
Abstract
Cocaine profoundly affects both cerebral blood vessels and neuronal activity in the brain. The vasoconstrictive effects of cocaine, concurrently with its effects on neuronal [Ca2+]i accumulation are likely to jeopardize neuronal tissue that in the prefrontal cortex (PFC) could contribute to impaired self-regulation and compulsive cocaine consumption. Here we used optical imaging to study the cerebrovascular and neuronal effects of acute cocaine (1 mg/kg i.v.) and to examine whether selective blockade of L-type Ca2+ channels by Nifedipine (NIF) (0.5 mg/kg i.v.) would alleviate cocaine’s effects on hemodynamics (measured with cerebral blood volume, HbT), oxygenation (measured with oxygenated hemoglobin, HbO2) and neuronal [Ca2+]i, which were concomitantly measured in the PFC of naive rats. Our results show that in the PFC acute cocaine significantly reduced flow delivery (HbT), increased neuronal [Ca2+]i accumulation and profoundly reduced tissue oxygenation (HbO2) and these effects were significantly attenuated by NIF pretreatment. They also show that cocaine-induced vasoconstriction is distinct from its increase of neuronal [Ca2+]i accumulation though both of them contribute to hypoxemia and both effects were attenuated by NIF. These results provide evidence that blockade of voltage-gated L-type Ca2+ channels might be beneficial in preventing vasoconstriction and neurotoxic effects of cocaine and give support for further clinical investigations to determine their value in reducing cocaine’s neurotoxicity in cocaine use disorders.
Similar content being viewed by others

Introduction
Cocaine is not only a highly addictive drug but also one associated with significant neurotoxicity, including cerebral strokes, transient ischemic attacks as well as seizures [1,2,3,4]. Clinical studies using brain imaging have documented profound reductions in cerebral blood flow (CBF) in the brain of cocaine users [5, 6], and preclinical optical imaging studies have reported similar findings in rodent models of cocaine exposure [7,8,9]. However, the mechanisms associated with cocaine-induced CBF reductions and ischemia are not well understood, but may result from direct vasoconstriction elicited by cocaine-induced [Ca2+]i increases in vascular smooth muscle cells [10] and/or by parallel increases in neuronal activity that exacerbate tissue hypoxemia [11]. Indeed cocaine alters neuronal ionic mechanisms [12], including neuronal K+ and Ca2+ conductance and influx/efflux [13]. In pyramidal neurons from the medial prefrontal cortex (PFC) cocaine increased voltage-sensitive calcium currents in response to membrane depolarization. Previously, we showed that in rodents chronically exposed to cocaine, an acute dose of cocaine triggered a long lasting reduction in CBF and persistent increases in deoxygenated hemoglobin ([HbR]) and in intracellular calcium concentrations ([Ca2+]i), which we measured in the somatosensory cortex and that were associated with behavioral manifestations of temporal paralysis [14, 15]. Acute cocaine also triggered vasoconstriction and ischemia in naive rats but these effects were shorter lasting than in the chronically exposed rats suggesting sensitization with repeated exposure. Thus cocaine’s vascular effects and its ability to increase neuronal [Ca2+]i is likely to contribute to its neurotoxicity.
The elevated intracellular [Ca2+]i accumulation resulting from cocaine exposures is associated with its actions on L-type Ca2+ channels (L-channels) [16]. Studies have shown that Ca2+ antagonists can modify cocaine-induced behavioral effects including motor activity and reinforcement [8, 17]. Acute cocaine abnormally increases Ca2+ influx in cortical neurons [18], most likely via L-channels, whereas chronic cocaine up-regulates L-channels in pyramidal cortical neurons [13]. Some have suggested that L-type Ca2+-channel blockers (CCBs) such as nimodipine, which prevented cocaine-induced motor stimulation, might reduce cocaine’s rewarding effects [19]. In vitro studies in the medial PFC showed that L-channel blockade abolished chronic cocaine-induced increases in Ca2+ influx through voltage-gated Ca2+ channels in pyramidal neurons [13]. Ca2+-channel blockers can also reduce negative outcomes from cocaine-induced cerebral ischemia and stroke by buffering cocaine-induced vasoconstriction [20]. Here, we hypothesize that blockade of L-channels would ameliorate cocaine-induced increases in neuronal [Ca2+]i and vasoconstriction. To test this, we used our multi-modality optical image technique to study the cerebrovascular and neuronal effects of acute cocaine in the PFC and examined the efficacy of the L-type Ca2+-channel blocker, Nifedipine (NIF) in alleviating cocaine’s effects on hemodynamics (measured by HbT and oxygenated hemoglobin, HbO2) and neuronal [Ca2+]i in vivo. We studied the PFC since this brain region is critical for executive function and its disruption is implicated in the impairments in self-regulation and compulsive drug consumption in cocaine addiction.
Material and method
Animals and experimental design
All experimental procedures were approved by the Institutional Animal Care and Use Committee at Stony Brook University. F344 adult male rats (n = 17) were used; female rats were not included to avoid the confounds from the effects of sex hormones on cerebral blood vessels [21] and on neuronal excitability in response to cocaine [22]. Animals were randomly assigned into four experimental groups as detailed in Table 1. Experimenters were unblind to the conditions as drugs needed to be prepared fresh to prevent dissolution. Rats were single-housed under a 12:12 h light/dark cycle, with ad libitum access to food and water.
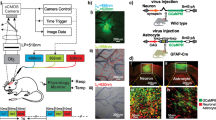
Virus infusion to express GCaMP6f in neurons in PFC
To measure changes in intracellular calcium ([Ca2+]i) in neurons, we used a genetically encoded calcium indicator, AAV1.Syn.GCaMP6f.WPRE.SV40 virus (Penn Vector Core). Animals were anesthetized with isofluorane, mounted on a stereotaxic frame, and their scalps opened. The GCaMP6f virus was then infused into the right PFC (A/P: +3; M/L: 0.8); two infusions of 0.5 μl were made (D/V: −1.4 and −1 mm from skull) at a rate of 0.2 μl/min, and the injector was left in place for 20 min following each infusion to allow for diffusion and then the scalp was closed. At the time of imaging, 3–4 weeks had elapsed since virus infusion to allow for GCaMP6f expression.
Surgical preparation
At 3–4 weeks after viral injection, an optical window was implanted over the PFC for imaging [23]. The rats were anesthetized with isofluorane, and a 16-gauge IV catheter was inserted into their trachea (Angiocath, BD) and attached to a respirator to control breathing. An incision was made proximal to the left hind limb to expose the left femoral artery and vein, and catheters (0.58 mm ID, 0.99 mm OD, Scientific Commodities Inc.) were inserted into the femoral artery to monitor arterial blood pressure, and into the femoral vein for drug delivery during imaging. Rats were mounted on a stereotaxic frame (Kopf 900) and a 4 × 6 mm2 portion of skull was removed above the frontal cortex (A/P: +1 to +5; M/L: −3 to +3). The dura was carefully removed to expose the brain surface and covered with 1.25% agarose gel and a cover glass (0.15 mm thick; VWR micro-cover glass) affixed to the skull using crazy glue (Gorilla Glue). The animal’s mean arterial blood pressure (MABP; mean = 82.28, SE = 1.88), body temperature (~37–38 °C) and respiration (~40–45 breaths/minute) were monitored and recorded (Small Animal Monitoring and Gating system, model 1025 L, SA Instruments Inc.).
Drugs and preparation
Cocaine-HCl was provided by the NIDA drug supply program and Nifedipine was purchased from MilloporeSigma (N-7634). Drug solutions were freshly prepared before imaging. Cocaine was dissolved in 0.9% saline solution. Nifedipine (NIF) was dissolve in DMSO (e.g., 50 mg/ml) and mixed with saline (0.5 mg/kg NIF administered in 0.25 ml i.v.). For the vehicle (VEH) we used 5% DMSO in saline (0.25 ml administered i.v.).
Optical imaging over PFC
For in vivo imaging of the PFC (A/P: +1 to +5; M/L: −3 to +3), we used a multi-modality image platform (MIP) developed in our laboratory using procedures previously described [11, 14, 24]. As illustrated in Fig. S1 (Supplementary Materials), the cranial window was sequentially illuminated by three LEDs (λ1 = 568 nm, λ2 = 630 nm and λexcitation = 488 nm; Spectra Light Engine, Lumicor), at a rate of 12.5 Hz/channel. Images were captured with MIP image probe connected to a sCMOS camera (pixel size: 6.5 μm; Zyla 4.3, Andor) using modified Solis software (version 4.26, Andor). The MIP allowed us to simultaneously image changes in Ca2+ fluorescence and in oxygenated- and deoxygenated-hemoglobin concentrations used to determine changes in total hemoglobin concentration (ΔHbT). Animals were excluded if the cortex was damaged during implantation, or GCaMP6f was insufficiently expressed to allow for accurate imaging.
We measured Ca2+ fluorescence and hemodynamics (ΔHbT) responses to cocaine after VEH and after Nifedipine (NIF) pretreatments. Animals in Group 3 and Group 4, were scanned twice: first when pretreated with VEH prior to a first cocaine injection, and then when pretreated with VEH (Group 3) or NIF (Group 4) prior to a second cocaine injection, with approximately 2 h between each cocaine infusion. Each scan consisted of a 10 min baseline, 10 min infusion of VEH or of NIF (0.5 mg/kg), then 10 min to establish a new baseline, followed by a 1 min cocaine infusion (1 mg/kg, 0.1 mL) after which imaging continued for 60 min post cocaine administration.
Image processing
The hemodynamic changes in the cortical tissue, i.e., the changes in oxygenated hemoglobin (ΔHbO2) and deoxygenated hemoglobin (ΔHbR), were calculated from λ1 and λ2 images obtained from MPI system according to the equation
where ελ1HbO2, ελ1HbR, ελ2HbO2, and ελ2HbR are the molar extinction coefficients for HbO2 and HbR at the two wavelengths, Rλ1(t) and Rλ2 (t) are the measured diffuse reflectance matrices (2-D images) at these wavelengths and Lλ1 (t) and Lλ2 (t) are the path lengths of light propagation [25, 26]. The sum of the changes in oxygenated hemoglobin (ΔHbO2) and deoxygenated hemoglobin (ΔHbR) produces the changes in the total hemoglobin (ΔHbT), which can be so-called as the changes in the cerebral blood volume within the cortex [24].
Changes in intracellular ([Ca2+]i) were calculated from fluorescence (λexcitation = 488 nm, λemission ≧ 510 nm). To minimize the effect of absorption changes within the tissue (due to blood flow changes) on the calcium fluorescence measurement, the fluorescence intensity in regions expressing GCaMP were normalized to the background intensity detected from the equivalent left hemisphere region, which had no expression (e.g., left side of PFC in the top panel of Fig. S1C). Results are presented as percent change from baseline to control for differences in GCaMP expression between animals.
To quantify cocaine-induced changes in neuronal [Ca2+]i fluorescence and hemodynamics (HbO2 and HbT) and to compare differences between VEH and NIF pretreatment, we selected regions of interest (ROIs) within the PFC where there were no visible blood vessels to minimize the light attenuation from high volume of blood passing through vessels. For each animal we extracted five ROIs in the GCaMP6f-expressed region (as demonstrated Fig. 1a0) and used the same ROI location for the hemodynamic channels (HbO2 in Fig. 2a0, HbT in Fig. 3a0) to ensure these multiple parameters were measured in the same region.
Statistical analysis
Results are reported as mean ± standard error (SE). Data was analyzed using two-way repeated measure ANOVAs, and Bonferroni corrected t-tests were used for post hoc analyses (SigmaStat, Systat Software, Inc.). Statistical tests were performed using SigmaStat software (Systat Software Inc.), and alpha levels were set at 0.05, with only significant effects reported.
Results
Optimization of Nifedipine (NIF) administration in vivo
To determine what doses of NIF would block intracellular Ca2+ in neurons while minimally affecting blood pressure we administered 0.1 mg/kg NIF every 10 min (shaded time periods in Fig. S2A, Supplementary Materials), while monitoring the changes in [Ca2+]i fluorescence. After the 5th infusion (i.e., 0.5 mg/kg NIF in total), the [Ca2+]i fluorescence signal did not change further for more than 10 min, indicative of stabilization, so this dose (0.5 mg/kg, i.v.) was selected for pretreatment before the cocaine challenge.
Figure S2B (Supplementary Materials) summarizes the changes in MABP as a function of time. After NIF (0.5 mg/kg, 0.25 ml i.v.) infusion (shaded time period in Fig. S2B), MABP decreased from 85.3 ± 1.45 mmHg at baseline to 66.3 ± 2.96 mmHg within 5–6 min of infusion and gradually recovered to 76.0 ± 2.08 mmHg at t = 12 min remaining stable thereafter for the time period of recording. Although 0.5 mg/kg NIF reduced MABP, it did not decrease it beyond the range for autoregulation [27], and prior to cocaine infusion it had stabilized at a level that did not differ from baseline (p = 0.43).
Nifedipine (NIF) decreased neuronal [Ca2+]i and increased blood volume in PFC
We hypothesize that NIF would diminish cocaine-induced [Ca2+]i increases in neurons and dilate vessels increasing blood supply to PFC. Figure S3 (Supplementary Materials) shows time courses of [Ca2+]i fluorescence and changes in cerebral blood volume (measured by changes in HbT) induced by NIF (0.5 mg/kg, i.v., over 10 min) following vehicle infusion. After 2–3 min of NIF infusion, mean neuronal [Ca2+]i gradually decreased, with a peak reduction of 3.66 ± 0.74% occurring after 15 min of NIF induction (i.e., at t = 20 min in Fig. S3), and plateauing thereafter. The decrease in [Ca2+]i with NIF differed significantly from baseline (p < 0.001, Fig. S3C). Meanwhile, significant increases in cerebral blood volume (ΔHbT) were observed following NIF that stabilized at 9.2 ± 1.65% over the baseline after 15 min of induction (i.e., at t = 22 min, p < 0.001, Fig. S3B). These results indicate that NIF concurrently triggered vascular and neuronal [Ca2+]i changes.
Nifedipine (NIF) reduced the duration of cocaine-induced changes in cortical activity
To compare cocaine’s effects on PFC with and without blockade of L-type Ca2+ channels by NIF each animal was administered cocaine twice, i.e., first with vehicle and then with NIF as pretreatment before cocaine administrations. To minimize lingering effects from the first cocaine dose we gave the second cocaine infusion 2 h after the first one. Figure S4 (Supplemental material) compares the hemodynamic responses (e.g., [HbO2], [HbR], and [HbT]) to cocaine (n = 4) between the first and the second cocaine challenge when given after vehicle (5% DMSO in saline) pretreatments, which indicates that, when rats were pretreated with vehicle twice, there was no significant difference in the PFC response to cocaine.

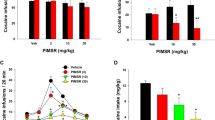
A Representative [Ca2+]i fluorescence images showing the spatial and dynamic changes in response to cocaine (1 mg/kg, i.v.) in PFCs after vehicle pretreatment (VEH, upper panel) and NIF-pretreatment (NIF, lower panel); It shows that NIF lowered baseline levels of Ca2+ activity even after cocaine. B Averaged dynamic changes for cocaine-induced [Ca2+]i fluorescence without (VEH) and with NIF pretreatment (0.5 mg/kg, i.v., NIF). C Comparison of mean peak changes in neuronal [Ca2+]i fluorescence (i.e., Δ[Ca2+]i) triggered by cocaine between vehicle and NIF pretreatment alongside the individual values for each animal (n = 5), showing the significant attenuation by NIF. D Averaged normalized dynamic change in [Ca2+]i following cocaine with vehicle and NIF pretreatment at time t = 0 min. Values in graphs correspond to mean and standard errors. ROIs: the regions of interest in the tissue that did not have visible blood vessels to minimize the light attenuation from blood passing through vessels. For each animal, five ROIs were selected in the GCaMP6f-expressed regions. *p < 0.05; NS: not significant.
Cocaine triggered significant changes in neuronal [Ca2+]i in PFC that differed when given after vehicle or after NIF pretreatment (Fig. 1). To obtain these measures we obtained a 10 min baseline scan, followed by a 10 min infusion of vehicle (5% DMSO in saline) or NIF (0.5 mg/kg i.v., 0.25 ml) scan, then a 10 min scan to establish a new baseline, followed by a 1 min cocaine infusion (1 mg/kg i.v., 0.1 mL) with imaging continuing for 60 min post cocaine administration. Figure 1Aa0, Ab0 shows representative images of GCaMP6f neuronal expression before and after NIF infusion, respectively. Figure 1Aa1–a7, Ab1–b7 shows the dynamic changes in GCaMP6f [Ca2+]i fluorescence in response to cocaine for VEH and NIF pretreatment, respectively. Figure 1Aa1–a7 shows that with VEH pretreatment cocaine increased neuronal Ca2+ fluorescence. Two-way repeated ANOVA showed a significant treatment and time effect [F(64, 256) = 2.99, p < 0.001] (n = 5, ROIs = 5/animal). Post hoc comparisons showed that in the VEH pretreated group [Ca2+]i fluorescence increased from 2 to 24 min (p < 0.05) and returned to baseline after 25 min (p > 0.05). In the NIF pretreated group, [Ca2+]i fluorescence increased from 3 to 12 min (p < 0.05) and returned to baseline after 13 min (p > 0.05). Figure 1B shows that NIF by itself reduced intracellular Ca2+ concentration, with an average of −3.70 ± 0.7% decrease from baseline (p < 0.01). Figure 1Ab2–b7 shows that even after cocaine, Ca2+ fluorescence with NIF remained lower (Fig. 1A and red curves in Fig. 1B,) than the vehicle baseline. The peak neuronal [Ca2+]i fluorescence increase induced by cocaine corresponded to 5.94 ± 0.7% for VEH and to 0.10 ± 1.5% for NIF pretreatment (p = 0.02) (Fig. 1C).
After normalizing the NIF baseline (NIF) to levels equivalent to the vehicle baseline (VEH t = 0) (Fig. 1D), one can observe that cocaine induced a short lasting increase in Ca2+ that by 25 min was lower than the baseline (Fig. 1D, [F(64, 256) = 10.31, p < 0.001]). NIF did not alter the peak amplitude of cocaine-induced increase in Ca2+ (4.0 ± 0.83%) but shortened its duration. A two-way repeated ANOVA on [Ca2+]i changes showed a significant drug and time interaction effect ([F(64, 256) = 2.99, p < 0.001]). Comparisons of [Ca2+]i changes for the same time points for the VEH and NIF pretreatment are shown in Fig. 1D and significant differences are summarized in Table S1 (Supplementary Materials). Analyses showed that, the normalized [Ca2+]i responses to cocaine did not differ between VEH and NIF during the time period of t = −4–17 min (p = 0.97~0.06) but the [Ca2+]i changes recovered much faster with NIF than with VEH, thus showing significant differences during t = 18–60 min (i.e., p = 0.04~0.004, Table S1A).
Nifedipine (NIF) did not change basal oxygenation but attenuated cocaine-induced [HbO2] reduction
Acute cocaine decreased [HbO2] after vehicle and NIF pretreatment attenuated this decrease and its duration (Fig. 2). Panels Aa1–a7 and Ab1–b7 (Fig. 2) show representative images of percent change in [HbO2] concentration over time following cocaine injection at t = 0 min with VEH and NIF pretreatment, respectively. Two-way ANOVA on the comparisons of Δ[HbO2] of VEH and NIF showed a significant interaction between pretreatment and time ([F(64, 256) = 3.07, p < 0.001]). For VEH, cocaine induced a persistent decrease that returned to baseline at 30 min (t = 1~32 min; p < 0.001, t = 33 min; p = 0.15) (Fig. 2B), whereas with NIF pretreatment, cocaine’s effect was reduced in amplitude by 13.21 ± 3.3%, (p < 0.001) and duration (<15 min) (p = 0.01) (Fig. 2B). NIF pretreatment did not cause a significant change in baseline [HbO2] (p = 0.17) (Fig. 2B). The peak decreases in [HbO2] by cocaine with VEH corresponded to 36.0 ± 1.5% and with NIF to 21.6 ± 1.4% (p < 0.05) (Fig. 2C). Figure 2D shows the normalized time courses of [HbO2] changes in response to cocaine with VEH or NIF pretreatment. Following VEH cocaine reduced [HbO2], which remained below baseline at 45 min post cocaine (p = 0.03), whereas after NIF the recovery was significantly faster (t = 31 ± 7.5 min post cocaine) (p = 0.008). The comparison of VEH and NIF responses to cocaine for the equivalent time points showed no differences in HbO2 changes from t = −4–1 min (p = 0.97~0.45, Supplementary Table S1B) and significant differences between t = 2 and 43 min (p < 0.001~p = 0.04,) followed by recovery to baseline with no significant differences from t = 44~60 min (p = 0.08~0.23, Table S1B).

A Representative images sowing dynamic changes in response to cocaine (1 mg/kg, i.v.) after vehicle pretreatment (VEH, upper panel) and after NIF-pretreatment (NIF, lower panel). B NIF pretreatment (0.5 mg/kg, i.v.) did not affect baseline [HbO2] but significantly attenuated cocaine-induced [HbO2] reductions. C Comparison of cocaine-induced peak decreases in tissue oxygenation (ΔHbO2) between vehicle and NIF pretreatment alongside the individual values (n = 5). D Normalized [HbO2] values (to baseline VEH) for baseline (t = 0) and after cocaine showing the attenuation of [HbO2] reductions with NIF pretreatment. Values in graphs correspond to mean and standard errors. ROIs: the regions of interest. For a given animal these ROIs were placed in the same location in hemodynamic channels to the Ca2+ fluorescence channel. *p < 0.05; NS: not significant.
Nifedipine (NIF) mitigates cocaine’s reduction of blood volume in PFC
Acute cocaine reduced cerebral blood volume as measured by changes in HbT (Fig. 3). Figure 3A (Aa1–a7) shows representative images for percent change in HbT (∆[HbT]) over time following cocaine injection from t = 0 min, illustrating the ∆[HbT] decreases. Figure 3Aa1, Ab1 shows ∆[HbT] images before and after NIF showing the increase in [HbT] after NIF (average increase of 6.9 ± 0.83% p = 0.003). Two-way repeated ANOVA for the comparisons between Veh-Coc and NIF-Coc showed a significant interaction of Pretreatment and Time ([F(64, 256) = 1.48, p = 0.02]) but the pretreatment effect was not significant (p = 0.39). Cocaine’s reduction of ∆ [HbT] was significantly shortened from 39.4 ± 5.8 min after VEH to 9.8 ± 2.1 min after NIF p = 0.004 (Fig. 3B). We also compared cocaine-induced changes (∆[HbT]) integrated over time before they returned to baseline and showed significant differences between VEH and NIF that corresponded to −14.45 ± 1.8% and −5.61 ± 1.1%, respectively (p = 0.003). Figure 3D shows the normalized dynamic changes in ∆[HbT] following cocaine at time t = 0 min, showing no significant differences between VEH and NIF (p > 0.05, Supplementary Table S1C).

A Representative images of the dynamic changes in cerebral blood volume (∆[HbT]) in response to cocaine (1 mg/kg, i.v.) in PFCs after vehicle (VEH, upper panel) and NIF-pretreatment (NIF, lower panel); Panels Aa0 and Ab0 show representative images for baseline before and after NIF (0.5 mg/kg, i.v.), respectively. B Dynamic measures of cocaine-induced cerebral blood volume (∆[HbT]) changes with vehicle and with NIF pretreatment (0.5 mg/kg, i.v.). C Comparison of the integrated decreases in ΔHbT (n = 5) between vehicle and NIF pretreatment after cocaine. D Normalized dynamic change in (∆[HbT]) following cocaine at time t = 0 min. These results show that NIF pretreatment (0.5 mg/kg, i.v.) dilates vessels, elevating baseline cerebral blood volume and attenuated the non-normalized cocaine-induced (∆[HbT]) decreases. Values in graphs correspond to mean and standard errors. ROIs: the regions of interest. For a given animal these ROIs were placed in the same location in hemodynamic channels to the Ca2+ fluorescence channel. *p < 0.05; NS: not significant.
Cocaine-induced hypoxemia was associated with both its reduction of blood volume and its neuronal Ca2+ increases, and NIF buffered these changes
In order to determine the extent to which the reductions in HbO2 with cocaine were due to reductions in blood volume (HbT) or due to increases in oxygen utilization from neuronal activation ([Ca2+]i) we computed the regression plots between ΔHbO2 and ΔHbT (Fig. 4A) and between ΔHbO2 and Δ[Ca2+]i (Fig. 4B) and compared these associations between VEH and NIF. The regression analyses were preformed across all animals including all ROI and time points (n = 5 and multiple ROIs (m = 5). The correlations between cocaine-induced changes in HbT and HbO2 were significant both for VEH (r = 0.73, p < 0.001) and for NIF (r = 0.92, p < 0.001). However, the regression slopes differed significantly (Z = −2.4, p = 0.01) such that for a given HbT reduction the HbO2 levels for VEH were markedly lower than for NIF indicating that an additional factor (neuronal activation) contributed to the reduced HbO2 (Fig. 4A). Supporting this, the regression plots showed significant correlation between cocaine-induced increases in [Ca2+]i and decreases in HbO2 both for VEH (r = −0.687, p < 0.001) and NIF (r = −0.861, p < 0.001), and though the slopes did not differ it showed an attenuated [Ca2+]i increase with NIF compared to VEH concomitantly with an attenuated reduction of HbO2 (Fig. 4B). Finally to assess if NIF prevented cocaine-induced dissociation of neuronal activity ([Ca2+]i) and blood delivery (HbT), we computed the regression plots between them (Fig. 4C). We showed that for both Δ[Ca2+]i increases were associated with ΔHbT decreases, both for vehicle (r = −0.77, p < 0.001) and NIF (r = −0.93, p < 0.001). However, their slopes differed (p = 0.04), showing a more constrained cocaine-induced reduction in HbT and increase in [Ca2+]i for NIF than for VEH indicating that NIF buffered cocaine’s effects both on blood vessels and on neuronal [Ca2+]i increases.

A Linear regression between cocaine-induced ΔHbT and ΔHbO2. B Linear regression between cocaine-induced Δ[Ca2+]i and ΔHbO2. C Linear regression between cocaine-induced Δ[Ca2+]i and ΔHbT. Green markers: with vehicle treatment; Orange markers: with NIF pretreatment.
Conclusion and discussion
Cocaine is one of the most addictive drugs and its use, which has expanded markedly in the past decade, is a major contributor of overdose fatalities in the United States [28]. While cocaine’s dopamine-enhancing properties are believed to underlie its rewarding and addictive effects, its vasoconstrictive effects [29] alongside neuronal Ca accumulation [11, 24] and disruption of neurovascular coupling [30] are likely to damage neuronal tissue and in the PFC contribute to impairments in executive function that facilitate compulsive drug taking [8]. In humans, chronic use of cocaine causes damages to the PFC disrupting CBF [5], metabolic activity [31, 32], structure [33, 34], function [35, 36], and neurotransmission [37,38,39]. In rodents, chronic cocaine exposure was associated with loss of neurons in the PFC [40], perhaps reflecting overactivation-induced neurotoxicity (excessive neuronal Ca2+ accumulation) associated with dysfunction of L-type Ca2+ channels [13, 41,42,43,44] that would be exacerbated by cocaine’s vasoconstriction effects. The increases in [Ca2+]i triggered by cocaine would make the tissue more vulnerable to ischemia secondary to decreases in CBF and to cell damage. Thus, we hypothesized that NIF by preventing cocaine-induced vasoconstriction and excitotoxicity from neuronal [Ca2+]i accumulation would minimize tissue hypoxemia.
Our results corroborated our hypothesis and showed that in the PFC acute cocaine significantly reduced flow delivery and increased neuronal [Ca2+]i accumulation profoundly reducing tissue oxygenation and these effects of cocaine were significantly attenuated by NIF pretreatment. Our results with NIF are consistent with prior studies that showed that the L-type calcium-channel blocker, isradipine reversed cocaine-induced cerebral ischemia and tissue damage in rodents exposed to chronic cocaine [1, 45, 46]. Interestingly, the antivasospastic effects of isradipine were attributed to blockade of dopamine release into cortical neurons that control the cerebral vasculature [17, 47]. Human studies using SPECT imaging reported that isradipine administered 60 min before cocaine (0.33 mg/kg i.v.) prevented both global and regional ischemia in dopamine-rich brain areas [48]. In the heart, clinical studies showed that pretreatment with NIF protected against cocaine’s depression of myocardial function and decrease in coronary blood flow [10, 49, 50] and intracoronary administration of a Ca-channel blocker reversed cocaine-induced microvascular dysfunction [51].
Our findings show that cocaine-induced vasoconstriction is distinct from its increase of neuronal Ca accumulation, and that NIF affected both processes. Specifically, we showed that while cocaine-induced decreases in blood delivery (ΔHbT) were significantly associated with the reduction in oxygenation (ΔHbO2) both for VEH and NIF for a given decrease in blood delivery the reductions in oxygenation were smaller for NIF than for VEH indicating that an additional effect of NIF (neuronal Ca accumulation) contributed to the reduced hypoxemia. Indeed, while cocaine-induced increases in [Ca2+]i and decreases in oxygenation were significantly correlated both for vehicle and NIF, the [Ca2+]i increase with NIF was attenuated concomitantly with an attenuation of hypoxemia. We also showed that NIF reduced cocaine-induced disruption of neurovascular coupling [11] as evidenced by a more constrained reduction in HbT concomitant to the increase in [Ca2+]i for NIF than for VEH, thus indicating that NIF buffered cocaine’s effects both on blood vessels and on the neuronal [Ca2+]i increases.
We had previously shown using a rodent model of chronic cocaine self-administration that animals that consumed high doses of cocaine showed sensitized responses to cocaine-induced vasoconstriction in PFC and the concomitant hypoxemia was associated with escalation of cocaine intake [8]. In this chronic model of cocaine administration that emulates human cocaine consumption, NIF pretreatment prevented cocaine’s vasoconstriction and neuronal [Ca2+]i increases while decreasing cocaine intake and blocking reinstatement [8]. Here we expand these finding to show that NIF prevents cocaine-induced vasoconstriction and neuronal [Ca2+]i accumulation, even in naive animals. Also by systematically studying the associations between cocaine’s effects in blood delivery, neuronal [Ca2+]i accumulation and hypoxemia in the same animal we are able to document that hypoxemia induced by cocaine is a function of both its vasoactive effects and its increase in neuronal [Ca2+]i content, and that NIF benefits likely stem from it preventing both of these effects.
The mechanism underlying the effects of cocaine on vasoconstriction as well as those underlying neuronal [Ca2+]i increases are not fully understood. Although the duration of neuronal [Ca2+]i response to cocaine with NIF pretreatment was significantly diminished (t > 17 min in Fig. 1D), the peak for the normalized neuronal [Ca2+]i increase did not differ from that of the VEH (t ≤ 17 min, Fig. 1D). Lack of a peak effect suggests that NIF did not interfere with cocaine-induced activation of NMDARs or their phosphorylation, which increases [Ca2+]i influx [52,53,54]. It could also indicate that NIF did not block the initial increase in intracellular Ca2+ release from the endoplasmic reticulum [55]. However, since the dose of NIF used (0.5 mg/kg) was relatively low compared to that used by others [56, 57], it is possible that higher doses might have reduced the peak. Moreover, it is likely that combined blockade of NMDARs and L-channels might have suppressed the increased peak.
Cocaine’s vasoconstriction effects are likely to reflect its sympathomimetic effects but its effects on L-type Ca channel’s function in blood vessels are also likely to contribute [58]. The mechanisms responsible for neuronal [Ca2+]i accumulation are also likely to involve cocaine’s effects on L-type [Ca2+]i channels in neurons, which are disrupted with chronic exposures [42, 44]. Additionally, chronic cocaine triggers neuroadaptations in glutamate neurotransmission [59] that could further worsen cocaine-induced neurotoxicity. Therefore, combined overactivation of L-channels and glutamate signaling with repeated use of cocaine could exacerbate neuronal [Ca2+]i dysregulation and excitotoxicity, as well as worsen vasoconstriction mediated by excessive [Ca2+]i in blood vessel smooth muscles. How and to what extent dysfunctional NMDA receptors contribute to cocaine-induced neuronal hyperactivity and [Ca2+]i dysregulation in PFC requires further investigation.
Our findings have clinical implication for they provide further evidence of the potential of L-type Ca-channel blockers for the treatment of cocaine use disorders. Further preclinical evidence is expanding on the beneficial effects of L-type Ca-channel blockers in preventing cocaine intake and relapse [17, 58, 60, 61]. To the extent that L-type Ca-channel blockers also block cocaine-induced vasoconstriction in the myocardium [62, 63], they could also help prevent cocaine’s lethality.
A limitation for this study is that we only studied male rats. Though we initially selected only males to avoid the confounds of sex hormones on cerebral blood vessels [21] and on neuronal excitability in response to cocaine [22], it is increasingly evident that sex differences in the response to cocaine are clinically relevant [64,65,66,67,68,69,70,71]. Thus future studies should evaluate the effects of NIF on female rats. Another limitation is the small sample size of our study, yet despite this the large and consistent effects of cocaine and its blockade by NIF enabled us to document significant effects. Also, the studies were done under anesthesia, which confounds our findings since we cannot rule out the possibility that some of cocaine’s effects reflect its interaction with the anesthetic agent [72, 73].
In summary, our findings indicate that blockade of L-channels may be beneficial in reducing cocaine-induced hypoxemia, which could help reduce damage to PFC, and potentially other brain regions and thus of therapeutic value in cocaine use disorder.
References
Johnson BA, Devous MD Sr, Ruiz P, Ait-Daoud N. Treatment advances for cocaine-induced ischemic stroke: focus on dihydropyridine-class calcium channel antagonists. Am J Psychiatry. 2001;158:1191–8.
Bartzokis G, Beckson M, Lu PH, Edwards N, Rapoport R, Bridge P, et al. Cortical gray matter volumes are associated with subjective responses to cocaine infusion. Am J Addict. 2004;13:64–73.
Bolouri MR, Small GA. Neuroimaging of hypoxia and cocaine-induced hippocampal stroke. J Neuroimaging. 2004;14:290–1.
Buttner A, Mall G, Penning R, Sachs H, Weis S. The neuropathology of cocaine abuse. Leg Med (Tokyo). 2003;5(Suppl 1):S240–2.
Volkow ND, Mullani N, Gould KL, Adler S, Krajewski K. Cerebral blood flow in chronic cocaine users: a study with positron emission tomography. Br J Psychiatry. 1988;152:641–8.
Bell KM, M. N, Lyons KP. Regional cerebral blood flow and cocaine abuse. West J Med Oct. 1994;161:412–3.
Chen W, Liu P, Volkow ND, Pan Y, Du C. Cocaine attenuates blood flow but not neuronal responses to stimulation while preserving neurovascular coupling for resting brain activity. Mol Psychiatry. 2016a;21:1408–16.
Du C, Volkow ND, You J, Park K, Allen CP, Koob GF, et al. Cocaine-induced ischemia in prefrontal cortex is associated with escalation of cocaine intake in rodents. Mol Psychiatry. 2020;25:1759–76.
Zhang A, Cheng TP, Altura BT, Altura BM. Acute cocaine results in rapid rises in intracellular free calcium concentration in canine cerebral vascular smooth muscle cells: possible relation to etiology of stroke. Neurosci Lett. 1996;215:57–9.
Zhang Q, You J, Volkow ND, Choi J, Yin W, Wang W, et al. Chronic cocaine disrupts neurovascular networks and cerebral function: optical imaging studies in rodents. J Biomed Opt. 2016;21:26006.
Allen CP, Park K, Li A, Volkow ND, Koob GF, Pan Y, et al. Enhanced neuronal and blunted hemodynamic reactivity to cocaine in the prefrontal cortex following extended cocaine access: optical imaging study in anesthetized rats. Addict Biol. 2019;24:485–97.
Trantham-Davidson H, Lavin A. Acute cocaine administration depresses cortical activity. Neuropsychopharmacology. 2004;29:2046–51.
Nasif FJ, Hu XT, White FJ. Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci. 2005a;25:3674–9.
Chen W, Park K, Volkow N, Pan Y, Du C. Cocaine-induced abnormal cerebral hemodynamic responses to forepaw stimulation assessed by integrated multi-wavelength spectroimaging and laser speckle contrast imaging. IEEE J Sel Top Quantum Electron. 2016b;22:146–53.
You J, Volkow ND, Park K, Zhang Q, Clare K, Du C, et al. Cerebrovascular adaptations to cocaine-induced transient ischemic attacks in the rodent brain. JCI Insight. 2017;2:e90809.
Kabir ZD, Martinez-Rivera A, Rajadhyaksha AM. From gene to behavior: L-type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics. 2017;14:588–613.
Swinford-Jackson SE, Pierce RC. Harmony and heresy of an L-type calcium channel inhibitor: suppression of cocaine seeking via increased dopamine transmission in the nucleus accumbens. Neuropsychopharmacology. 2018;43:2335–6.
Du C, Yu M, Volkow ND, Koretsky AP, Fowler JS, Benveniste H. Cocaine increases the intracellular calcium concentration in brain independently of its cerebrovascular effects. J Neurosci. 2006;26:11522–31.
Pani L, Kuzmin A, Diana M, De Montis G, Gessa GL, Rossetti ZL. Calcium receptor antagonists modify cocaine effects in the central nervous system differently. Eur J Pharm. 1990;190:217–21.
Kosten TR. Pharmacotherapy of cerebral ischemia in cocaine dependence. Drug Alcohol Depend. 1998;49:133–44.
Duckles SP, Krause DN. Cerebrovascular effects of oestrogen: multiplicity of action. Clin Exp Pharm Physiol. 2007;34:801–8.
Wissman AM, McCollum AF, Huang GZ, Nikrodhanond AA, Woolley CS. Sex differences and effects of cocaine on excitatory synapses in the nucleus accumbens. Neuropharmacology. 2011;61:217–27. https://doi.org/10.1016/j.neuropharm.2011.04.002.
Park K, Volkow ND, Pan Y, Du C. Chronic cocaine dampens dopamine signaling during cocaine intoxication and unbalances D1 over D2 receptor signaling. J Neurosci. 2013;33:15827–36.
Yuan Z, Luo Z, Volkow ND, Pan Y, Du C. Imaging separation of neuronal from vascular effects of cocaine on rat cortical brain in vivo. Neuroimage. 2011;54:1130–9.
Dunn AK, Devor A, Dale AM, Boas DA. Spatial extent of oxygen metabolism and hemodynamic changes during functional activation of the rat somatosensory cortex. Neuroimage. 2005;27:279–90.
Yuan S, Devor A, Boas DA, Dunn AK. Determination of optimal exposure time for imaging of blood flow changes with laser speckle contrast imaging. Appl Opt. 2005;44:1823–30.
Gozzi A, Ceolin L, Schwarz A, Reese T, Bertani S, Crestan V, et al. A multimodality investigation of cerebral hemodynamics and autoregulation in pharmacological MRI. Magn Reson Imaging. 2007;25:826–33.
Kariisa M, Scholl L, Wilson N, Seth P, Hoots B. Drug overdose deaths involving cocaine and psychostimulants with abuse potential - United States, 2003–2017. MMWR Morb Mortal Wkly Rep. 2019;68:388–95.
Ren H, Du C, Yuan Z, Park K, Volkow ND, Pan Y. Cocaine-induced cortical microischemia in the rodent brain: clinical implications. Mol Psychiatry. 2012;17:1017–25.
Chen W, Volkow ND, Li J, Pan Y, Du C. Cocaine Decreases Spontaneous Neuronal Activity and Increases Low-Frequency Neuronal and Hemodynamic Cortical Oscillations. Cereb Cortex. 2019;29:1594–1606.
Volkow ND, Fowler JS, Wolf AP, Hitzemann R, Dewey S, Bendriem B, et al. Changes in brain glucose metabolism in cocaine dependence and withdrawal. Am J Psychiatry. 1991;148:621–6.
Volkow ND, Hitzemann R, Wang GJ, Fowler JS, Wolf AP, Dewey SL, et al. Long-term frontal brain metabolic changes in cocaine abusers. Synapse. 1992;11:184–90.
Matochik JA, London ED, Eldreth DA, Cadet JL, Bolla KI. Frontal cortical tissue composition in abstinent cocaine abusers: a magnetic resonance imaging study. Neuroimage. 2003;19:1095–102.
Moreno-López L, Catena A, Fernández-Serrano MJ, Delgado-Rico E, Stamatakis EA, Pérez-García M, et al. Trait impulsivity and prefrontal gray matter reductions in cocaine dependent individuals. Drug Alcohol Depend. 2012;125:208–14.
Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–52.
Kaufman JN, Ross TJ, Stein EA, Garavan H. Cingulate hypoactivity in cocaine users during a GO-NOGO task as revealed by event-related functional magnetic resonance imaging. J Neurosci. 2003;23:7839–43.
Ke Y, Streeter CC, Nassar LE, Sarid-Segal O, Hennen J, Yurgelun-Todd DA, et al. Frontal lobe GABA levels in cocaine dependence: a two-dimensional, J-resolved magnetic resonance spectroscopy study. Psychiatry Res. 2004;130:283–93.
Licata SC, Renshaw PF. Neurochemistry of drug action: insights from proton magnetic resonance spectroscopic imaging and their relevance to addiction. Ann N Y Acad Sci. 2010;1187:148–71.
Volkow ND, Fowler JS, Wang GJ. The addicted human brain: insights from imaging studies. J Clin Invest. 2003;111:1444–51.
George O, Mandyam CD, Wee S, Koob GF. Extended access to cocaine self-administration produces long-lasting prefrontal cortex-dependent working memory impairments. Neuropsychopharmacology. 2008;33:2474–82.
Ford KA, Wolf ME, Hu XT. Plasticity of L-type Ca2+ channels after cocaine withdrawal. Synapse. 2009;63:690–697.
Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharm Exp Ther. 2005b;312:1305–13.
Wayman WN, Chen L, Hu XT, Napier TC. HIV-1 transgenic rat prefrontal cortex hyper-excitability is enhanced by cocaine self-administration. Neuropsychopharmacology. 2016;41:1965–73.
Wayman WN, Chen L, Napier TC, Hu XT. Cocaine self-administration enhances excitatory responses of pyramidal neurons in the rat medial prefrontal cortex to human immunodeficiency virus-1 Tat. Eur J Neurosci. 2015;41:1195–206.
Sauter A, Rudin M. Calcium antagonists reduce the extent of infarction in rat middle cerebral artery occlusion model as determined by quantitative magnetic resonance imaging. Stroke. 1986;17:1228–34.
Sauter A, Rudin M, Wiederhold KH. Reduction of neural damage in irreversible cerebral ischemia by calcium antagonists. Neurochem Pathol. 1988;9:211–36.
Sauter A, Rudin M, Wiederhold KH, Hof RP. Cerebrovascular, biochemical, and cytoprotective effects of isradipine in laboratory animals. Am J Med. 1989;86:134–46.
Johnson B, Barron B, Fang B, Lamki L, Wagner L, Wells L, et al. Isradipine prevents global and regional cocaine-induced changes in brain blood flow: a preliminary study. Psychopharmacol (Berl). 1998;136:335–41.
Hale SL, Alker KJ, Rezkalla SH, Eisenhauer AC, Kloner RA. Nifedipine protects the heart from the acute deleterious effects of cocaine if administered before but not after cocaine. Circulation. 1991;83:1437–43.
Rezkalla SH, Hale S, Kloner RA. Cocaine-induced heart diseases. Am Heart J. 1990;120:1403–8.
Chokkalingam Mani B, Fischman DL, Savage MP. Cocaine-induced microvascular dysfunction and its reversal by administration of intracoronary calcium-channel blocker. J Invasive Cardiol. 2016;28:E120–1.
Cepeda C, Levine MS. Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev Neurosci. 1998;20:1–18. https://doi.org/10.1159/000017294.
Missale C, Fiorentini C, Busi C, Collo G, Spano PF. The NMDA/D1 receptor complex as a new target in drug development. Curr Top Med Chem. 2006;6:801–8. https://doi.org/10.2174/156802606777057562.
Rolland B, Karila L, Geoffroy PA, Cottencin O. Shared vulnerability between seizures and psychosis in cocaine addiction?. Epilepsy Behav. 2011;22:596–8. https://doi.org/10.1016/j.yebeh.2011.08.004.
Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharm Sci. 2006;27(Feb):78–84.
Fleckenstein A, Frey M, Zorn J, Fleckenstein-Grün G. Amlodipine, a new 1,4-dihydropyridine calcium antagonist with a particularly strong antihypertensive profile. Am J Cardiol. 1989;64:21I–34I. https://doi.org/10.1016/0002-9149(89)90957-0.
Grundy JS, Eliot LA, Foster RT. Extrahepatic first-pass metabolism of Nifedipine in the rat. Biopharm Drug Dispos. 1997;18:509–22. https://doi.org/10.1002/(sici)1099-081x(199708)18:6<509::aid-bdd38>3.0.co;2-5.
Addy NA, Nunes EJ, Hughley SM, Small KM, Baracz SJ, Haight JL, et al. The L-type calcium channel blocker, isradipine, attenuates cue-induced cocaine-seeking by enhancing dopaminergic activity in the ventral tegmental area to nucleus accumbens pathway. Neuropsychopharmacology. 2018;43:2361–72.
D’Souza MS. Glutamatergic transmission in drug reward: implications for drug addiction. Front Neurosci. 2015;9:404.
Degoulet M, Stelly CE, Ahn KC, Morikawa H. L-type Ca2+ channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug-associated contextual memory. Mol Psychiatry. 2016;21:394–402.
Bavley CC, Fetcho RN, Burgdorf CE, Walsh AP, Fischer DK, Hall BS, et al. Cocaine- and stress-primed reinstatement of drug-associated memories elicit differential behavioral and frontostriatal circuit activity patterns via recruitment of L-type Ca2+ channels. Mol Psychiatry. 2020;25:2373–91.
Knuepfer MM, Branch CA. Calcium channel antagonists reduce the cocaine-induced decrease in cardiac output in a subset of rats. J Cardiovasc Pharm. 1993;21:390–6.
Richards JR, Garber D, Laurin EG, Albertson TE, Derlet RW, Amsterdam EA, et al. Treatment of cocaine cardiovascular toxicity: a systematic review. Clin Toxicol (Philos). 2016;54:345–64.
Becker JB, Hu M. Sex differences in drug abuse. Front Neuroendocrinol. 2008;29:36–47.
Griffin WC III, Middaugh LD. The influence of sex on extracellular dopamine and locomotor activity in C57BL/6J mice before and after acute cocaine challenge. Synapse 2006;59:74–81.
Kuppers E, Krust A, Chambon P, Beyer C. Functional alterations of the nigrostriatal dopamine system in estrogen receptor-alpha knockout (ERKO) mice. Psychoneuroendocrinology 2008;33:832–8.
van den Buuse M, Halley P, Hill R, Labots M, Martin S.Altered N-methyl-D-aspartate receptor function in reelin heterozygous mice: male-female differences and comparison with dopaminergic activity.Prog Neuropsychopharmacol Biol Psychiatry. 2012;37:237–46.
Martinez LA, Gross KS, Himmler BT, Emmitt NL, Peterson BM, Zlebnik NE. et al. Estradiol facilitation of cocaine self-administration in female rats requires activation of mGluR5. eNeuro. 2016;3:ENEURO.0140-16.2016.
Peterson BM, Mermelstein PG, Meisel RL. Estradiol mediates dendritic spine plasticity in the nucleus accumbens core through activation of mGluR5. Brain Struct Funct. 2015;220:15–22.
Di Paolo T. Modulation of brain dopamine transmission by sex steroids. Rev Neurosci. 1994;5:27–41.
Clare K, Pan C, Kim G, Park K, Zhao J, Volkow ND, et al. Cocaine Reduces the Neuronal Population While Upregulating Dopamine D2-Receptor-Expressing Neurons in Brain Reward Regions: Sex-Effects. Front. Pharmacol. 2021. https://doi.org/10.3389/fphar.2021.624127.
Du C, Tully M, Volkow ND, Schiffer WK, Yu M, Luo Z, et al. Differential effects of anesthetics on cocaine’s pharmacokinetic and pharmacodynamic effects in brain. Eur J Neurosci. 2009;30:1565–75.
Park K, Chen W, Volkow ND, Allen CP, Pan Y, Du C. Hemodynamic and neuronal responses to cocaine differ in awake versus anesthetized animals: optical brain imaging study. Neuroimage. 2019;188:188–97.
Acknowledgements
This work was supported in part by National Institutes of Health (NIH) grants 2R01 DA029718 (CD, YP), RF1DA048808 (YP & CD), R21DA042597 (CD); R01NS084817, R01DA044552 and DA044552-03S1 (X-TH) and NIH’s Intramural Program of NIAAA (NDV). The authors would like to thank the NIDA drug supply program for providing the cocaine used in this study.
Author information
Authors and Affiliations
Contributions
C.D., X-T.H., N.D.V., and Y.P. designed the research; C.A. carried out the in vivo experiments; K.P. participated in the animal preparation; K.P. and C.A. conducted image processing and analysis; C.D., X-T.H., N.V.D., and Y.P. contributed significantly to data interpretation, result discussions, and the manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, C., Park, K., Allen, C.P. et al. Ca2+ channel blockade reduces cocaine’s vasoconstriction and neurotoxicity in the prefrontal cortex. Transl Psychiatry 11, 459 (2021). https://doi.org/10.1038/s41398-021-01573-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01573-7
This article is cited by
-
Astrocytes modulate cerebral blood flow and neuronal response to cocaine in prefrontal cortex
Molecular Psychiatry (2024)
-
Cocaine’s cerebrovascular vasoconstriction is associated with astrocytic Ca2+ increase in mice
Communications Biology (2022)
