 59. Springer. doi:https://doi.org/10.1007/978-3-319-24043-5
Weigert F, Müller A, Häusler I, Geißler D, Skroblin D, Krumrey M, Unger W, Radnik J, Resch-Genger U (2020) Combining HR-TEM and XPS to elucidate the core-shell structure of ultrabright CdSe/CdS semiconductor quantum dots. Sci Rep 10(1):20712
Saleh MI, Rühle B, Wang S, Radnik J, You Y, Resch-Genger U (2020) Assessing the protective effects of different surface coatings on NaYF4:Yb3+, Er3+ upconverting nanoparticles in buffer and DMEM. Sci Rep 10(1):19318
Dietrich PM, Hennig A, Holzweber M, Thiele T, Borcherding H, Lippitz A, Schedler U, Resch-Genger U, Unger WES (2014) Surface analytical study of poly(acrylic acid)-grafted microparticles (beads): characterization, chemical derivatization, and quantification of surface carboxyl groups. J Phys Chem C 118(35):20393–20404
Fairclough SM, Tyrrell EJ, Graham DM, Lunt PJB, Hardman SJO, Pietzsch A, Hennies F, Moghal J, Flavell WR, Watt AAR, Smith JM (2012) Growth and characterization of strained and alloyed type-II ZnTe/ZnSe Core-shell nanocrystals. J Phys Chem C 116(51):26898–26907
Hardman SJO, Graham DM, Stubbs SK, Spencer BF, Seddon EA, Fung HT, Gardonio S, Sirotti F, Silly MG, Akhtar J, O'Brien P, Binks DJ, Flavell WR (2011) Electronic and surface properties of PbS nanoparticles exhibiting efficient multiple exciton generation. Phys Chem Chem Phys 13(45):20275–20283
Page RC, Espinobarro-Velazquez D, Leontiadou MA, Smith C, Lewis EA, Haigh SJ, Li C, Radtke H, Pengpad A, Bondino F, Magnano E, Pis I, Flavell WR, O'Brien P, Binks DJ (2015) Near-unity quantum yields from chloride treated CdTe colloidal quantum dots. Small 11(13):1548–1554
Wang Y-C, Engelhard MH, Baer DR, Castner DG (2016) Quantifying the impact of nanoparticle coatings and nonuniformities on XPS analysis: gold/silver core–shell nanoparticles. Anal Chem 88(7):3917–3925
Torelli MD, Putans RA, Tan YZ, Lohse SE, Murphy CJ, Hamers RJ (2015) Quantitative determination of ligand densities on nanomaterials by X-ray photoelectron spectroscopy. ACS Appl Mater Interfaces 7(3):1720–1725
ISO 18118:2015(en) Surface chemical analysis—Auger electron spectroscopy and X-ray photoelectron spectroscopy - Guide to the use of experimentally determined relative sensitivity factors for the quantitative analysis of homogeneous materials (2015). International Organization for Standardization (ISO)
Seah MP (1995) A system for the intensity calibration of electron spectrometers. J Electron Spectros 71(3):191–204
Hesse R, Streubel P, Szargan R (2005) Improved accuracy of quantitative XPS analysis using predetermined spectrometer transmission functions with UNIFIT 2004. Surf Interface Anal 37(7):589–607
Walton J, Fairley N (2006) A traceable quantification procedure for a multi-mode X-ray photoelectron spectrometer. J Electron Spectros 150(1):15–20
Graf N, Lippitz A, Gross T, Pippig F, Holländer A, Unger WES (2010) Determination of accessible amino groups on surfaces by chemical derivatization with 3,5-bis(trifluoromethyl)phenyl isothiocyanate and XPS/NEXAFS analysis. Anal Bioanal Chem 396(2):725–738
Gross T, Pippig F, Merz B, Merz R, Vohrer U, Mix R, Steffen H, Bremser W, Unger WES (2010) Determination of OH groups at plasma oxidised poly(propylene) by TFAA chemical derivatisation XPS: an inter-laboratory comparison. Plasma Process Polym 7(6):494–503
Jasieniak J, Smith L, van Embden J, Mulvaney P, Califano M (2009) Re-examination of the size-dependent absorption properties of CdSe quantum dots. J Phys Chem C 113(45):19468–19474
Smekal W, Werner WSM, Powell CJ (2005) Simulation of electron spectra for surface analysis (SESSA): a novel software tool for quantitative Auger-electron spectroscopy and X-ray photoelectron spectroscopy. Surf Interface Anal 37(11):1059–1067
Chudzicki M, Werner WSM, Shard AG, Wang YC, Castner DG, Powell CJ (2015) Evaluating the internal structure of Core-Shell nanoparticles using X-ray photoelectron intensities and simulated spectra. J Phys Chem C 119(31):17687–17696
Kalbe H, Rades S, Unger WES Determining shell thicknesses in stabilised CdSe@ZnS core-shell nanoparticles by quantitative XPS analysis using an infinitesimal columns model. J Electron Spectros 2016, 212:34–43
Powell CJ, Werner WSM, Kalbe H, Shard AG, Castner DG (2018) Comparisons of analytical approaches for determining shell thicknesses of core–shell nanoparticles by X-ray photoelectron spectroscopy. J Phys Chem C 122(7):4073–4082
Sarma DD, Santra PK, Mukherjee S, Nag A (2013) X-ray photoelectron spectroscopy: a unique tool to determine the internal Heterostructure of nanoparticles. Chem Mater 25(8):1222–1232
Acquafredda P (2019) XRF technique. Phys Sci Rev 4(8):20180171
Adams C, Brand C, Dentith M, Fiorentini M, Caruso S, Mehta M (2020) The use of pXRF for light element geochemical analysis: a review of hardware design limitations and an empirical investigation of air, vacuum, helium flush and detector window technologies. Geochem-Explor Env A 20(3):366–380
Lai MS, Xiang LW, Lin JM, Li HF (2013) Quantitative analysis of elements (C, N, O, Al, Si and Fe) in polyamide with wavelength dispersive X-ray fluorescence spectrometry. Sci China Chem 56(8):1164–1170
Dietrich PM, Streeck C, Glamsch S, Ehlert C, Lippitz A, Nutsch A, Kulak N, Beckhoff B, Unger WES (2015) Quantification of Silane molecules on oxidized silicon: are there options for a traceable and absolute determination? Anal Chem 87(19):10117–10124
Fischer T, Dietrich PM, Streeck C, Ray S, Nutsch A, Shard A, Beckhoff B, Unger WES, Rurack K (2015) Quantification of variable functional-group densities of mixed-silane monolayers on surfaces via a dual-mode fluorescence and XPS label. Anal Chem 87(5):2685–2692
Reinhardt F, Osan J, Torok S et al (2012) Reference-free quantification of particle-like surface contaminations by grazing incidence X-ray fluorescence analysis. J Anal At Spectrom 27(2):248–255
Nowak SH, Reinhardt F, Beckhoff B, Dousse JC, Szlachetko J (2013) Geometrical optics modelling of grazing incidence X-ray fluorescence of nanoscaled objects. J Anal At Spectrom 28(5):689–696
Meder F, Kaur S, Treccani L, Rezwan K (2013) Controlling mixed-protein adsorption layers on colloidal alumina particles by tailoring carboxyl and hydroxyl surface group densities. Langmuir 29(40):12502–12510
Ederer J, Janos P, Ecorchard P et al (2017) Determination of amino groups on functionalized graphene oxide for polyurethane nanomaterials: XPS quantitation vs. functional speciation. RSC Adv 7(21):12464–12473
Huynh J, Palacio R, Safizadeh F, Lefèvre G, Descostes M, Eloy L, Guignard N, Rousseau J, Royer S, Tertre E, Batonneau-Gener I (2017) Adsorption of uranium over NH2-functionalized ordered silica in aqueous solutions. ACS Appl Mater Interfaces 9(18):15672–15684
van de Waterbeemd M, Sen T, Biagini S, Bruce IJ (2010) Surface functionalisation of magnetic nanoparticles: quantification of surface to bulk amine density. Micro Nano Lett 5(5):282–285
Bakhshaei S, Kamboh MA, Nodeh HR, Md Zain S, Mahmad Rozi SK, Mohamad S, Mohammed Mohialdeen IA (2016) Magnetic solid phase extraction of polycyclic aromatic hydrocarbons and chlorophenols based on cyano-ionic liquid functionalized magnetic nanoparticles and their determination by HPLC-DAD. RSC Adv 6(80):77047–77058
Mansfield E (2015) Recent advances in thermal analysis of nanoparticles: Methods, models and kinetics. In:Tewary VK, Zhang Y (eds) Modeling, characterization, and production of Nanomaterials - Electronics, Photonics and Energy Applications. Woodhead Publishing, pp 167–178. https://doi.org/10.1016/b978-1-78242-228-0.00006-5
Steinhäuser KG, Sayre PG (2017) Reliability of methods and data for regulatory assessment of nanomaterial risks. NanoImpact 7:66–74
Das D, Yang Y, O'Brien JS et al (2014) Synthesis and physicochemical characterization of mesoporous SiO2 nanoparticles. J Nanomater 2014:176015
Sebby KB, Mansfield E (2015) Determination of the surface density of polyethylene glycol on gold nanoparticles by use of microscale thermogravimetric analysis. Anal Bioanal Chem 407(10):2913–2922
Mansfield E, Tyner KM, Poling CM, Blacklock JL (2014) Determination of nanoparticle surface coatings and nanoparticle purity using microscale thermogravimetric analysis. Anal Chem 86(3):1478–1484
Schirowski M, Hauke F, Hirsch A (2019) Controlling the degree of functionalization: in-depth quantification and side-product analysis of Diazonium chemistry on SWCNTs. Chemistry 25(55):12761–12768
Clausen PA, Kofoed-Sorensen V, Norgaard AW, Sahlgren NM, Jensen KA (2019) Thermogravimetry and mass spectrometry of extractable organics from manufactured nanomaterials for identification of potential coating components. Materials 12(22):3657
Lagarrigue P, Soulié J, Grossin D, Dupret-Bories A, Combes C, Darcos V (2020) Well-defined polyester-grafted silica nanoparticles for biomedical applications: synthesis and quantitative characterization. Polymer 211:123048
Demin AM, Mekhaev AV, Kandarakov OF et al (2020) L-lysine-modified Fe3O4 nanoparticles for magnetic cell labeling. Colloid Surface B 190:110879
Kunc F, Kodra O, Brinkmann A, Lopinski GP, Johnston LJ (2020) A multi-method approach for quantification of surface coatings on commercial zinc oxide nanomaterials. Nanomaterials 10(4):678
Jang E, Kim Y, Won YH, Jang H, Choi SM (2020) Environmentally friendly InP-based quantum dots for efficient wide color gamut displays. ACS Energy Lett 5(4):1316–1327
Bajaj M, Wangoo N, Jain DVS, Sharma RK (2020) Quantification of adsorbed and dangling citrate ions on gold nanoparticle surface using thermogravimetric analysis. Sci Rep 10(1):8213
You Z, Nirmalananthan-Budau N, Resch-Genger U, Panne U, Weidner SM (2020) Separation of polystyrene nanoparticles bearing different carboxyl group densities and functional groups quantification with capillary electrophoresis and asymmetrical flow field flow fractionation. J Chromatogr A 1626:461392
Giusti A, Atluri R, Tsekovska R, Gajewicz A, Apostolova MD, Battistelli CL, Bleeker EAJ, Bossa C, Bouillard J, Dusinska M, Gómez-Fernández P, Grafström R, Gromelski M, Handzhiyski Y, Jacobsen NR, Jantunen P, Jensen KA, Mech A, Navas JM et al (2019) Nanomaterial grouping: existing approaches and future recommendations. NanoImpact 16:100182
Acknowledgements
The authors would like to thank Dr. Jörg Radnik (BAM, Division 6.1 - Surface Analysis and Interfacial Chemistry) for his valuable input to the XPS part.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was financially supported by the Federal Institute for Materials Research and Testing (BAM) through an MI type 3 project, by the German Federal Ministry for Economic Affairs and Energy (BMWi) through the WIPANO project “AquaFunkNano” (03TNK005A), and by the European Metrology Programme for Innovation and Research (EMPIR) as part of the projects 18HLT01”MetVes II” and 18HLT02 “AeroTox”. The EMPIR initiative is co-funded by the European Union’s Horizon 2020 research and innovation programme and by the EMPIR participating states.
Author information
Authors and Affiliations
Contributions
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no competing of interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Geißler, D., Nirmalananthan-Budau, N., Scholtz, L. et al. Analyzing the surface of functional nanomaterials—how to quantify the total and derivatizable number of functional groups and ligands. Microchim Acta 188, 321 (2021). https://doi.org/10.1007/s00604-021-04960-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-021-04960-5