
Abstract
Natural Killer (NK) cells are critical members of the innate immunity lymphocytes and have a critical role in host defense against malignant cells. Adoptive cell therapy (ACT) using chimeric antigen receptor (CAR) redirects the specificity of the immune cell against a target-specific antigen. ACT has recently created an outstanding opportunity for cancer treatment. Unlike CAR-armored T cells which hadnsome shortcomings as the CAR-receiving construct, Major histocompatibility complex (MHC)-independency, shorter lifespan, the potential to produce an off-the-shelf immune product, and potent anti-tumor properties of the NK cells has introduced NK cells as a potent alternative target for expression of CAR. Here, we aim to provide an updated overview on the current improvements in CAR NK design and immunobiology and describe the potential of CAR-modified NK cells as an alternative “off-the-shelf” carrier of CAR. We also provide lists for the sources of NK cells in the process of CAR NK cell production, different methods for transduction of the CAR genetic sequence to NK cells, the differences between CAR T and CAR NK, and CAR NK-targeted tumor antigens in current studies. Additionally, we provide data on recently published preclinical and clinical studies of CAR NK therapy and a list of finished and ongoing clinical trials. For achieving CAR NK products with higher efficacy and safety, we discuss current challenges in transduction and expansion of CAR NK cells, CAR NK therapy side effects, and challenges that limit the optimal efficacy of CAR NK cells and recommend possible solutions to enhance the persistence, function, safety, and efficacy of CAR NK cells with a special focus on solid tumors.
Graphical abstract

Similar content being viewed by others
Introduction
Conventional treatment of cancer including radiotherapy, chemotherapy, and surgery is associated with poor efficacy and significant side effects [1, 2]. Therefore, novel strategies with higher efficacy and fewer complications, such as immunotherapy, have been developed. Immunotherapy is the modification and enhancement of the host immune system to combat different pathologies, such as cancer. Adoptive cell therapy (ACT) is a type of immunotherapy that includes the application of immune cells to treat cancer. One of the most attractive emerging areas in the ACT is the development of modified genetic receptors, chimeric antigen receptors (CARs). Genetic engineering of T cells to express CAR (CAR T cell therapy) has become a major milestone in the treatment of cancer. CAR-engineered T cell therapy has demonstrated unprecedented results in the treatment of B-lineage hematologic malignancies and to a lesser extent solid tumors. Two CAR T-based drugs, Kymriah and Yescarta, have been approved for the treatment of CD-19+ B-cell lineage hematological cancers [3, 4]. Very recently, a new drug named Breyanzi (Lisocabtegene maraleucel) has been approved by the United States Food and Drug Administration for the treatment of refractory/relapsed large B-cell lymphoma [5] (https://www.fda.gov/drugs/dr ug-approvals-and-databases/FDA-approves-lisocabtagene-maraleucel-relapsed-or-refractory-large-b-cell-lymphoma).
Natural killer (NK) cells are lymphocytes and a member of the innate lymphoid cell type-I (ILC-I) group that recognize the target antigen independent of major histocompatibility complex I (MHC-I). NK cells are frontline anti-cancer effectors of innate immunity and have a critical role in eliminating malignant and stressed cells [6]. Modifying NK cells to express the CAR can enhance the anti-tumoral function of NK cells, promote the cancer-recognizing properties of NK cells, and overcome the immune evasion of cancer [7]. The potential of CAR-modified NK therapy has opened a promising window to develop an alternative and effective anti-cancer immunotherapeutic method. In this article, we aim to review the biological features, structure, production, and recent preclinical and clinical studies of CAR NK cells. We also discuss the challenges of CAR NK cell therapy and possible future approaches to enhance the safety and efficacy of CAR-modified NK cells against cancer.
Natural Killer (NK) Cells
NK cells consist of 5–15% of the systemic circulation leukocytes and are CD3−CD56+NKp46+ ILC-Is. Produced in the bone marrow (BM) and maturated in both BM and secondary lymphoid organs, including uterus, liver, and tonsil, NK cells act as a member of the innate immunity [8]. Mature NK cells eliminate viral-contaminated and stressed cells, such as cancer cells, and are of great importance in defeating malignancies. In contrast to T cells, NK cells do not demand the previous encounter with the antigen (Ag) to exert their cytotoxic effect. Moreover, NK cell recognition of the target cell is independent of MHC-expression. Identification of the target cell by NK cells is regulated by NK cell receptors (NCRs). The primary activation of the NK receptors, such as NKp46, NKp44, and NKp30, can lead to the activation or inhibition of the NK cells. Contact with dendritic cells (DCs), binding to antibody complexes, contact with MHC-I-negative cells, activation of NK group 2 member D (NKG2D), activation of CD226 (DNAM-1) through interaction with CD112, activation of killer cell immunoglobulin-like receptors (KIRs) such as KIR2DS1, KIR2DS4, and KIR2DL4, immune-stimulating cytokines such as interleukin (IL)-1, 2, 12, 15, 18, and 21, and type I interferon (IFN), and contact with tumor-associated molecules/patterns are the factors that can trigger the activation of NK cells. These activatory signals activate DAP12, CD3ζ, or high-affinity receptor for IgE (Fc3RIγ), which are members of the immunoreceptor tyrosine-based activation motif (ITAM). This process finally activates the NK cell function against the target cell [9,10,11]. Inhibitory signals including NK group 2 member A (NKG2A), C-type lectin-like receptor superfamily (CTLR) such as Lectin-like transcript 1 (LLT1), and identification of MHC-I-negative cells by KIRs and leukocyte immunoglobulin-like receptor (LIRs), are the most common factors that inhibit the activation of NK cells [12]. CD56+ NK cells are divided into two subsets, CD56-dim and CD56-bright. CD56-bright cells are the primary effector NK cells that exert their cytotoxic effect by producing perforin and granzyme [13]. However, CD56-dim NK cells act as the late immune cells that regulate the immune function by producing pro-inflammatory mediators such as IFN-γ, IL-22, IL-17, and tumor-necrosis factor-α (TNF-α) [14].
After identification of the malignant cell by NK cells, they exert a critical role in inhibiting the expansion, metastasis, and proliferation of the malignancy by multiple methods. The first is the direct cytotoxicity of NK cells on tumor cells by producing perforin/granzyme. The second mechanism is the induction of death receptor-mediated apoptosis by Fas/Fas-L interaction. The third is the Ab-dependent cellular cytotoxicity (ADCC), by binding to the CD16 to the Ag-Ab complex. And the last mechanism is the production of immune-stimulating cytokines, such as IL-5, 10, and 13, granulocyte-monocyte colony-stimulating factor (GM-CSF), and TNF-α. These immune-stimulating cytokines activate dendritic cells (DCs) and macrophages that inhibit the proliferation and invasion of the malignancy [15].
As members of innate immunity, NK cells have a major role in the destruction of the target malignant cells. Studies have shown that the presence of the NK cells in the TME is associated with the outcome of the solid tumor-bearing host. The tumor-killing potential of the NK cells in the TME is less than circulating NK cells, which react against extravasated tumor cells. This is caused by the immune-suppressive effects of the TME induced by tumor-promoting T cells, DCs, macrophages, MDSCs, fibroblasts, tumor cells, and cytokines [16]. These NK-suppressing mechanisms lead to the inhibition of the activatory receptors or stimulation of the inhibitory receptors of the Nk cells [17,18,19]. As discussed later, understanding the interaction between NK cells and the TME can help us to develop superior immunotherapeutic strategies against the tumor. In addition, NK cells also have a role in the process of rejection/tolerance after transplantation. Transplantation of the solid organ activates the resident alloreactive NK cells of the host which can then induce the subversion of the transplanted cells by inducing the ADCC or secreting chemokines/cytokines. These immune-mediating cytokines further induce the chemotaxis of the immune cells, such as DCs, activate the Th1, which ends in rejection of the allograft transplant, or Th2/Tregs, which results in tolerance [20, 21]. Accordingly, the significant anti-tumor potentials of NK cells have made them an attractive target for the immunotherapy of cancer [22].
Chimeric Antigen Receptor (CAR)
Initially introduced in the 1980s [23], CAR-based immunotherapy has attracted significant attention for the treatment of cancer. CAR is a genetically recombinant receptor that can be transduced to different immune cells, such as NK cells and T lymphocytes. The CAR redirects the specificity of the T/NK cell against a specific antigen on tumor cells. Unlike T cell receptor (TCR) that identifies MHC-I on the target cell, CAR-function is independent of MHC. Therefore, CAR can identify diverse target antigens including carbohydrates, proteins, glycolipids, etc. [3]. The structure of the CAR includes four domains. The extracellular section is mostly generated from a single-chain variable fragment (scFv) of an antibody specific for a target antigen [24]. In addition to scFv-based extracellular domains, another ectodomain named “universal CAR” has been recently introduced as a novel structure for target recognition of the CAR. Universal CAR can be redirected to a specific antigen by binding its avidin to the biotinylated monoclonal antibody (mAb) bonded to the target antigen. Moreover, universal CAR can also be redirected against a specific antigen by its FITC-specific scFv that binds to the labeled mAb-FITC complex. Therefore, the universal CAR has the superiority to target a wide spectrum of antigens without requiring multiple infusions [25]. Non-scFv extracellular domains, such as variable lymphocyte receptor (VLR), nanoantibody (VHH), and designed ankyrin repeat protein (DARP), can be used as the extracellular domain of the CAR [26]. The extracellular domain is connected to the transmembrane domain using spacer/hinge, which is commonly manufactured by CD4, the Fc region of IgG, or CD8, and has shown a major contribution in the activation and signaling of the CAR. The intracellular domain of the CAR is responsible for the transmission of the activatory signals into the cell that finally leads to the anti-cancer function of the modified cell. The intracellular domain of the CAR includes CD3ζ, the intracellular subset of the conventional TCR, as the main domain. The CAR can also include costimulatory domains such as CD28, 4-1BB, CD27, or OX-40, which can boost the activation and cytokine production of the CAR T cell [4]. Considering CAR NK cells, 2B4 has been introduced as a novel NK cell-specific costimulatory receptor and has been reported to increase the anti-tumor efficacy, proliferation, and cytokine production of CAR NK cells [27]. CARs are T-cell-specific constructs and are not specified to NK cells. Therefore, the development of NK-specific CAR constructs is necessary which has been discussed in further sections.
Multiple generations of CAR-engineered T cells with different designs have been produced. First-generation CARs included the scFv and CD3ζ which did not exhibit adequate anti-tumor efficacy due to poor persistence and inadequate cytokine production [28]. Second-generation CARs with one costimulatory domain (4-1BB or CD28) were designed which showed higher efficacy and cytokine releasing potential due to stronger intracellular signaling. The addition of the costimulatory domain to the CAR increased the tumor elimination and cytokine-production capabilities of the CAR T cells. Third-generation CARs included two costimulatory domains, most commonly 4-1BB and CD28. The fourth generation of CAR T cells, also named “T cell redirected for universal cytokine-mediated killing (TRUCK)” includes a nuclear factor of activated T cell (NFAT) that enables the cell to express proteins and cytokines. TRUCK T cells have shown encouraging preclinical and clinical results in previous studies [29]. Newer generations of CARs such as SUPRA CAR, Tandem CAR, Dual CAR, physiological CAR, split CAR, and SynNotch CARs have also been developed and are being investigated [25, 30].
CAR NK Cell vs CAR T Cell (Table 2)
CAR T cell therapy has demonstrated unprecedented results in the treatment of hematologic and solid malignancies; however, this method is also associated with shortcomings and adverse effects. Cytokine release syndrome (CRS) is the most frequent toxicity secondary to CAR T therapy which occurs due to uncontrolled secretion of pro-inflammatory mediators [31, 32]. “On-target, off-tumor” toxicity is another adverse effect that occurs secondary to the importance of CAR-modified T cells to distinguish healthy cells from target tumor cells which can cause severe damage to different organs [33]. Graft-versus-host disease (GvHD) [34], tumor lysis syndrome [35], and neurologic side effects such as acute cerebral edema and immune effector cell-associated neurologic syndrome (ICANS) [36] are other CAR T therapy complications that can occur in patients receiving CAR T cells [37]. In an attempt to overcome these shortcomings and taking advantage of the anti-cancer potentials of NK cells, scientists have thought to transfect the CAR transgene to NK cells. Identical to CAR T cells, the CAR sequence is transduced to the NK cell using a retroviral/lentiviral vector. CAR-modified NK cells have a shorter lifespan and limited cytokine secretion profile and therefore can be a potent substitute for CAR T cells [38]. Modification of NK to express CAR has the advantage of targeting the CAR-specific antigen, the potential to target multiple antigens using the CAR, double CAR-dependent and NK receptor-dependent function of CAR NK cells, and better safety profile. Moreover, the CAR NK cells can overcome the immune evasion mechanisms of the tumor, such as downregulation of human leukocyte antigen (HLA) and proteasomal antigen processing, and the potential of remote control to activate or deactivate the CAR NK cells in case of severe toxicities [39].
In comparison to CAR T cells, CAR NK cells possess superiorities in fighting the malignant cells (Table1). Since conventional T cells require HLA-matching to recognize the target cell, GvHD is one of the prevalent toxicities of CAR T cell therapy. Since CAR NK cells act independently of the HLA-matching to recognize the target cell, GVHD has rarely been reported in clinical and preclinical studies of CAR-modified NK therapy [40]. Nevertheless, in animal models, NK cells have even been reported to restrain GvHD by inhibiting alloreactive T cells [41]. CRS, the most common side effect of CAR T therapy, is caused by the production of enormous amounts of IFN-γ, TNF-α, IL-6, and IL-1 by CAR T cells. In comparison to CAR-transduced T cells, CAR NK cells produce other cytokines with fewer immune-stimulating properties, such as GM-CSF, IL-3, and IFN-γ, and therefore, do not cause CRS [42]. Moreover, the limited life-span, the controlled in-vivo persistence of CAR NK cells, and the inability of CAR-modified NK cells to form memory cells (except in some viral diseases) are the factors that reduce the need to control CAR NK cells using suicide genes or other approaches [43,44,45]. The other advantage of CAR NK therapy is the simplicity of its production and lower production costs. Unlike CAR T cells which are mostly produced from an autologous cell source, CAR NK cells can be conveniently produced using peripheral blood mononuclear cells (PBMCs), induced pluripotent stem cells (iPSCs), hematopoietic stem cells (hPSCs), NK cell lines (such as NK-92), and umbilical cord blood (UCB) NK cells as the primary source for CAR NK production. Due to unlimited proliferation potential and lesser sensitivity to multiple cycles of freezing, most clinical trials have used the NK-92 cell line as the primary source of NK cells [46]. One of the main elements that reduce the success of CAR T therapy is tumor antigen loss. In the long term, this process leads to tumor relapse/recurrence. However, since intracellular signaling of NK cells (NKG2D and NDAM1) can contribute to the identification of the target cell independent of CAR, antigen-loss does not affect the efficacy of CAR-modified NK cells [30, 47]. Moreover, the lower expression of programmed death-1 (PD-1), an important immunosuppressive factor expressed by the tumor microenvironment, is another privilege of CAR NK therapy, especially in fighting solid malignancies [48]. Therefore, CAR NK cell therapy can be considered as an efficacious and safe alternative immunotherapeutic carrier for CAR in the long-term treatment of malignancies [30].
The CAR construct of the CAR NK cell and CAR T cell are common in some features; however, there are some differences in co-stimulatory and transmembrane domains of the NK-specific CAR constructs. Similar to CAR T cells, CD3ζ and 4-1BB are conserved in CAR NK cells. DNAX-activation protein-10 (DAP10), DAP12, 2B4, and CD137 are domains that can be applied as co-stimulatory domains in NK-specific CAR construct, in addition to 4-1BB [49]. 2B4 is a subset of the signaling lymphocytic activation molecule (SLAM) family that contributes to the intracellular signaling of the NK cell through CD48 and immunoreceptor tyrosine-based switch motif (ITSM). In a study, the application of 2B4 as the co-stimulatory domain led to enhanced proliferation and cytokine production and diminished apoptosis of the CAR NK cells [27]. In a study by Li et al., ten groups of iPSC-derived anti-mesothelin CAR NK cells were produced using different signaling domains. In this study, anti-mesothelin-NKG2D-2B4 CARs showed the best results in terms of anti-tumor response, which showed the potential of 2B4 as the co-stimulatory domain of NK-specific CAR [50]. DAPs are a subset of NK cell-membrane receptors that participate in the NK cell intracellular signaling. DAP10 and DAP12 can both be used as an alternative for CD3ζ or as co-stimulatory domains of the CAR construct [51]. In a study, DAP12 increased the anti-tumor efficacy compared to CD3ζ [52]. Unlike CAR T cells that included CD8/CD28 as the transmembrane domain, NK-specific CAR construct can include NKG2D, NKp46, NKp44, or CD16 as the transmembrane domain. NKG2D is a subset of the natural killer activating receptors (NKGs) that activates the intracellular signaling of the NK cell by phosphorylating the DAP10 and has been used as the transmembrane domain of the NK-specific CAR [53]. A phase I study is assessing the anti-tumor effect of off-the-shelf FT596 CAR NK cells that include 2B4 as the co-stimulatory domain and NKG2D as the transmembrane domain in B cell lymphoma and chronic lymphocytic leukemia (NCT04245722) (Table 2, Figs. 1 and 2).

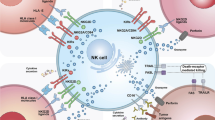
Structure of the natural killer cell receptors and diferent generations of CARs in NK cells. Created by Esmaeiladeh et al.

Recent developments of CAR NK cells to increase the efficacy and safety of cancer treatment. Created by Esmaeiladeh et al. CAR: chimeric antigen receptor, NK: natural killer cell, TAA: tumor-associated antigen, IL-4: interleukin-4, IL-7: interleukin-7, TGF-ΒRII: transforming growth factor beta receptor II, IL-15: interleukin-15, KIR: killer inhibitory receptor, iC9: inducible caspase 9, ICR: inverted cytokine receptor, SynNotch: synthetic Notch, scFv: single-chain variable fragment
Production of CAR NK Cell
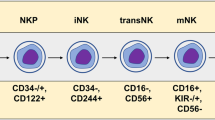
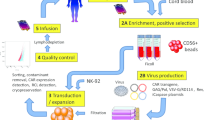
The process of CAR NK cell production is similar to the process of CAR T cell production but there are some differences. To produce CAR NK cells an appropriate source of NK cells is required. NK cell origins for the production of the CAR NK and the properties of each method are described in Table 1. PBMC-derived NK cells are extracted by leukapheresis, sorting, isolation, and CD56 selection of the blood sample from a donor [54]. NK cell lines, especially NK-92, are among the latest evolved sources for manufacturing CAR NK cells that have solved the challenge of requiring high numbers of engineered NK cells. Since NK-92 is a derivative of the non-Hodgkin's lymphoma cell line, it should be precariously used for developing CAR NK strategies and should be irradiated before transduction of the CAR genome to reduce the risk of GVHD and tumorigenicity [55,56,57]. NK-92 has recently been used to produce CAR-modified NK cells in clinical, as well as preclinical studies [58, 59]. UCB-derived NK cells are extracted by induction of the cord blood stem cells through lymphocytic lineage. hPSC cells can be differentiated from the NK cell line using specific cytokines and enriched media [60]. iPSC-derived NK cells are another source for the production of engineered NK cells in high numbers but are also associated with teratogeniity and especially the development of teratoma [50, 61,62,63]. Multiple strategies, such as deletion of the Cytokine-inducible SH2-containing protein (CISH) [63], can be applied to overcome the shortcomings of the iPSC-derived NK cell therapy, which are discussed in further sections. After extraction of the NK cells, they must be stimulated and enriched using CD56+ beads. Then, the CAR genome sequence is transfected to the NK cells using a vector. There are various methods to transduce the CAR transgene to NK cells (Table 3). The most commonly used method is viral vectors, such as retroviral and lentiviral vectors. The transfected NK cells must be expanded using cytokines that stimulate the expansion of NK cells, such as IL-2, 15, 18, 21, and 12 [43]. When the CAR NK cells have reached the appropriate number, they are cryopreserved and the quality-control processes are performed to ensure the expression of CAR by NK cells. All steps of the CAR NK manufacturing process must be performed in good-manufacturing practice (GMP)-grade clinical condition [64]. After the production of CAR NK cells, the last step is to inject the product into the patient; however, chemotherapy and bone-marrow depletion before injection can increase the efficacy of CAR NK therapy [65].
Preclinical Results of CAR NK Therapy
CAR NK cells have been introduced as a revolutionary immunotherapeutic method for the immunotherapy of cancer. Preclinical studies have been performed to evaluate the anti-tumor efficacy of CAR NK cells in hematologic and solid cancers. Most of the studies have targeted CD19+ B cell malignancies. Romanski and et al. explored the anti-tumor function of NK-92-derived CD19 targeting CAR NK cells on CD19+ leukemia cell lines which demonstrated that CD19-redirected CAR NK cells could effectively lyse the leukemia malignant cells [66]. Several other studies have targeted B-lineage malignancies using CD-19-directed CAR NK cells [58, 66, 67]. In a study, anti-CD20 resistant and sensitive cell lines of lymphoma were treated with anti-CD19-CAR-NK92 cells. This resulted in increased production of IFN-γ and superior apoptotic activity through secretion of IL-10, CCL3, and FAS-L in anti-CD20 resistant lymphoma cells [68]. Therefore, CD19 CAR NK therapy has shown potent anti-tumor activity against CD19+ B-cell malignancies. CD20 is the second most studied target in B-lineage hematological malignancies. In a study on the rituximab-resistant mice model of Burkitt lymphoma, second-generation 4-IBB-CD3ζ anti-CD20 CAR-modified NK cells cultured with K562-mb-IL15-41BBL demonstrated increased anti-tumor cytotoxicity and enhanced the mice survival [69]. Muller et al. designed NK-92-derived CD20-CAR NK cells for the treatment of CD20+ leukemia/lymphoid cells. This study showed that CD20-redirected CAR NK cells were able to invade NK cell-resistant leukemia/lymphoid cells and exhibited potent anti-tumor function [70]. In another study, Biossel et al. compared the efficacy of CD20-targeting CAR NK cells with CD20-blocking monoclonal antibodies. Compared with anti-CD20 antibodies, anti-CD20 CAR-engineered NK cells showed to have a superior antitumor activity in the chronic lymphocytic leukemia (CLL) cells and mice model [71]. CAR NK cells targeting multiple other antigens such as CD7 [72], CD5 [27], CD4 [43], CS1 [73], CD138 [74], and CD33 [75] have been used in the treatment of leukemia/lymphoma mice models. In another study, CD138 CAR NK cells increased survival and demonstrated a potent anti-tumor effect in a murine model of multiple myeloma [74]. In another study, simultaneous injection of CD138 and CD19 CAR NK cells demonstrated sufficient in-vitro cytotoxicity and targeted tumor killing [76]. In an orthotopic model of multiple myeloma, NK-92-derived second-generation CAR NK cells that targeted CS1 could restrict tumor progression, increase survival, and enhance the secretion of IFN-γ [73].
CAR NK cells have also been studied in solid tumors. Neuroblastoma is a brain cancer with a poor prognosis that mostly occurs in children. In a preclinical study, NK-92-GD2-CAR NK cells were used to treat multidrug-resistant neuroblastoma cell line and a xenograft model of neuroblastoma which showed substantial anti-tumor efficacy [77]. Moreover, the application of second-generation CD276-NK-92 CAR NK cells could effectively lyse 3D and monolayer spheroids of neuroblastoma. This study supported the CAR NK-92 product as an “off-the-shelf” option for treating neuroblastoma [78].
Glioblastoma is a malignant brain tumor with a poor prognosis. Treatment of glioblastoma in a tumor-bearing mice model with EGFR-CD28- CD3ζ CAR NK cells demonstrated potent cytotoxic effects and increased murine survival [79]. Other studies have targeted CD73 [80], EGFRvIII [81], and HER2 [82] in glioblastoma cell lines which have shown significant anti-glioblastoma activity and tumor lysis. According to the immunosuppressive microenvironment and heterogeneity of the GBM, targeting more than one moieties could improve the efficacy of CAR-modified immune cells. In a human glioblastoma cell line, dual targeting of glioblastoma using EGFR-EGFRvIII-dual CAR NK cell showed significant immune-evasion and cytotoxicity. Moreover, the application of EGFR-EGFRvIII-dual CAR NK cell in a xenograft mice model of glioblastoma reduced the immune escape of the tumor and showed more efficacy compared to monotherapy using EGFRvIII or EGFR targeting CAR NK cell [83]. In another study, dual-specific GD2-NKG2DL-CAR NK cells could recognize the third moiety of tumor-responsive local secretion of anti-CD73 antibody. The injection of this multifunctional CAR NK product demonstrated anti-tumor function and repressed the production of adenosine. Further evaluation showed that pharmacologic impairment of autophagy can enhance the sensitivity of glioblastoma to the CAR-modified NK cells [84]. The results of these studies demonstrated that multi-targeted CAR NK cells can be an effective strategy to treat glioblastoma by inhibiting tumor heterogeneity and immune suppression.
Prostate stem cell antigen (PSCA) is a cell surface antigen expressed by prostate cancer cells. PB-NK and YPS-NK derived PSCA-DAP12-CAR NK cells exhibited significant tumor-lysis against PSCA+ prostate cancer cells [51] PSMA (prostate-specific membrane antigen) is a tumor-associated antigen that is overexpressed on malignant cells of the prostate gland. In-vitro studies demonstrated that Anti-PSMA CAR NK-92 cells exhibited potent IFN-γ production and highly specific lytic activity against prostate cancer cells. Infusion of the PSMA-CAR NK-92 cells to the prostate-cancer-bearing mice effectively limited tumor growth and increased survival [85]. Mesothelin is expressed by ovarian cancer cells and has been studied for targeted therapy of ovarian cancer. In a mice model of ovarian cancer, subcutaneous and intraperitoneal application of anti-MSLN-CAR NK cells resulted in specific anti-tumor immune response and increased survival in intraperitoneally tumor-grafted mice [86]. Moreover, iPSC-derived CAR-modified NK cells were shown to repress the proliferation of ovarian malignant cells in a murine model of ovarian cancer [50].
CAR-modified NK therapy has also been considered to be used for breast malignancies. In a study, ErbB2-5-28ζ-CAR-NK-92 cells were used to treat breast cancer cells. The in-vitro analysis showed that ErbB2-CAR-NK-92 cells successfully destroyed ErbB2+ malignant cells. In-vivo administration of these NK-tailored cells showed favorable immune function and tumor-infiltration in a murine xenograft model of breast cancer [87]. Triple-negative breast cancer (TNBC) is resistant to the majority of current treatment options. Therefore, Hu et al. thought to investigate the anti-tumor function of CAR NK cells targeting tissue factor (TF-CAR-NK-92 cells) on TNBC cell line and a xenograft model of TNBC. This study showed that TF-CAR-NK-92 cells successfully lysed TNBC cells in in-vitro and in the xenograft model of TNBC [88]. EGFR is a potential target that is overexpressed by breast cancer. In a study by Liu et al., using a lentiviral vector, two different ScFvs were used to produce EGFR-CAR-NK cells against TNBC cells. The produced EGFR-CAR NK cells showed accurately induced in-vitro lysis of the TNBC cells and restrained tumor growth in a murine model [89]. It can be concluded that breast cancer is a potent target for CAR NK therapy and further and this immunotherapeutic method could improve the clinical results of breast cancer treatment in the future.
Hepatocellular carcinoma (HCC) is the most lethal form of liver malignancies. Huang et al. investigated the anti-tumor activity of anti-GPC3 CAR NK cells containing 2B4/DNAM1 or CD28 as their costimulatory domain against HCC. This study demonstrated that GPC3-2B4/DNAM1-CAR-NK-92 cells exhibited a stronger immune response with enhanced cytotoxicity against hepatocellular carcinoma (HCC) cell line [90]. In another study, NKG2D-DAP10-CD3ζ-CAR-NK cells demonstrated potent anti-tumor immune response against cell lines of osteosarcoma, pancreatic cancer, breast cancer, and HCC [38]. Colorectal cancer includes malignancies of the colon and rectal epithelial tissue that mostly occur in elderlies and are associated with poor prognosis. Zhang et al. transduced the EpCAM-CAR to the NK-92 cell line using a lentiviral vector. The EpCAM+ colorectal cancer cells were treated with anti-EpCAM-CAR-NK-92 cells which showed potent anti-tumor cytotoxicity and cytokine secretion. Injection of anti-EpCAM-CAR-NK-92 cells to the colorectal cancer xenograft model in combination with regorafenib, a protein kinase inhibitor, effectively suppressed tumor growth [91].
Folate receptor alpha (αFR) is overexpressed on malignant cells of the ovary. In a study, three generations of αFR-CAR were produced using lentiviral a vector (αFR-ζ, αFR-28ζ, αFR-28BBζ CAR NK-92 cells). In-vitro studies demonstrated that although all constructs showed favorable efficacy, the third generation CAR construct had higher cytokine secretion, proliferation, and antigen-specific cytotoxicity than others. In the xenograft model of ovarian cancer, these CAR NK-92 cells could effectively lyse αFR+ cancer cells and increase survival. This study showed that applying third-generation CAR NK cells against solid tumors may have superior efficacy than other generations [92].
In conclusion, the preclinical results of CAR NK therapy demonstrate that CAR NK therapy can be introduced as promising immunotherapy against hematological and solid cancers. However, further clinical studies, and specifically large-scale clinical studies are still required to prove the efficacy of CAR NK therapy against these malignancies (Table 4).
Clinical Results of CAR NK Therapy (Table 5).
Promising results of preclinical CAR NK therapy have opened the way to translate CAR NK therapy into clinical application. In previous years, the number of clinical trials investigating the therapeutic efficacy of CAR NK cells has significantly increased. As of April 2021, 27 clinical trials have been registered in clinicaltrials.gov, of which, 17 clinical trials target hematological and 10 target solid malignancies. All trials are in phase I/II and the most targeted malignancy is B-cell lineage leukemia/lymphoma targeting CD19 antigen. Other clinical trials target CD22, CD33, BCMA, NKG2DL, and CD7 in different lymphoproliferative malignancies.
Clinical Application of CAR NK Therapy in Hematologic Malignancies
Immunotherapy using non-engineered NK cells has shown promising results especially in acute myeloid leukemia (AML). Genetic engineering of NK cells with CARs can enhance the clinical efficacy of this method. CD33 and CD19 are the most common targets of CAR-modified NK therapy in clinical studies on hematologic malignancies [58]. In a first-in-man trial by Tang et al. on three R/R AML patients, third-generation NK-92-derived CAR-modified cells were transduced with CD33-CAR. Each patient received 5 × 109 CD33-CAR-NK-92 cells of which three of the patients experienced mild CRS and fever that was relieved by the next day. Although no permanent therapeutic responses were obtained, this study showed the transfusion of CAR NK-92 cells to be safe [42]. The initial large-scale study of CAR-modified NK therapy was conducted on 11 chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL) patients in MD Anderson cancer center. All patients were treated with lymphodepleting chemotherapy before infusion. Patients received cord blood-extracted CD19-CD28-CD3ζ-CAR NK cells that also contained suicidal gene switch and the genetic sequence for production of IL-15. The expression of IL-15 by CAR-modified cells enhances the survival, proliferation, and persistence of the engineered NK cells in-vivo. 7 out of 11 patients responded well and experienced sustained complete remission (CR) up to 13.8 months. The injected CAR NK cells were reported to persist in the blood for more than a year. This study exhibited the administration of cord blood-derived anti-CD19 CAR NK therapy in B-cell lineage malignancies to be efficacious and safe. None of these patients represented any significant side effects that needed intensive-level care which encourages the potential of CAR NK cells to be used as an “off-the-shelf” product for the treatment of malignancies [91]. FT596 is an iPSC-derived off-the-shelf CAR NK product that includes NKG2D as the transmembrane and 2B4 as the co-stimulatory Sect. [93]. It also includes a genetic sequence of IL15/IL15R to improve the persistence and survival of CAR NK cells. A clinical trial (NCT04245722) is investigating the efficacy of FT596 monotherapy and a combination of FT596 with a CD20-targeting monoclonal antibody in chronic lymphocytic leukemia and B cell lymphoma in a phase I trial. In conclusion, these studies demonstrate the clinical safety and applicability of CAR NK cells and the potential of this method to become one of the cornerstone treatments of hematological cancers. Moreover, iPSC-derived and UCB-derived CAR NK cells can be considered as appropriate sources for the manufacturing of “off-the-shelf” CAR NK products. The lower cost of CAR-engineered NK production compared to CAR T cells encourages the wide application of off-the-shelf CAR NK cells in a clinical scale in forthcoming years.
Clinical Application of CAR NK Therapy in Solid Malignancies
Preclinical results have shown the potent anti-tumor function of CAR NK cells; however, clinical application of CAR NK cells in solid tumors is in its infancy. Available data of clinical results of CAR NK cell therapy are limited. Ten clinical trials are currently investigating the clinical efficacy of CAR-modified NK therapy in multiple solid tumors, such as neuroblastoma, prostatic cancer, glioblastoma, etc. (Table 5). GD-2, mucin-1 (MUC-1), mesothelin, ROBO-1, HER-2, and NKG2DL are the targeted antigens that are being studied in clinical trials. FT596 is an iPSC-derived off-the-shelf CAR NK product that includes NKG2D as its TM and 2B4 as its co-stimulatory domain. FT596 also includes membrane-tethered IL-15 functioning as the autocrine IL15-signaling function [93]. Off-the-shelf availability and IL-15-mediated signaling are two factors that render FT596 as a target of great potential to be used for immunotherapy against solid tumors. iNKT cells are a specific subgroup of T lymphocytes that comprise the typical features of NK cells. NKT cells express both αβ-TCR and NK-lineage immune markers, such as CD16 and CD56. The TCR of the NKT cells recognizes the CD1d expressed by antigen-presenting cells (APCs) and contributes to the recognition of lipid antigens. After recognition of the target cell, NKT cells stimulate the innate and adaptive immune responses by producing immune-enhancing cytokines [94]. Therefore, iNKT cells have been considered as a potential target for transduction of the CAR (iNKT-CAR). GD2-specific-iNKT-CARs have shown promising results in preclinical studies on the neuroblastoma mouse model and a phase I clinical trial has been designed to investigate the anti-tumor function of GD2-iNKT-CAR in patients with neuroblastoma (NCT03294954). In conclusion, CAR NK cells have a great potential for clinical application and further research is being inducted to translate the preclinical results of CAR NK therapy to clinical implications.
Challenges and Future Approaches
CAR-modified NK therapy is a recently developed immunotherapeutic method for cancer treatment. Despite promising results of CAR NK therapy in preclinical studies, the clinical application of CAR NK therapy is still in its infancy and is encountered with several challenges. Recognition of the challenges is necessary for reprogramming CAR NK cells and enhance the anti-tumor efficacy of this approach. Here, we aim to discuss current challenges and future approaches to improving the therapeutic outcome of NK therapy in the clinical setting.
CAR NK Transduction Challenges
CAR structure was first designed for transduction to T cells. The structural and functional differences of T and NK cells can influence the efficacy of CAR transduction and function in NK cells. Therefore, it is necessary to specify CAR for NK cells using NK-specific CARs. One of the most important issues in CAR NK production is the strategy of CAR transduction to the NK cells. There are several methods for transferring the CAR genetic sequence to the host NK cell which is described in Table 3. One of the main challenges of CAR NK production is that, in comparison to T cells, the success rate of gene transduction is lower in NK cells. Each method for CAR transduction has its advantages and disadvantages. Choosing the appropriate method for transduction of the CAR is necessary to achieve favorable clinical results. Lentiviral and retroviral transduction methods are the most used approaches. The success rate of CAR transduction by retroviral transfer is 43–93% based on different studies and has recently entered clinical trials. The expression of CAR by retroviral transmission is a short time [95, 96]. However, it has been shown that the addition of the IL-15 membrane-binding site can increase the viability and cytotoxicity of the retrovirally-transduced CAR NK cell [96]. Lentiviral transduction of CAR is another highly used method. The success rate of CAR transduction by lentiviral vector has been reported to be 29% [97]. However, this success rate was increased up to 50% by IL-2 and IL-12 coculturing [98]. The highest efficacy was achieved by using the phytohaemagglutinin (PHA) which increased the success rate of transduction up to 80% [98]. Therefore, it could be concluded that the addition of PHA can tremendously increase the transduction efficacy of lentiviral CAR transfer to the NK cells.
Nucleofection and DNA electroporation has shown diverse transfer success (1–90%) based on NK cell source, the culture media, and the CAR transgene construct [98]. The viability of NK cells after nucleofection and DNA electroporation is 45–97%. Nevertheless, mRNA electroporation has shown an acceptable success rate (80–90%), except in UCB and PB-derived NK cells which were less than 10% [98, 99]. Therefore, mRNA transduction is not proposed to be used in UCB and PB-derived NK cells. In a study, mRNA transduction of CAR to NK cells exhibited higher cytotoxicity aginst CLL than lentiviral-transfected NK cells. An updated approach is using eGFP-mRNA that can improve the transduction efficacy up to 86%. A 3D nanochannel-electroporation (NEP) method which has been recently developed has a 70% success rate in NK cell transduction [99]. Novel vaccinia and adenoviral transduction methods have also been studied for CAR transfection. Using this approach is encountered with two important shortcomings. The first is the phenotype change in target cells which decreases the cytotoxic capabilities of NK cells [100]. And the second obstacle is the lower success rate of CAR transduction [101]. Application of CAR genetic sequence by adenoviral and vaccinia vector does not seem to be a favorable method for CAR NK production and further studies are required for solving transfection obstacles of this approach. The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (CRISPR/Cas9) is another approach that has recently been used for the transduction of CAR to the NK cells. The CRISPR/Cas9 can insert the CAR genetic sequence into the NK genome after delivery to the target cell. Different methods such as an adeno-associated virus (AAV) and electroporation can be used for delivering the CRISPR/Cas9. In this method, AAV is used as a carrier for gene delivery and insertion of the CAR into the NK cell genome which has shown 68% efficacy [102, 103]. Another method for CRISPR/Cas9 insertion in NK cell is electroporation by Lonza 4D nucleofector system that has some advantages such as less toxic effect on the target cell, less rejection by host immunity, and less anti-regulatory effect in the target cell. Using gold nano-particles as the carrier of CRISP/Cas9 to target cells for DNA modification is another successful method [104]. In conclusion, CAR transduction can be optimized based on transferring method and available pieces of equipment.
CAR NK Expansion Challenges
One of the important steps of CAR NK production is the expansion of transduced cells. A large amount of CAR NK cells are required to achieve optimal clinical-scale responses. The low dividing capacity of NK cells is a factor that limits achieving an adequate number of CAR NK cells. The length of the telomere is an important factor that determines the dividing capacity of the dividing cell. NK cell expansion encounters telomer shortening that limits the potential of NK cells to divide. This obstacle can be resolved with the addition of the IL-21 to the culture media [105]. K562 feeder cells are specific cells that are used to improve the proliferation and expansion of different cell lines. A study showed that co-culturing NK cells with IL-15-expressing, 4-1BB ligand, and major histocompatibility complex class I chain-related protein A (MICA) feeder cells resulted in increased proliferation and survival of NK cells [106]. In another study, membrane-bound IL-21 human B-lymphoblastoid cell-line 721.221 (hereinafter, 221)-based artificial feeder (221-mIL-21) cells enhanced the cytotoxicity and expansion capability of NK and CAR-transduced NK cells [107].
Cytokines can be used to enhance the ex-vivo expansion of CAR-transduced NK cells. Infusion of the IL-2 diphtheria toxin (IL-2DT) to the NK culture media increases the expansion of anti-AML haploidentical NK cells [108,109,110]. Moreover, the addition of multiple cytokine cocktails, including IL-2, 15, and 18, and anti-CD3 and CD52 beads, accelerated the expansion of ex-vivo NK cells [108, 111]. Since IL-15 does not stimulate the activation of regulatory T cells, exclusive application of IL-15 without IL-2 has been reported to increase the cytotoxicity and expansion of NK cells [52, 111, 112]. IL-21 is another cytokine that has been shown to enhance the expansion of NK cells, especially when combined with feeder cells [60]. In conclusion, achieving an adequate amount of CAR-transduced NK cells is one of the challenges of adoptive immunotherapy using CAR NK cells. Developing novel ex-vivo expansion methods can enable clinicians to reach an adequate amount of CAR NK cells for clinical application.
Side Effects of CAR NK Therapy
Similar to other treatment methods, CAR-modified NK therapy is accompanied by some side effects. CAR T therapy has shown multiple side effects of which cytokine release syndrome (CRS) is the most frequent and deleterious one. “On-target, off-tumor” toxicity, neurotoxicity, tumor-lysis syndrome (TLS), and Graft-versus-host disease (GvHD) are other important side effects of CAR T therapy [3]. Shorter lifetime and lesser production of cytokines by NK cells are the two major causes that decrease the risk of side effects. Unlike CAR T cells, CAR-modified NK cells include a shorter lifespan and do not invade normal tissues such as the lung and liver. Therefore, CAR NK therapy has shown a favorable safety profile with lower GvHD and CRS; nevertheless, CAR NK cells have been reported to cause GvHD in one study [113]. The most common side effects of CAR NK therapy are fever and fatigue which are caused by increased serum levels of C-reactive protein (CRP) and IL-6. Application of CD33-CAR-NK92 cells in r/r AML caused fever (up to 40 ֯C) and grade I CRS with increased serum levels of IL-10 and 17 [42]. CAR NK therapy has not been reported to cause neurotoxicity and TLS in a different study. The transient reversible hematologic toxic effect has been reported by a study [111]. Consequently, the favorable safety profile encourages the application of CAR NK therapy. Furthermore, the limited life span of CAR NK cells can provide the opportunity for multiple CAR NK infusions which can lead to a better outcome. Despite few side effects, some methods, such as inducible suicide gene, can be applied to increase the safety of CAR-modified NK therapy, which are described in furthur sections.
Improving CAR NK Therapy Safety and Efficacy, With a Focus on Solid Tumors
CAR NK cells have a better safety profile than CAR T cells in case of side effects [39]. However, more preclinical and clinical studies are still required to completely evaluate its potential toxicities. Reviewing the current results on CAR NK therapy can demonstrate the barriers and shortcomings of this approach which can open the way to the development of novel CAR NK constructs with higher cytotoxic capabilities. Furthermore, CAR NK therapy of solid tumors has been less successful compared to hematological malignancies [4]. Several limitations reduce the efficacy of CAR NK therapy in solid tumors. The heterogeneity of antigen expression, barriers limiting the trafficking of CAR NK cells to the solid tumor site, excretion of the matrix metalloproteinases that disrupt the ADCC-mediated tumor killing, hypoxia and metabolic-deficiencies of the TME, and the immunosuppressive TME secondary to secretion of inhibitory cytokines by M2 macroaphages, regulatory T cells, and MDSCs, are examples of CAR NK therapy limitations in solid malignancies [17, 114]. Here, we discuss recent approaches to improve the CAR NK therapy outcome, especially in solid malignancies, and further approaches to increase the safety and efficacy of CAR-modified NK therapy.
Suicide Gene
Suicide genes have been successfully applied to improve CAR T cell safety in different studies [115,116,117]. Identical to CAR T therapy, to increase the safety of CAR NK cell therapy, some studies have suggested using suicide genes. Integration of a suicide gene to CAR NK cell construct can be executed by several methods, such as using the HSV-TK/GCV suicide system and Fas/Caspase9 apoptotic pathway [118]. The suicide gene system can be activated using pharmacological or non-pharmacological methods. In a preclinical report by Liu et. al, CAR NK cells were transfected with a construct including CD19-CAR, inducible caspase-9 (iC9), and a transgene encoding IL-15. After administration of the pharmacologic component, the activation of the iC9 led to the successful elimination of the CAR NK cells [119]. This study demonstrated that suicide gene therapy can be considered an effective method to enhance the safety of CAR-modified NK cells. Future studies are required to investigate the potency of suicide genes for the elimination of engineered NK cells in clinical investigations. Nevertheless, the limited lifespan of CAR NK cells might reduce the need for the application of the suicide genes [120].
Inverted Cytokine Receptor (ICR)/SynNotch CARs
Inverted cytokine receptor (ICR) or Synthetic Notch (SynNotch) receptors are synthetic transmembrane receptors that were first described by Lim et al. [121]. SynNotch receptors are activated when a tissue-specific ligand is linked to the cell surface. The stimulation of the synNotch receptor results in induction of the intracellular transcriptional pathway that leads to the expression of a CAR specific for a target antigen. Therefore, SynNotch receptors can improve the precision of tumor recognition by CAR NK cells and can improve its efficacy [122]. Studies have shown the precise tumor retargeting using synNotch receptor in iPSC-derived CAR T cells [123]. SynNotch receptors can also be combined with other novel methods of CAR-engineering, such as TRUCK T cells. Luo et al. engineered NK-92 cells using a synNotch receptor targeting GPC3 that also included the NFAT for IL-12 secretion (GPC3-Syn-IL12-NK92). In-vivo studies demonstrated that the target-specific expression of CAR and IL-12 by these engineered T cells resulted in target-dependent production of IL-12 and increased the efficacy of CAR T therapy [124]. Thus, synNotch CAR could be introduced as a novel approach to decrease the off-target side effects and improve the safety of CAR NK therapy to be used in further clinical and preclinical studies.
Bispecific CARs
Bispecific CARs contain two different scFvs that can recognize two distinct target antigens on the same CAR structure. The engineered cell is only activated when both antigens are present, and can thus improve its precise tumor recognition. Bispecific CAR engineering has been investigated in CAR T cells. Kloss et al. co-transduced T cells with a bispecific structure that expressed both PSMA and PSCA antigens which successfully eliminated prostate cancer cells that co-expressed both antigens [125]. Other studies have also shown the improved safety and efficacy of bispecific CAR T cells in hematological tumors [76, 126,127,128]. Bispecific cancer retargeting is to be evaluated in CAR NK design. Recently, Li et al. transduced a retroviral bispecific CAR (PD1-DAP10/NKG2D) to the NK-92 cell line. In-vitro and in-vivo treatment of SGC-7901 gastric cancer cells with PD1-DAP10/NKG2D-NK92 cells resulted in strong anti-tumor cytotoxicity and apoptosis of the gastric cancer cells [129]. In conclusion, bispecific CARs can promote the function of the CAR-engineered NK cells by redirecting the dual tumor-specific activity of the engineered NK cells.
TRUCK NK Cells
TRUCKs are engineered T cells that are integrated with a transgenic payload that enables them to secrete proteins and other cellular components upon CAR activation. TRUCKs are specifically engineered to enhance CAR T therapy efficacy against solid malignancies [130]. The most studied immune-enhancing transgene is IL-12 which can reprogram the tumor microenvironment and increase CAR T therapy efficacy [131, 132]. CAR NK cells could also be an appropriate target for transduction of the transgene encoding NK-stimulating cytokines, such as IL-15. In a study, CD19-IL15-iCas9-CAR NK cells were engineered to secrete IL-15 after recognition of the CD19 by the NK cells. The investigation of these cells on a Raji lymphoma murine model resulted in better efficacy and prolonged the survival of the mice. Moreover, the pharmacological activation of iCas9 showed that iCas9 could be applied as a safety switch in CAR-modified NK cells [119]. Therefore, TRUCK NK cells with the potential of immune-stimulatory cytokine secretion can be considered as the next generation of CAR-engineered NK cells to be investigated in future studies.
Armored CAR NK cells
Armored CAR NK cells are engineered NK cells that are designed to co-express other factors such as cytokines and enzymes. These cytokines and enzymes could have immune-stimulating characteristics that increase the function of the NK cells and/or promote the infiltration of the immune cells into the tumor microenvironment [133]. Armored CAR T cells have shown improved tumor infiltration and stronger immune response in recent studies [29, 131]. The promising results of armored CAR T therapy have prompted the development of armored CAR NK cells. Armored CAR NK cells could be engineered in combination with immune checkpoint inhibitory strategies. In a recent study by Rezvani et al., armored cord blood NK cells secreting IL-15 were modified to co-target the cytokine-inducible Src homology 2-containing (CIS) and CD19. CIS is a negative regulator of the IL-15 and an important intracellular immune checkpoint of the NK cells. The CIS protein is produced via its gene named CISH. Utilizing the CRISPER/Cas9 gene-editing system, the CISH gene of the cord blood NK cells was knocked out. This gene edition along with the secretion of the IL-15 led to an improved cytotoxic function and improved the in-vivo persistence of the CAR NK cells [134]. Armored CAR NK cells can produce a wide range of cytokines/proteins/enzymes and thus are a potential tool for the development of newer generations of CAR NK cells with improved anti-tumor efficacy.
Combination Therapy
CAR NK cells have lesser expansion and persistency in comparison to CAR T cells and the long-term anti-tumor efficacy of CAR NK therapy is still a major dilemma. The mixed application of CAR-modified NK cells with other therapeutic methods can improve the long-term anti-tumor effect of CAR NK cell therapy. Combination of CAR NK immunotherapy with lymphodepleting chemotherapy (e.x. cyclophosphamide or Fludarabine) reduces the rejection rate of the injected CAR NK cells, shrinks the tumor size, and decreases MDSCs and T regulatory cells in the tumor. In brief, lymphodepletion chemotherapy decreases TME-induced immunosuppressive effect on immune cells and can improve the outcome of immunotherapy [135, 136]. Using Romidepcin as a combination of chemotherapy with anti-CD20-CAR NK cells has shown favorable therapeutic outcomes in the immunodeficient mice model of Burkitt lymphoma [69]. One of the main barriers of chemotherapeutic agents is the improper distribution of the drug to the tumor location and off-target side effects. CAR NK cells have the potential of functioning as biologic carriers to deliver cytotoxic/biologic drugs to the tumor site and can thus increase the intracellular levels of the chemotherapeutic drug. Siegler et al. engineered CAR NK-92 cells that contained Paclitaxel (PTX)-loaded nanoparticles. These CAR NK cells successfully delivered the chemotherapeutic agent to the tumor and showed promising anti-tumor results. This drug delivery method also reduced the off-target toxicities of the chemotherapeutic drug in Her2/CD19-overexpressing malignant models [137].
One of the major anti-tumor mechanisms of NK cells is their ADCC-mediated anti-tumor effects. In this method, CD16+ NK cells recognize the antibody linked to the tumor cell. This leads to the triggering of the immune system against the malignant cell. Application of antibodies specific for tumor-associated antigen promotes the cytotoxic function of NK cells [138]. The main shortcoming of this method is the downregulation of CD16 by NK cells which occurs due to its cleavage after a while. The possible solution to this obstacle is the application of non-cleavable CD16 on the NK cells. Non-cleavable CD16 is a promoter for ADCC-dependent activation of NK cells in the tumor site. Previous studies have shown that concomitant application of tumor-specific antibodies (such as anti-GD2 monoclonal antibody in neuroblastoma and anti-CD20 antibody in lymphoma) with allogeneic NK cells with non-cleavable CD16 has shown promising synergistic effects in inhibiting tumor growth [69, 139]. FT595 is an iPSC-derived off-the-shelf CAR NK product that has shown to have a multi-antigen targeting function in r/r B cell lymphoma [140]. A phase I clinical trial (NCT04245722) is evaluating the synergistic effects of FT595 with non-cleavable CD16 + NK cells and anti-CD20 monoclonal antibodies (Rituximab and Obinutuzumab) in r/r chronic lymphocytic leukemia and B-cell lymphoma. Another approach to increase the anti-tumor efficacy of immunotherapy using monoclonal antibodies is modifying NK cells to express the Fc receptor of CD16 and CD64. NK-92MI is an IL-2-independent NK92-derived NK cell line that lacks Fc receptor CD16. Therefore, Chen et al. developed third-generation CAR NK-92MI cells that included the Fc receptor for CD16/64. In the presence of rituximab, these cells exhibited an improved tumor lysis against NHL cells [141]. Therefore, modification of NK cells to express CD16/64 enhances the anti-tumor efficacy of immunotherapy using monoclonal antibodies.
Radiotherapy is a conventional palliative/curative treatment for multiple cancers, especially solid tumors. In previous studies, radiotherapy has been shown to have synergistic therapeutic effects in combination with immunotherapeutic methods, such as immune checkpoint inhibition [142]. The possible synergistic mechanisms are the shrinkage of the tumor size along with the higher expression of tumor antigens secondary to DNA damage [143]. The combination of radiotherapy with ACT has been studied. In a glioblastoma model, radiation therapy and NKG2D-CAR T cells synergistically improved the anti-tumor efficacy and results. The DNA damage caused by radiotherapy increased the expression of NKG2D by tumor cells that led to improved activation of the CAR T cells and improved the outcome [144]. In another study on the xenograft triple-negative breast cancer model, ex-vivo expanded NK cells and radiation therapy synergistically led to long-term anti-metastatic and anti-tumor results [145]. In conclusion, considering the synergistic effects of radiotherapy and immunotherapy, the combination of radiation therapy with CAR NK therapy could be proposed as an alternative approach to improve the clinical results in further studies.
Oncolytic viruses have been recently developed and have shown encouraging results in the treatment of cancer [146]. Chen et al. studied the combinatorial results of the oncolytic herpes simplex virus 1 (oHSV-1) and CAR NK-92 cells targeting EGFR on brain metastases of breast cancer. The results demonstrated that the combination of an oncolytic virus with EGFR-CAR-NK92 cells had a higher cytolytic effect and prolonged survival compared to monotherapy with either of them [147, 148].
Immune Checkpoint Inhibition/Modification
Inhibitory immune checkpoints are immune-regulatory receptors that are expressed by immune cells and play a critical role in the immune tolerance mechanism. Some tumors suppress the anti-tumor immune response by overexpressing the inhibitory checkpoints. Multiple inhibitory checkpoint receptors can inhibit the activation of NK cells in the tumor TME, of which B7-H3, TIM3, PD-1, TIGIT, LAG3, and CTLA-4 are the most important ones [149,150,151]. Previous studies have shown that inhibition of these inhibitory checkpoints can improve the CAR T cell and NK-mediated anti-tumor immune response and yield persistent immunity against tumors [12, 152, 153]. The application of anti-immune checkpoint agents such as anti-PD-1, anti-CTLA-4, and anti-TIGIT antibodies is among the latest strategies to improve the anti-tumor function of CAR NK cells. Inhibition of PD-1 in NK cells and CAR T cells can be administrated by systemic/local injection of anti-PD-1 agents and CRISPR/Cas9 engineering of PD-1-knock out CAR T cells [154,155,156]. Another developing strategy for evading the immune suppression of inhibitory checkpoints is targeting the checkpoint using a chimeric costimulatory converting receptor (CCCR). In a study, targeting PD-1 improved the activation of the CAR NK cells by reversing the suppressor effect of PD-1 to activate the CAR NK cell-mediated toxicity. NK-92 cells with PD1-NKG2D-4-1BB CAR exhibited enhanced anti-tumor response against H1299 lung cancer cells by inducing potent pyroptosis in cancer cells [157]. B7-H3 is an immune inhibitory checkpoint that inhibits the activation of NK cells. In a study, lentiviral transduction of the anti-B7-H3 CAR to the NK cells showed to potently inhibit the tumor growth in the non-small cell lung cancer cell line and xenograft model [158].
GD2 is a potential target for the immunotherapy of Ewing sarcoma. In a study, anti-GD2 CAR NK cells showed potent in-vitro tumor-lysis capacity against Ewing sarcoma malignant cells. Nevertheless, these cells were unable to exert their anti-tumor effect in in-vivo studies. The histopathologic investigations demonstrated the upregulation of the immunosuppressive checkpoint, HLA-G, by Ewing sarcoma cells which inhibited the activation of CAR NK cells. The authors introduced HLA-G as an inhibitory immune checkpoint to be studied for immunotherapy in Ewing sarcoma [159]. NKG2A is an inhibitory receptor on NK cells that inhibits the activation of the NK cells upon binding to HLA-E. A study showed that NKG2A-null NK cells had higher anti-tumor function compared to NKG2A+ NK cells since they overcame the immune resistance of the NKG2A [160]. Similarly, CAR NK cells can be engineered to overcome the immune evasion of NKG2A by using anti-NKG2A agents or genetic modification of NK cells. In conclusion, immune checkpoint inhibition/modification using inhibitory receptor blockers, modifying NK cells by CRISPR/Cas9, and redirecting the specificity of the NK cell against an inhibitory checkpoint can be used to increase the efficacy of CAR NK cells, specifically against solid tumors.
Sequential Therapy
CAR NK cells have rapid and potent cytotoxic effects on cancer cells and have a higher safety profile in comparison with CAR T cells [39]. Nevertheless, CAR NK therapy still has some shortcomings. Lower in-vivo persistence and expansion is the main shortcoming of CAR NK therapy. Unlike CAR NK cells, CAR T cells have shown higher expansion and persistence in-vivo. To investigate the anti-tumor effect of the combination of CAR NK and CAR T therapy, Li et al. transduced NK and T cells with a retroviral construct targeting CD19 (αCD19 scFv-CD8αTM-OX40-CD3ζ). The results of the study demonstrated that combining CAR NK and CAR T cell therapy promoted in-vivo persistence of the CD19-CAR NK cells, reduced the production of CRS-associated cytokines, enhanced the efficacy of treatment and mice survival, and prevented tumor relapse for several months [161]. Therefore, using NK cells with T cells sequentially as a therapeutic protocol has been proposed to be more effective than using any of them as monotherapy. Moreover, using NK cells before T cells lead to less neurotoxicity, reduced cytokine release syndrome, durable persistence of CAR T cells, and reduces the burden of the tumor, and can lead to a better outcome [49].
Proteome detector CAR NK cell (TCR-CARs)
NK cell-mediated recognition of the target cell is independent of MHC-I. T cell receptors (TCRs) are T cell-specific cell surface receptors that play a major role in the recognition of MHC-I-expressing target cells by T cells [162]. Engineering NK cells with TCRs enables the recognition of MHCs by NK cells. Producing CAR NK cells with TCRα and TCRβ extracellular domain ables NK cells to detect tumor cells base on their proteome expression on the cell surface. Walseng et al. transduced NK-92 cells with a TCR-CAR that showed to be able to react gainst MHC-I-positive tumor cells [163]. Another study has similarly designed TCR-CD3ζ-NK92 cells and showed that these TCR-redirected NK cells exhibit proper anti-tumor function by targeting MHC-I-expressing cancer cells [164]. In brief, integration of the TCR with CAR NK cells improves the tumor-recognition potential of CAR NK cells and inhibits the tumor escape by malignant cells. Nevertheless, future studies are still necessary to determine the efficacy and safety of TCR-CARs in the clinical setting.
Inhibitory KIRs Silencing
Inhibitory KIRs are NK-surface receptors that restrict the cytotoxic activity of the NK cells after exposure to MHC-I on healthy cells. One of the main mechanisms that reduce GVHD by NK ce lls is inhibitory KIRs, such as KIR2DL1, 2, and 3 [165]. Silencing inhibitory KIRs can lead to enhanced cytotoxic function of the CAR NK cells. One approach to silence inhibitory KIR is using KIR-blocking antibodies. Irilumab is an anti-IgG4 monoclonal antibody that exerts its anti-tumor effects by blocking the inhibitory KIR2D family. Irilumab has been shown to enhance NK-mediated immune response against solid and hematological tumors [166]. In another study, inhibition of inhibitory KIRs using Irilumab in combination with Rituximab has been shown to enhance the anti-tumor efficacy of NK cells in a syngeneic murine model of lymphoma [167]. Therefore, silencing the inhibitory KIRs can lead to improved recognition of the tumor cell and could be considered to be used in combination with CAR NK therapy. Future studies are mandatory to investigate the effects of KIR inhibition on improving the function of CAR NK cells against the tumor.
Converter CARs
“Converter” CARs are capable of converting the undesirable TME to a sensible environment for the immune effector cell. Converter CAR NK cells can convert TME adverse effects to a favorable immune-promoting environment. Converter CARs are produced by fusion of the tumor-promoting ectodomain of a molecule to the immune-stimulating endodomain of another one. For example, combining the IL-4 ectodomain with IL-7 endodomain converts detrimental effects of IL-4 to beneficial effects of IL-7 on immune cells. This approach has been shown to enhance the anti-tumor function of CAR T cells by repressing the immunosuppressive function of the TME in a pancreatic cancer model [168]. Another example is the fusion of the TGF-βRII ectodomain, a T cell-inhibitory ligand, with NKG2D endodomain, which is an activating ligand. The application of this method enhanced the anti-tumor function of NK-92 cells [169]. Therefore, converter CARs possess a promising potential for developing novel generations of CAR NK cells and CAR T cells with the potential of reversing the tumor microenvironment and improving outcomes, especially in solid tumors.
Dual signaling CARs
Despite promising results of CAR NK therapy in several studies, in some cases, the tumor becomes resistant to the treatment. Moreover, off-tumor side effects occur secondary to the expression of the targeted antigen by the healthy tissue [81]. A potential solution to this problem is using “dual” CARs, CARs that have two different ScFvs with one intracellular signaling domain. A study thought to investigate the therapeutic efficacy of CD1D-expressing lymphoma using invariant NK T (iNKT) cells by co-targeting CD19 and CD1d. Results indicated that dual targeting of CD19 and CD1d by CAR-iNKT cells had enhanced cytotoxicity and improved survival [170]. Another study transduced NK-92 cells with dual EGFR-EGFRvIII-CD28-CD3ζ. Dual targeting of glioblastoma using a second-generation EGFR-EGFRvIII-CAR NK cell product overcame the antigen heterogeneity of the tumor and further immune escape and improved the anti-tumor function and survival in murine models of glioblastoma [83]. As a result, dual targeting of tumors using CAR NK cells can be used as a powerful tool to defeat the antigen heterogeneity and tumor escape of the solid tumors.
CRISPR/Cas9
The CRISPR/Cas9 system is a technology that has recently been developed for gene editing with high on-target specificity. It has recently been shown to improve the production of CAR T and NK cells that are resistant to tumor-induced exhaustion [171,172,173,174]. CRISPR/Cas9 can also be used to transduce the CAR genetic construct to the target cell [173]. The CRISPR/Cas9 can be applied to knock in the immune cell-activator genes or to knock out the inhibitory genes. Programmed death-1 (PD-1) is one of the immunosuppressive mediators of the TME that facilitates the progression of tumor growth by inducing the exhaustion of the T cells. In a study, CRISPR/Cas9 was utilized to disrupt the encoding gene for PD-1 in EGFRvIII-CAR T cells. This showed to enhance the inhibition of the glioblastoma cell line [175]. One of the immunosuppressive components of the TME is TGF-β. The receptor for TGF-β is expressed by NK cells [176]. In a study, the authors used CRISPR/Cas9 to knock out the gene encoding this receptor (TGFβR2). This led to an improvement in the resistance of the NK cells to the immunosuppressive microenvironment of the AML [177].
CRISPR/Cas9 has the potential to act on different regions of the genome and thus can be used to eliminate multiple genes. An example is a study that studied the anti-tumor effectiveness of CD19 CAR T cells that were knocked out of three genes, PD-1, B2M, and TRAC. None of the studies reported any change in the phenotype or effector function of the immune cells after CRISPR/Cas9 gene editing [178]. Another potential of CRISPR/Cas9 is the option to delete inhibitory receptors and signaling in NK cells. Moreover, CRISPR/Cas9 can be used to knock in specific activatory genes. The authors would like to propose the investigation of engineering NK cell-inhibitory or activatory signaling using CRISPR/Cas9. Furthermore, the multiplex gene editing potential of CRISPR/Cas9 can be used to knock out multiple inhibitory signals in NK cells which may enhance the function of the NK cells.
CAR-Engineered Stem Cells
Stem cells are a possible source for the production of CAR NK products [179]. However, stem cells can be transfected with the genetic sequence to express the CAR. CAR-engineered stem cell designing is a recently developed method that can subsequently differentiate into CAR NK or CAR T cells. The potential of hematopoietic/progenitor stem cells (HPSCs) to CAR T differentiation has been recently shown against human immunodeficiency virus (HIV) infection [180]. Li et al. transduced iPSCs with a CAR genetic sequence including NKG2D as the transmembrane, CD3ζ, 2B4 as the co-stimulatory domain as the intracellular signaling domain (NK-CAR-iPSC-NK cells). In comparison with iPSC-derived CAR-modified T cells and non-CAR-bearing immune cells, iPSC-derived CAR NK cells exhibited a prolonged survival and improved anti-tumor function in the xenograft model of ovarian cancer [50]. A recent study has reported the significant anti-tumor function of iPSC-derived anti-GPC3 CAR-modified NK/ILC cells and prolonged survival in an ovarian cancer model with no acute systemic toxicity [181]. The self-renewing potential of stem cells might reduce the need for multiple doses of the immunotherapeutic product. Another advantage of CAR-engineered stem cells is their potential to produce large amounts of off-the-shelf and universal CAR NK and CAR T cell products, which can also reduce the costs of immune cell production [182]. Moreover, gene-engineering tools, such as CRISPR/Cas9, can be used for improving the anti-cancer potential of CAR-engineered stem cells. Since these cells are non-differentiated, the genomic alterations will be present in differentiated immune cells [182, 183]. In general, the development of CAR-engineered stem cells has a great potential to produce GMP-grade clinical-scale CAR NK products with lower cost and improved genetic engineering methods.
Trans-Signaling CARs (Inducible CARs)
“Inducible CAR” are a recently-developed generation of CARs that express the CAR after exposure to a specific drug. Inducible CARs are activated in the presence of the specific drug and can thus improve the safety of the immunotherapeutic approach [39]. Inducible CAR T cells have been previously studied and have shown successful expression of CAR by T cells after administration of the drug [184]. Inducible MyD88/CD40 (iMC) is a transcellular protein switch that is activated after exposure to rimiducid. iMC activation contributes to the proliferation and activation of T cells. A study was thought to evaluate the effect of coupled ectopic-IL15/iMC on cytokine secretion and cytotoxicity of anti-CD123/BCMA CAR NK cells. Activation of iMC/IL-15 improved the cytotoxicity and persistence of the CAR NK cells. Moreover, the application of rapamycin-regulated Caspase-9 (iRC9) led to successful pharmacologic elimination of the engineered cells [185]. The results of this study demonstrated that dual-switch CAR NK cells can be modified to exert an improved anti-tumor activity with enhanced efficacy and safety. Inducible CAR NK cells can be proposed as a novel method for improving the efficacy and safety of CAR NK cells in further studies.
Targeting/Stimulation of Activatory NK Receptors
NK cells express activatory receptors, such as activating KIRs, CD16, and NKG2D, that stimulate the activation and function of NK cells upon binding to a specific ligand [186]. Overexpression of NKG2D by CAR NK cells and/or NKG2D-ligand by tumor cells leads to activation of the engineered NK cells and improves NK-mediated cytotoxicity. Histone deacetylase inhibitors have shown to upregulate the expression/secretion of MICA, MICB, and ULBP2, which act as ligands for NKG2D, by the tumor cells [187]. In a study, the application of valproic acid, a histone deacetylase inhibitor, improved NK-mediated killing of hepatocellular carcinoma cells by upregulating the expression of MICA/B [188]. Another method for taking advantage of activatory NK receptors is the engineering of NKG2D-targeting CARs. In a study, mRNA electroporation was used for the transduction of an anti-NKG2D-CAR to NK cells. The results of this study demonstrated that NKG2D-targeting engineered NK cells had potent cytotoxic activity and therapeutic efficacy in preclinical and clinical administration against colorectal cancer [189]. Similar results of NKG2D-targeting CAR NK cells were observed in multiple myeloma [190] and neuroblastoma [190]. Therefore, targeting NKG2D using CAR-engineered NK cells and stimulation of NKG2D using pharmacologic components might be potential methods for developing CAR NK cells with higher anti-tumor efficacy.
Universal CAR NK Cells
NK cells are potent alternative tools for producing off-the-shelf immunotherapeutic products. Universal CARs are chimeric receptors with the potential to target various antigen epitopes. Universal CAR-engineering of T cells has significantly improved the treatment of cancer [25]; however, the development of universal CAR NK cells is still in its infancy. Currently, two studies have reported the feasibility of universal CAR NK production and the efficacy of this approach. The first study targeted various gp160 epitopes by application of anti-gp160 antibodies that were conjugated to 2,4-dinitrophenyl (DNP) as the adaptor section. These universal CAR NK cells exhibited anti-HIV efficacy in HIV-infected cells [191]. The second study used a tumor-specific target module (TM) for binding to the antigen and contained an IgG4 or ScFv-based antibody for targeting GD2-positive malignant cells [192]. In conclusion, the universal CAR structure can be introduced as a potential and flexible platform for the production of off-the-shelf CAR NK cells.
Conclusion
Recent advances and promising results of CAR T therapy have set the pace for the establishment of CAR NK cell therapy. CAR-modified NK cells are a recently-developed adoptive cell therapy platform. The most important advantage of the CAR-engineered NK cell is its suitable safety profile that increases the chances to produce available off-the-shelf CAR NK productions for the immunotherapy of cancer. Despite promising results, CAR NK therapy has encountered some challenges. Novel gene manipulating strategies, such as using CRISPR/Cas9, has improved the engineering of novel CAR NK cells with enhanced function and better safety profile. Moreover, a combination of CAR NK cells with other immunotherapies, such as immune checkpoint inhibition and CAR T therapy, is another approach to improve the persistence of engineered NK cells and improve the therapeutic outcome against solid tumors. Furthermore, novel approaches for modulation of the TME could ameliorate the outcome of using CAR NK therapy against solid tumors. With the development of novel generations of CAR NK cells in preclinical and clinical studies, this method is expected to show revolutionary advances in cancer cell therapy and improve the outcome of patients in the near future.
Data Availability
The data are immediately required upon request by the reviewers.
References
Pearce A, Haas M, Viney R, Pearson S-A, Haywood P, Brown C, et al. Incidence and severity of self-reported chemotherapy side effects in routine care: A prospective cohort study. 2017;12(10):e0184360.
Di Maio, M., Basch, E., & Bryce, J. (2016). Perrone FJNrCo. Patient-reported outcomes in the evaluation of toxicity of anticancer treatments., 13(5), 319–325.
Elahi, R., Khosh, E., Tahmasebi, S., & Esmaeilzadeh, A. (2018). Journal of Frontiers in immunology. Immune cell hacking: Challenges and clinical approaches to create smarter generations of chimeric antigen receptor T cells., 9, 1717.
Tahmasebi, S., Elahi, R., & Esmaeilzadeh, A. (2019). Journal of Stem Cell Reviews and Reports. Solid tumors challenges and new insights of CAR T cell engineering., 15(5), 619–636.
Squibb FB-M. Lisokabtagen-maraleucel vid refraktära eller relapserande B-cells-non-Hodgkins lymfom.
Jo H, Cha B, Kim H, Brito S, Kwak BM, Kim ST, et al. (2021) α-Pinene Enhances the Anticancer Activity of Natural Killer Cells via ERK/AKT Pathway. 22(2):656.
Shin MH, Kim J, Lim SA, Kim J, Kim S-J, Lee K-MJIn (2020) NK cell-based immunotherapies in cancer. 20(2).
Viola D, Dona A, Caserta E, Troadec E, Besi F, McDonald T, et al. (2021) Daratumumab induces mechanisms of immune activation through CD38+ NK cell targeting. 35(1):189–200
Lanier LLJNi (2008) Up on the tightrope: natural killer cell activation and inhibition. 9(5):495–502
Lanier LLJIr (2009) DAP10‐and DAP12‐associated receptors in innate immunity. 227(1):150–60.
Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJJNi. (2003) NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. 4(6):557–64.
Khan M, Arooj S, Wang HJFii (2020) NK cell-based immune checkpoint inhibition. 11.
de Jonge K, Ebering A, Nassiri S, Maby-El Hajjami H, Ouertatani-Sakouhi H, Baumgaertner P, et al. (2019) Circulating CD56 bright NK cells inversely correlate with survival of melanoma patients. 9(1):1–10.
Cichocki F, Cooley S, Davis Z, DeFor TE, Schlums H, Zhang B, et al. (2016) CD56 dim CD57+ NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. 30(2):456–63.
Wu S-Y, Fu T, Jiang Y-Z, Shao Z-MJMC. Natural killer cells in cancer biology and therapy. 2020;19(1):1-26.
Larsen SK, Gao Y, Basse PHJCRiO. (2014) NK cells in the tumor microenvironment. 19(1–2).
Melaiu O, Lucarini V, Cifaldi L, Fruci DJFii (2020) Influence of the tumor microenvironment on NK cell function in solid tumors. 10: 3038.
Ben-Shmuel A, Biber G, Barda-Saad MJFii. Unleashing Natural Killer Cells in the Tumor Microenvironment–The Next Generation of Immunotherapy? 2020;11:275.
Vitale, M., Cantoni, C., Pietra, G., & Mingari, M. C. (2014). Moretta LJEjoi. Effect of tumor cells and tumor microenvironment on NK-cell function., 44(6), 1582–1592.
Benichou, G., Yamada, Y., & Aoyama, A. (2011). Madsen JCJCoiot. Natural killer cells in rejection and tolerance of solid organ allografts., 16(1), 47.
Pontrelli, P., Rascio, F., Castellano, G., Grandaliano, G., & Gesualdo, L. (2020). Stallone GJFiI. The role of natural killer cells in the immune response in kidney transplantation., 11, 1454.
Mantesso S, Geerts D, Spanholtz J, Kučerová LJFiI (2020) Genetic Engineering of Natural Killer Cells for Enhanced Antitumor Function. 11.
Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. (1987) Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. 149(3):960-8.
Esmaeilzadeh A, Tahmasebi S, Athari SS, JB (2020). Chimeric antigen receptor-T cell therapy: Applications and challenges in treatment of allergy and asthma. Pharmacotherapy, 123:109685.
Tahmasebi S, Elahi R, Khosh E, Esmaeilzadeh A, JC, Oncology T. (2020) Programmable and multi-targeted CARs: a new breakthrough in cancer CAR-T cell therapy. :1–17.
Guedan, S., Calderon, H., & Posey, A. D., Jr. (2019). Maus MVJMT-M, Development C. Engineering and design of chimeric antigen receptors., 12, 145–156.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. 2019;12(1):1–13.
Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. 2017;66(11):1425–36.
Yeku OO, Brentjens RJJBST. (2016) Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. 44(2):412-8.
Klingemann HJO. Are natural killer cells superior CAR drivers? 2014;3(4):e28147.
Chen H, Wang F, Zhang P, Zhang Y, Chen Y, Fan X, et al. (2019) Management of cytokine release syndrome related to CAR-T cell therapy. 13(5):610-7.
Giavridis T, van der Stegen SJ, Eyquem J, Hamieh M, Piersigilli A, Sadelain MJNm. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. 2018;24(6):731–8.
Bueno C, Velasco-Hernandez T, Gutiérrez-Agüera F, Zanetti SR, Baroni ML, Sánchez-Martínez D, et al. CD133-directed CAR T-cells for MLL leukemia: on-target, off-tumor myeloablative toxicity. 2019;33(8):2090–125.
Liu J, Zhong JF, Zhang X, Zhang CJJoh, oncology. Allogeneic CD19-CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. 2017;10(1):1–8.
Azoulay E, Darmon M, Valade SJICM. (2020) Acute life-threatening toxicity from CAR T-cell therapy. ;46(9):1723-6.
Santomasso, B., Bachier, C., Westin, J., & Rezvani, K. (2019). Shpall EJJASoCOEB. The other side of CAR T-cell therapy: Cytokine release syndrome, neurologic toxicity, and financial burden., 39, 433–444.
Bonifant, C. L., Jackson, H. J., & Brentjens, R. J. (2016). Curran KJJMT-O. Toxicity and management in CAR T-cell therapy., 3, 16011.
Lin C, Zhang JJ (2018). Reformation in chimeric antigen receptor based cancer immunotherapy: Redirecting natural killer cell. BeBA-RoC, 1869(2):200-15.
Yilmaz A, Cui H, Caligiuri MA, Yu JJJoH (2020) Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. Oncology, 13(1):1-22.
Wang, W., & Jiang, J. (2020). Wu CJCl. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects., 472, 175–180.
Olson, J. A., Leveson-Gower, D. B., Gill, S., Baker, J., Beilhack, A., & Negrin, R. S. J. B. (2010). NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects., 115(21), 4293–4301.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. 2018;8(6):1083.
Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. 2017;8(68):112783.
Glienke, W., Esser, R., Priesner, C., Suerth, J. D., Schambach, A., Wels, W. S., et al. (2015). Advantages and applications of CAR-expressing natural killer cells., 6, 21.
Sun JC, Lanier LLJCSHpib. Is there natural killer cell memory and can it be harnessed by vaccination? NK cell memory and immunization strategies against infectious diseases and cancer. 2018;10(10):a029538.
Kloess S, Oberschmidt O, Dahlke J, Vu X-K, Neudoerfl C, Kloos A, et al. Preclinical Assessment of Suitable Natural Killer Cell Sources for Chimeric Antigen Receptor Natural Killer–Based “Off-the-Shelf” Acute Myeloid Leukemia Immunotherapies. 2019;30(4):381–401.
Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3‐positive B‐ALL and inhibit in vivo leukemia growth. 2019;145(7):1935–45.
Alvarez M, Simonetta F, Baker J, Morrison AR, Wenokur AS, Pierini A, et al. Indirect impact of PD-1/PD-L1 blockade on a murine model of NK cell exhaustion. 2020;11:7.
Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen JJE. CAR-NK cells: A promising cellular immunotherapy for cancer. 2020;59:102975.
Li Y, Hermanson DL, Moriarity BS, Kaufman DSJCsc. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. 2018;23(2):181–92. e5.
Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. 2015;194(7):3201–12.
Imai, C., Iwamoto, S., & Campana, D. J. B. (2005). Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells., 106(1), 376–383.
Rezvani, K., Rouce, R., Liu, E., & Shpall, E. J. M. T. (2017). Engineering natural killer cells for cancer immunotherapy., 25(8), 1769–1781.
Ng YY, Tay JC, Wang SJMT-O. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. 2020;16:75–85.
Zhang J, Zheng H, Diao YJIjoms. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. 2019;20(2):317.
Klingemann H, Boissel L, Toneguzzo FJFii. Natural killer cells for immunotherapy–advantages of the NK-92 cell line over blood NK cells. 2016;7:91.
Li H-K, Hsiao C-W, Yang S-H, Yang H-P, Wu T-S, Lee C-Y, et al. A Novel off-the-Shelf Trastuzumab-Armed NK Cell Therapy (ACE1702) Using Antibody-Cell-Conjugation Technology. 2021;13(11):2724.
Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. 2017;19(2):235–49.
Wang W-n, Zhou G-y, Zhang W-lJI. NK-92 cell, another ideal carrier for chimeric antigen receptor. 2017;9(9):753–65.
Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of human NK cell manufacturing: fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. 2017;28(10):897–913.
Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. 2020;12(568).
Miller J, Cichocki F, Ning J, Bjordahl R, Davis Z, Tuininga K, et al. 155 iPSC-derived NK cells mediate robust anti-tumor activity against glioblastoma. BMJ Specialist Journals; 2020.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. 2020;27(2):224–37. e6.
Morgan MA, Büning H, Sauer M, Schambach AJFiI. Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK Cells. 2020;11:1965.
Klapdor, R., Wang, S., Hacker, U., Büning, H., Morgan, M., Dörk, T., et al. (2017). Improved Killing of Ovarian Cancer Stem Cells by Combining a Novel Chimeric Antigen Receptor-Based Immunotherapy and Chemotherapy., 28(10), 886–896.
Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, et al. CD 19‐CAR engineered NK‐92 cells are sufficient to overcome NK cell resistance in B‐cell malignancies. 2016;20(7):1287–94.
Herrera L, Santos S, Vesga M, Anguita J, Martin-Ruiz I, Carrascosa T, et al. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. 2019;9(1):1–10.
Ravi D, Sarkar S, Purvey S, Passero F, Beheshti A, Chen Y, et al. Interaction kinetics with transcriptomic and secretory responses of CD19-CAR natural killer-cell therapy in CD20 resistant non-hodgkin lymphoma. 2020;34(5):1291–304.
Chu Y, Yahr A, Huang B, Ayello J, Barth M, S. Cairo MJO. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. 2017;6(9):e1341031.
Müller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann H-G, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. 2008;57(3):411–23.
Boissel L, Betancur M, Lu W, Krause D, Van Etten R, Wels W, et al. Retargeting NK-92 cells by means of CD19-and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. 2013;2(10):e26527.
You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. 2019;9(1):64.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. 2014;28(4):917–27.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. 2014;8(2):297–310.
Rafiq S, Purdon TJ, Schultz L, Klingemann H, Brentjens RJJC. NK-92 cells engineered with anti-CD33 chimeric antigen receptors (CAR) for the treatment of Acute Myeloid Leukemia (AML). 2015;17(6):S23.
Zhao S, Han Z, Ji C, An G, Meng H, Yang LJEJoI. The research significance of concomitant use of CAR-CD138-NK and CAR-CD19-NK to target multiple myelomas. 2018;16:2058739218788968.
Seidel, D., Shibina, A., Siebert, N., Wels, W. S., Reynolds, C. P., Huebener, N., et al. (2015). Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma., 64(5), 621–634.
Grote S, Chan KCH, Baden C, Bösmüller H, Sulyok M, Frauenfeld L, et al. CD276 as a novel CAR NK‐92 therapeutic target for neuroblastoma. 2021;4(1):e105.
Han, J., Chu, J., Chan, W. K., Zhang, J., Wang, Y., Cohen, J. B., et al. (2015). CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells., 5(1), 1–13.
Wang J, Lupo KB, Chambers AM, Matosevic SJJfioc. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. 2018;6(1):1–14.
Murakami, T., Nakazawa, T., Natsume, A., Nishimura, F., Nakamura, M., Matsuda, R., et al. (2018). Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells., 38(9), 5049–5056.
Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. 2016;108(5):djv375.
Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. 2016;5(4):e1119354.
Wang J, Torregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, et al. Tumor-responsive, multifunctional CAR-NK cells cooperate with impaired autophagy to infiltrate and target glioblastoma. 2020.
Montagner IM, Penna A, Fracasso G, Carpanese D, Pietà AD, Barbieri V, et al. Anti-PSMA CAR-engineered NK-92 cells: an off-the-shelf cell therapy for prostate cancer. 2020;9(6):1382.
Cao B, Liu M, Wang L, Liang B, Feng Y, Chen X, et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. 2020;524(1):96–102.
Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. 2015;23(2):330–8.
Hu ZJSr. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. 2020;10(1):1–13.
Liu Y, Zhou Y, Huang KH, Fang X, Li Y, Wang F, et al. Targeting epidermal growth factor‐overexpressing triple‐negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. 2020;53(8):e12858.
Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng JJCM, et al. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. 2020;12:3247.
Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. 2018;2018.
Ao X, Yang Y, Li W, Tan Y, Guo W, Ao L, et al. Anti-αFR CAR-engineered NK-92 cells display potent cytotoxicity against αFR-positive ovarian cancer. 2019;42(8):284.
Goodridge, J. P., Mahmood, S., Zhu, H., Gaidarova, S., Blum, R., Bjordahl, R., et al. (2019). FT596: Translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. American Society of Hematology Washington.
Kriegsmann K, Kriegsmann M, von Bergwelt‐Baildon M, Cremer M, Witzens‐Harig MJEjoh. NKT cells—New players in CAR cell immunotherapy? 2018;101(6):750–7.
Streltsova MA, Barsov E, Erokhina SA, Kovalenko EIJJoim. Retroviral gene transfer into primary human NK cells activated by IL-2 and K562 feeder cells expressing membrane-bound IL-21. 2017;450:90–4.
Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. 2014;124(7):1081–8.
Tran, J. (2007). Kung SKJMT. Lentiviral vectors mediate stable and efficient gene delivery into primary murine natural killer cells., 15(7), 1331–1339.
Micucci, F., Zingoni, A., Piccoli, M., Frati, L., Santoni, A., & Galandrini, R. JEh. (2006). High-efficient lentiviral vector-mediated gene transfer into primary human NK cells., 34(10), 1344–1352.
Chang L, Gallego-Perez D, Zhao X, Bertani P, Yang Z, Chiang C-L, et al. Dielectrophoresis-assisted 3D nanoelectroporation for non-viral cell transfection in adoptive immunotherapy. 2015;15(15):3147–53.
Savan, R., Chan, T., & Young, H. A. (2010). Lentiviral gene transduction in human and mouse NK cell lines (pp. 209–221). Springer.
Danthinne, X. (2000). Imperiale MJGt. Production of first generation adenovirus vectors: A review., 7(20), 1707–1714.
Kararoudi MN, Likhite S, Elmas E, Yamamoto K, Schwartz M, Sorathia K, et al. CRISPR-Targeted CAR Gene Insertion Using Cas9/RNP and AAV6 Enhances Anti-AML Activity of Primary NK Cells. 2021.
Kararoudi MN, Likhite S, Elmas E, Schwartz M, Sorathia K, Yamamoto K, et al., editors. CD33 targeting primary CAR-NK cells generated by CRISPR mediated gene insertion show enhanced anti-AML activity. Blood; 2020: AMER SOC HEMATOLOGY 2021 L ST NW, SUITE 900, WASHINGTON, DC 20036 USA.
Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. 2017;1(11):889–901.
Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. 2012;7(1):e30264.
Gong W, Xiao W, Hu M, Weng X, Qian L, Pan X, et al. Ex vivo expansion of natural killer cells with high cytotoxicity by K562 cells modified to co‐express major histocompatibility complex class I chain‐related protein A, 4‐1BB ligand, and interleukin‐15. 2010;76(6):467–75.
Yang, Y., Badeti S,Tseng H-c, Ma MT, Liu T, Jiang J-G, , et al. (2020). Superior expansion and cytotoxicity of human primary NK and CAR-NK cells from various sources via enriched metabolic pathways., 18, 428–445.
Naume B, Gately M, Espevik TJTJoI. A comparative study of IL-12 (cytotoxic lymphocyte maturation factor)-, IL-2-, and IL-7-induced effects on immunomagnetically purified CD56+ NK cells. 1992;148(8):2429–36.
Trinchieri G, Matsumoto-Kobayashi M, Clark S, Seehra J, London L, Perussia BJTJoem. Response of resting human peripheral blood natural killer cells to interleukin 2. 1984;160(4):1147–69.
Martín-Antonio, B., Suñe, G., Perez-Amill, L., & Castella, M. (2017). Urbano-Ispizua AJIjoms. Natural killer cells: Angels and devils for immunotherapy., 18(9), 1868.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. 2020;382(6):545–53.
Wagner J, Pfannenstiel V, Waldmann A, Bergs JW, Brill B, Huenecke S, et al. A two-phase expansion protocol combining interleukin (IL)-15 and IL-21 improves natural killer cell proliferation and cytotoxicity against rhabdomyosarcoma. 2017;8:676.
Shah, N. N., Baird, K., Delbrook, C. P., Fleisher, T. A., Kohler, M. E., Rampertaap, S., et al. (2015). Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood, 125(5), 784–792.
Navin, I., Lam, M. T., & Parihar, R. J. C. (2020). Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment., 12(12), 3871.
Casucci, M. (2011). Bondanza AJJoC. Suicide gene therapy to increase the safety of chimeric antigen receptor-redirected T lymphocytes., 2, 378.
Gargett T, Brown MPJFip. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. 2014;5:235.
Philip, B., Kokalaki, E., Mekkaoui, L., Thomas, S., Straathof, K., Flutter, B., et al. (2014). A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy., 124(8), 1277–1287.
Vassaux, G. (2004). Martin-Duque PJEoobt. Use of suicide genes for cancer gene therapy: Study of the different approaches., 4(4), 519–530.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. 2018;32(2):520–31.
Jones, B. S., Lamb, L. S., & Goldman, F. (2014). Di Stasi AJFip. Improving the safety of cell therapy products by suicide gene transfer., 5, 254.
Morsut, L., Roybal, K. T., Xiong, X., Gordley, R. M., Coyle, S. M., Thomson, M., et al. (2016). Engineering customized cell sensing and response behaviors using synthetic notch receptors., 164(4), 780–791.
Roybal, K. T., Rupp, L. J., Morsut, L., Walker, W. J., McNally, K. A., Park, J. S., et al. (2016). Precision tumor recognition by T cells with combinatorial antigen-sensing circuits., 164(4), 770–779.
Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S-i, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. 2013;12(1):31–6.
Luo H, Wu X, Sun R, Su J, Wang Y, Dong Y, et al. Target-Dependent expression of IL12 by synNotch Receptor-Engineered NK92 cells increases the antitumor activities of CAR-T cells. 2019;9:1448.
Kloss, C. C., Condomines, M., Cartellieri, M., & Bachmann, M. (2013). Sadelain MJNb. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells., 31(1), 71–75.
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. 2020;13:1–10.
Daver, N. J. B. (2020). The Journal of the American Society of Hematology. A bispecific approach to improving CAR T cells in AML., 135(10), 703–704.
Huang C, Zhang Hc, Ho Jy, Liu Rx, Wang L, Kuang N, et al. Dual specific CD19/CD22‑targeted chimeric antigen receptor T‑cell therapy for refractory diffuse large B‑cell lymphoma: A case report. 2020;20(4):1-.
Li M, Zhi L, Yin M, Guo C, Zhang H, Lu C, et al. A novel bispecific chimeric PD1-DAP10/NKG2D receptor augments NK92-cell therapy efficacy for human gastric cancer SGC-7901 cell. 2020;523(3):745–52.
Chmielewski M, Hombach AA, Abken HJIr. Of CAR s and TRUCK s: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. 2014;257(1):83–90.
Yeku, O. O., Purdon, T. J., Koneru, M., & Spriggs, D. (2017). Brentjens RJJSr. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment., 7(1), 1–14.
Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJJJotm. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16 ecto directed chimeric antigen receptors for recurrent ovarian cancer. 2015;13(1):1–11.
Carlsten, M. (2015). Childs RWJFii. Genetic manipulation of NK cells for cancer immunotherapy: Techniques and clinical implications., 6, 266.
Daher, M., Basar, R., Gokdemir, E., Baran, N., Uprety, N., Nunez Cortes, A. K., et al. (2021). Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells., 137(5), 624–636.
Miller, J. S., Soignier, Y., Panoskaltsis-Mortari, A., McNearney, S. A., Yun, G. H., Fautsch, S. K., et al. (2005). Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer., 105(8), 3051–3057.
Restifo, N. P., Dudley, M. E., & Rosenberg, S. A. J. N. R. I. (2012). Adoptive immunotherapy for cancer: Harnessing the T cell response., 12(4), 269–281.
Siegler, E. L., Kim, Y. J., Chen, X., Siriwon, N., Mac, J., Rohrs, J. A., et al. (2017). Combination cancer therapy using chimeric antigen receptor-engineered natural killer cells as drug carriers., 25(12), 2607–2619.
Cheng, M., Chen, Y., Xiao, W., Sun, R., & Tian, Z. J. C. (2013). immunology m. NK cell-based immunotherapy for malignant diseases., 10(3), 230–252.
Modak S, Le Luduec J-B, Cheung IY, Goldman DA, Ostrovnaya I, Doubrovina E, et al. Adoptive immunotherapy with haploidentical natural killer cells and Anti-GD2 monoclonal antibody m3F8 for resistant neuroblastoma: Results of a phase I study. 2018;7(8):e1461305.
Bachanova V, Cayci Z, Lewis D, Maakaron JE, Janakiram M, Bartz A, et al., editors. Initial Clinical Activity of FT596, a First-in-Class, Multi-Antigen Targeted, Off-the-Shelf, iPSC-Derived CD19 CAR NK Cell Therapy in Relapsed/Refractory B-Cell Lymphoma. Blood; 2020: AMER SOC HEMATOLOGY 2021 L ST NW, SUITE 900, WASHINGTON, DC 20036 USA.
Chen Y, You F, Jiang L, Li J, Zhu X, Bao Y, et al. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-ζ or CD64-BB-ζ receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. 2017;8(23):37128.
Theelen WS, Peulen HM, Lalezari F, van der Noort V, De Vries JF, Aerts JG, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non–small cell lung cancer: results of the PEMBRO-RT phase 2 randomized clinical trial. 2019;5(9):1276–82.
Gasser S, Orsulic S, Brown EJ, Raulet DHJN. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. 2005;436(7054):1186–90.
Weiss T, Weller M, Guckenberger M, Sentman CL, Roth PJCr. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. 2018;78(4):1031–43.
Kim, K. W., Jeong, J.-U., Lee, K.-H., Uong, T. N. T., Rhee, J. H., Ahn, S.-J., et al. (2020). Combined NK cell therapy and radiation therapy exhibit long-term therapeutic and antimetastatic effects in a human triple negative breast cancer model., 108(1), 115–125.
Osali A, Zhiani M, Ghaebi M, Meymanat M, Esmaeilzadeh AJPR. Multidirectional Strategies for Targeted Delivery of Oncolytic Viruses by Tumor Infiltrating Immune Cells. 2020:105094.
Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. 2016;7(19):27764.
Kochneva, G., Sivolobova, G., Tkacheva, A., Gorchakov, A., & Kulemzin, S. J. M. B. (2020). Combination of oncolytic virotherapy and car t/nk cell therapy for the treatment of cancer., 54(1), 1–12.
Stojanovic, A. (2018). Cerwenka AJNi. Checkpoint inhibition: NK cells enter the scene., 19(7), 650–652.
Cao, Y., Wang, X., Jin, T., Tian, Y., Dai, C., Widarma, C., et al. (2020). Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy., 5(1), 1–19.
Seo H, Kim B-S, Bae E-A, Min BS, Han YD, Shin SJ, et al. IL21 Therapy Combined with PD-1 and Tim-3 Blockade Provides Enhanced NK Cell Antitumor Activity against MHC Class I–Deficient Tumors. 2018;6(6):685–95.
Grosser, R., Cherkassky, L., Chintala, N., & Adusumilli, P. S. J. C. C. (2019). Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors., 36(5), 471–482.
Beldi-Ferchiou, A. (2017). Caillat-Zucman SJIjoms. Control of NK cell activation by immune checkpoint molecules., 18(10), 2129.
Zhu H, You Y, Shen Z, Shi LJP, Research O. EGFRvIII-CAR-T cells with PD-1 knockout have improved anti-glioma activity. 2020;26(4):2135–41.
Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. 2019;68(3):365–77.
Oyer JL, Gitto SB, Altomare DA, Copik AJJO. PD-L1 blockade enhances anti-tumor efficacy of NK cells. 2018;7(11):e1509819.
Lu C, Guo C, Chen H, Zhang H, Zhi L, Lv T, et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. 2020;122:200–6.
Yang S, Cao B, Zhou G, Zhu L, Wang L, Zhang L, et al. Targeting B7-H3 Immune Checkpoint With Chimeric Antigen Receptor-Engineered Natural Killer Cells Exhibits Potent Cytotoxicity Against Non-Small Cell Lung Cancer. 2020;11.
Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. 2017;6(1):e1250050.
Kamiya T, Seow SV, Wong D, Robinson M, Campana DJTJoci. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. 2019;129(5):2094–106.
Li G, Wu X, Chan IH, Trager JB. A combination of CAR-NK and CAR-T cells results in rapid and persistent anti-tumor efficacy while reducing CAR-T cell mediated cytokine release and T-cell proliferation. American Association for Cancer Research; 2020.
Korenevskii A, Milyutina YP, Zhdanova A, Pyatygina K, Sokolov D, Sel’kov SJBoeb, et al. Mass-spectrometric analysis of proteome of microvesicles produced by NK-92 natural killer cells. 2018;165(4):564–71.
Walseng, E., Köksal, H., Sektioglu, I. M., Fåne, A., Skorstad, G., Kvalheim, G., et al. (2017). A TCR-based chimeric antigen receptor., 7(1), 1–10.
Mensali, N., Dillard, P., Hebeisen, M., Lorenz, S., Theodossiou, T., Myhre, M. R., et al. (2019). NK cells specifically TCR-dressed to kill cancer cells., 40, 106–117.
Mahaweni NM, Ehlers FA, Bos GM, Wieten LJFii. Tuning natural killer cell anti-multiple myeloma reactivity by targeting inhibitory signaling via KIR and NKG2A. 2018;9:2848.
Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. 2018;9(25):17675.
Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. 2014;123(5):678–86.
Mohammed, S., Sukumaran, S., Bajgain, P., Watanabe, N., Heslop, H. E., Rooney, C. M., et al. (2017). Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer., 25(1), 249–258.
Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-βR II and NKG2D. 2017;66(4):537–48.
Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. 2018;34(4):596–610. e11.
Ren J, Zhao YJP, cell. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. 2017;8(9):634–43.
Ureña-Bailén G, Lamsfus-Calle A, Daniel-Moreno A, Raju J, Schlegel P, Seitz C, et al. CRISPR/Cas9 technology: towards a new generation of improved CAR-T cells for anticancer therapies. 2020;19(3):191–200.
Afolabi LO, Adeshakin AO, Sani MM, Bi J, Wan XJI. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. 2019;158(2):63–9.
Rautela J, Surgenor E, Huntington NDJB. Efficient genome editing of human natural killer cells by CRISPR RNP. 2018:406934.
Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, et al. Effect of CRISPR/Cas9-mediated PD-1-disrupted primary human third-generation CAR-T cells targeting EGFRvIII on in vitro human glioblastoma cell growth. 2020;9(4):998.
Slattery K, Gardiner CMJFii. NK cell metabolism and TGFβ–implications for immunotherapy. 2019;10:2915.
Daher M, Basar R, Shaim H, Gokdemir E, Uprety N, Kontoyiannis A, et al. The TGF-β/SMAD signaling pathway as a mediator of NK cell dysfunction and immune evasion in myelodysplastic syndrome. 2017;130(Supplement 1):53-.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. 2017;27(1):154–7.
Knorr, D. A., Ni, Z., Hermanson, D., Hexum, M. K., Bendzick, L., Cooper, L. J., et al. (2013). Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy., 2(4), 274–283.
Zhen, A., Kamata, M., Rezek, V., Rick, J., Levin, B., Kasparian, S., et al. (2015). HIV-specific immunity derived from chimeric antigen receptor-engineered stem cells., 23(8), 1358–1367.
Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non–clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell‐derived anti–glypican‐3 chimeric antigen receptor‐expressing natural killer/innate lymphoid cells. 2020;111(5):1478–90.
Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg K-J, editors. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Seminars in immunopathology; 2019: Springer.
Lee JMJIjoms. When CAR Meets Stem Cells. 2019;20(8):1825.
Drent E, Poels R, Mulders MJ, van de Donk NW, Themeli M, Lokhorst HM, et al. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. 2018;13(5):e0197349.
Wang X, Jasinski DL, Medina JL, Spencer DM, Foster AE, Bayle JHJBa. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. 2020;4(9):1950–64.
Sentman CL, Meehan KRJCj. NKG2D CARs as cell therapy for cancer. 2014;20(2):156.
Bhat J, Dubin S, Dananberg A, Quabius ES, Fritsch J, Dowds CM, et al. Histone deacetylase inhibitor modulates NKG2D receptor expression and memory phenotype of human gamma/delta T cells upon interaction with tumor cells. 2019;10:569.
Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell–mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. 2005;65(14):6321–9.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. 2019;27(6):1114–25.
Leivas, A., Rio, P., Mateos, R., Paciello, M. L., Garcia-Ortiz, A., Fernandez, L., et al. (2018). NKG2D-CAR transduced primary natural killer cells efficiently target multiple myeloma cells. American Society of Hematology Washington.
Lim RM, Rong L, Zhen A, Xie JJACB. A universal CAR-NK cell targeting various epitopes of HIV-1 gp160. 2020;15(8):2299–310.
Mitwasi N, Feldmann A, Arndt C, Koristka S, Berndt N, Jureczek J, et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. 2020;10(1):1–16.
Hu Y, Tian Z-g, Zhang CJAPS. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. 2018;39(2):167–76.
Ziyun AY, editor The Advances and Challenges of CAR-NK Cells for Tumor Immunotherapy. E3S Web of Conferences; 2019: EDP Sciences.
Shaim, H., & Yvon, E. J. C. (2015). Cord blood: A promising source of allogeneic natural killer cells for immunotherapy., 17(1), 1–2.
Veluchamy, J. P., Kok, N., van der Vliet, H. J., Verheul, H. M., & de Gruijl, T. D. (2017). Spanholtz JJFii. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: Recent innovations and future developments., 8, 631.
Altvater, B., Landmeier, S., Pscherer, S., Temme, J., Schweer, K., Kailayangiri, S., et al. (2009). 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clinical Cancer Research, 15(15), 4857–4866.
Liu, S., Galat, V., Galat, Y., Lee, Y. K. A., Wainwright D,Wu JJJoH, , et al. (2021). NK cell-based cancer immunotherapy: From basic biology to clinical development., 14(1), 1–17.
Fang, F., Wang, W., Chen, M., & Tian, Z. (2019). Xiao WJCb, medicine. Technical advances in NK cell-based cellular immunotherapy., 16(4), 647.
Siegler, E. L., Zhu, Y., & Wang, P. (2018). Yang LJCsc. Off-the-shelf CAR-NK cells for cancer immunotherapy., 23(2), 160–161.
Wang, K., Han, Y., & Cho, W. C. (2019). Zhu HJEoobt. The rise of human stem cell-derived natural killer cells for cancer immunotherapy., 19(2), 141–148.
Hu, W., Wang, G., Huang, D., & Sui, M. (2019). Xu YJFii. Cancer immunotherapy based on natural killer cells: Current progress and new opportunities., 10, 1205.
Zenere, G., Olwenyi, O. A., & Byrareddy, S. N. (2019). Braun SEJDdt. Optimizing intracellular signaling domains for CAR NK cells in HIV immunotherapy: A comprehensive review., 24(4), 983–991.
Karakostas P, Panoskaltsis N, Mantalaris A, Georgiadis MCJC, Engineering C. Optimization of CAR T-cell therapies supply chains. 2020;139:106913.
Ye C, Brand D, Zheng SGJSt, therapy t. Targeting IL-2: an unexpected effect in treating immunological diseases. 2018;3(1):1–10.
Matosevic SJJoir. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. 2018;2018.
Pfefferle, A., & Huntington, N. D. J. C. (2020). You have got a fast CAR: Chimeric antigen receptor NK cells in cancer therapy., 12(3), 706.
Hartmann J, Schüßler‐Lenz M, Bondanza A, Buchholz CJJEmm. Clinical development of CAR T cells—challenges and opportunities in translating innovative treatment concepts. 2017;9(9):1183–97.
Wang W-n, Zhou G-y, Zhang W-l. NK-92 cell, another ideal carrier for chimeric antigen receptor. 2017;9(9):753–65.
Papathanasiou MM, Stamatis C, Lakelin M, Farid S, Titchener-Hooker N, Shah NJCgt. Autologous CAR T-cell therapies supply chain: challenges and opportunities? 2020;27(10):799–809.
Rafei H, Daher M, Rezvani K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity.n/a(n/a).
Basar R, Daher M, Rezvani K. Next-generation cell therapies: the emerging role of CAR-NK cells. Hematology 2014, the American Society of Hematology Education Program Book. 2020;2020(1):570–8.
Theoharis S. Current State of the Art of Allogeneic CAR Approaches–Pile ‘Em High and Sell’Em Cheap. Journal of Pharmaceutical Sciences. 2021.
Jamali, A., Hadjati, J., Madjd, Z., Mirzaei, H. R., Thalheimer, F. B., Agarwal, S., et al. (2020). Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Frontiers in immunology., 11, 2028.
Li, L.-L., Yuan, H.-L., Yang, Y.-Q., & Wang, L. (2020). Zou R-CJJoC. A brief review concerning Chimeric Antigen Receptors T cell therapy., 11(18), 5424.
Wang, W., Jiang, J., & Wu, C. (2020). CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer letters., 472, 175–180.
Morgan, M. A., Büning, H., Sauer, M., & Schambach, A. (2020). Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK Cells. Frontiers in Immunology., 11, 1965.
Zah E, Nam E, Bhuvan V, Tran U, Ji BY, Gosliner SB, et al. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. 2020;11(1):1–13.
Karadimitris, A. (2020). Cord blood CAR-NK cells: Favorable initial efficacy and toxicity but durability of clinical responses not yet clear. Cancer Cell, 37(4), 426–427.
Barry, K. C., Hsu, J., Broz, M. L., Cueto, F. J., Binnewies, M., Combes, A. J., et al. (2018). A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nature Medicine., 24(8), 1178–1191.
Fabian KP, Padget MR, Donahue RN, Solocinski K, Robbins Y, Allen CT, et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. Journal for immunotherapy of cancer. 2020;8(1).
Mohanty, R., Chowdhury, C. R., Arega, S., Sen, P., Ganguly, P., & Ganguly, N. (2019). CAR T cell therapy: A new era for cancer treatment. Oncology reports., 42(6), 2183–2195.
Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. 2009;15(15):4857–66.
Shin MH, Kim J, Lim SA, Kim J, Kim S-J, Lee K-M. NK cell-based immunotherapies in cancer. Immune network. 2020;20(2).
Herrera, L., Santos, S., Vesga, M., Anguita, J., Martin-Ruiz, I., Carrascosa, T., et al. (2019). Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Scientific reports., 9(1), 1–10.
Hoogstad-van Evert, J. S., Bekkers, R., Ottevanger, N., Jansen, J. H., Massuger, L., & Dolstra, H. (2020). Harnessing natural killer cells for the treatment of ovarian cancer. Gynecologic oncology., 157(3), 810–816.
Mehta, R. S., Randolph, B., Daher, M., & Rezvani, K. (2018). NK cell therapy for hematologic malignancies. International journal of hematology., 107(3), 262–270.
Schmidt, P., Raftery, M. J., & Pecher, G. (2021). Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. Frontiers in Immunology., 11, 3404.
Ding L, Hu Y, Huang H. Novel progresses of chimeric antigen receptor (CAR) T cell therapy in multiple myeloma. Stem Cell Investigation. 2021;8.
Sheth VS, Gauthier J. Taming the beast: Crs and icans after car t-cell therapy for all. Bone Marrow Transplantation. 2020:1–15.
Gust, J., Ceppi, F., & Turtle, C. J. (2020). neurotoxicities after CAR T-cell immunotherapy (pp. 83–105). Elsevier.
Gutierrez, C., Brown, A. R. T., Herr, M. M., Kadri, S. S., Hill, B., Rajendram, P., et al. (2020). The chimeric antigen receptor-intensive care unit (CAR-ICU) initiative: Surveying intensive care unit practices in the management of CAR T-cell associated toxicities. Journal of critical care., 58, 58–64.
Zhang, H., Zhao, P., & Huang, H. J. E. H. (2020). Oncology. Engineering better chimeric antigen receptor T cells., 9(1), 1–13.
Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann HJLr. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. 2009;33(9):1255–9.
Schmidt P, Raftery MJ, Pecher GJFiI. Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. 2021;11:3404.
Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. (2019) Human CAR NK cells: a new non-viral method allowing high efficient transfection and strong tumor cell killing. 10:957.
Arias J, Yu J, Varshney M, Inzunza J, Nalvarte IJSctm. HSC and iPS cell‐derived CAR‐NK cells as reliable cell‐based therapy solutions. 2021.
Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. 2020;26(11):1686–90.
Sait S, Modak SJEroat. Anti-GD2 immunotherapy for neuroblastoma. 2017;17(10):889–904.
Zhang G, Liu R, Zhu X, Wang L, Ma J, Han H, et al. Retargeting NK‐92 for anti‐melanoma activity by a TCR‐like single‐domain antibody. 2013;91(10):615–24.
Table1 Different sources of NK cells for CAR NK cell production
Acknowledgements
As members of the healthcare system, the authors would like to deeply appreciate all healthworkers around the world who are fighting COVID-19 and hope that all scholars be safe from this pandemic. The rapid vaccination could provide the scientific community with the opportunity to return to cancer-targeted research to develop novel treatments for cancer patients.
Author information
Authors and Affiliations
Contributions
R.E., A.H., and K.H. contributed to the data gathering, preparing the primary manuscript, and designing the tables. R.E. and A.H. contributed to revising the final manuscript. R.E. contributed to the designing of the figures. A.E. contributed to the hypothesis, correspondence, revising the final manuscript, scientific and structural editing, and confirming the manuscript before submission.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent to Publish
Not applicable.
Disclosure of Potential Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be considered as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Elahi, R., Heidary, A.H., Hadiloo, K. et al. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev and Rep 17, 2081–2106 (2021). https://doi.org/10.1007/s12015-021-10246-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-021-10246-3